
Abstract
Molecular genetic testing for congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21-OHD) is offered worldwide and is of importance for differential diagnosis, carrier detection and adequate genetic counseling, particularly for family planning. In 2008 the European Molecular Genetics Quality Network (EMQN) for the first time offered a European-wide external quality assessment scheme for CAH (due to 21-OH deficiency). The interest was great and over the last years at about 60 laboratories from Europe, USA and Australia regularly participated in that scheme. These best practice guidelines were drafted on the basis of the extensive knowledge and experience got from those annually organized CAH-schemes. In order to obtain the widest possible consultation with practicing laboratories the draft was therefore circulated twice by EMQN to all laboratories participating in the EQA-scheme for CAH genotyping and was updated by that input. The present guidelines address quality requirements for diagnostic molecular genetic laboratories, as well as criteria for CYP21A2 genotyping (including carrier-testing and prenatal diagnosis). A key aspect of that article is the use of appropriate methodologies (e.g., sequencing methods, MLPA (multiplex ligation dependent probe amplification), mutation specific assays) and respective limitations and analytical accuracy. Moreover, these guidelines focus on classification of variants, and the interpretation and standardization of the reporting of CYP21A2 genotyping results. In addition, the article provides a comprehensive list of common as well as so far unreported CYP21A2-variants.
Similar content being viewed by others

Introduction—CAH (21-OH deficiency)
Best practice guidelines
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder, caused in more than 90% of cases by variants of CYP21A2 impairing the function of 21-hydroxylase [1,2,3,4]. Since CYP21A2 genotyping is of importance for differential diagnosis, carrier detection and adequate genetic counseling, it is performed worldwide and completes 21-OH deficiency newborn screening offered in many countries. Encouraged by the European Molecular Genetics Quality Network (EMQN) and its European-wide external quality assessment scheme for CAH (due to 21-OH deficiency) best practice guidelines were drafted on the basis of the extensive knowledge and experience got from those annually organized CAH-schemes.
In order to obtain the widest possible consultation with practicing laboratories the draft was therefore circulated twice by EMQN to all laboratories participating in the EQA-scheme for CAH genotyping and was updated by that input. Nevertheless, the authors are aware that due to the wide range of 21-OHD phenotypes, the complex genetic background and different legal obligations, it will be difficult to achieve consensus for all issues addressed in this paper.
In deficiency of 21-hydroxylase (21-OHD), a cytochrome P450 enzyme (P450C21) involved in cortisol biosynthesis, 21-hydroxylation is impaired in the zona fasciculata of the adrenal cortex so that 17α-hydroxyprogesterone (17α-OHP) and progesterone are not converted to 11-deoxycortisol and 11-deoxycorticosterone, respectively. Owing to the resulting diminished plasma concentrations of aldosterone and cortisol, ACTH levels increase, resulting in overproduction and accumulation of cortisol precursors (particularly 17α-OHP), which are finally diverted to androsterone and testosterone [5,6,7]. Variable degrees of prenatal and/or postnatal androgen excess as well as diminished cortisol and aldosterone synthesis reflect the broad clinical range of CAH due to 21-OHD.
From the clinical point of view 21-OHD is categorized into classic (including life threatening salt-wasting as well as simply virilizing), non-classic or so-called cryptic forms of the disease [1,2,3], the latter often not brought to medical attention. Key features of classic 21-OHD in newborns are ambiguous genitalia in females, neonatal salt loss, failure to thrive and potentially fatal hypovolemia and shock [8, 9]. In the following years classical CAH is characterized by rapid postnatal growth, sexual precocity, different signs of hyperandrogenism, reduced fertility and a variety of additional health problems [10,11,12].
In contrast to the classical salt-wasting and simple virilizing forms of 21-OHD, the non-classical forms are milder [13, 14], present without adrenal insufficiency and show variable symptoms of postnatal androgen excess (premature pubarche, hirsutism, acne, menstrual abnormalities, unfulfilled pregnancy) or are without clinical symptoms (cryptic CAH). Recent data suggest a reduced quality of life and cognition in CAH patients due to the chronic illness and excess exposure to androgens and corticosteroids [15, 16]. Thus, early diagnosis and adequate treatment is of major importance for mild as well as severe cases of 21-OHD, particularly for children in order to omit unnecessary suffering and to reduce future health problems in adult life [3, 4, 10, 17, 18].
On the basis of neonatal screening programs [8, 9, 19], which are performed in many countries to avoid life threatening salt wasting crises in newborns, the classical forms of CAH are suggested to occur with an incidence of about 1:16,000 in most populations, with a higher incidence reported in Germany (≈1:12,000 for 2005–2007, National Screening Report, www.screening-dgns.de). Non-classic forms of the disease are assumed to be more common [9, 20] and to occur in 0.1–0.2% of the normal Caucasian population. In that context it is of note that recent findings suggest a higher carrier frequency in the general population (of 15% in a Spanish [20] and of 10% in a middle European [21] population) than previously deduced from neonatal screening programs.
Functional impairment of the 21-hydroxylase is caused by variants affecting the function of the CYP21A2 gene, which is located on chromosome 6p21.33 (Genome assembly GRCh 38). 21-OHD’s broad clinical range reflects the fact that the majority of patients are compound heterozygous for at least two of the more than 200 disease causing CYP21A2 variants identified so far. In general there is a good phenotype–genotype correlation, the milder variant determining the phenotype [22,23,24]. Of note, however, recent data suggest that variants in the promoter as well as in the 3′UTR could influence the phenotype [25, 26]. Comprehensive genotyping is therefore of importance for adequate genetic counseling for further pregnancies, the patients’ offspring and family planning.
CYP21A2 genotyping is error-prone in particular due to the presence of the closely located highly homologous pseudogene CYP21A1P, as well as complex duplications, deletions and rearrangements within chromosome 6p21.3. CYP21A2 genotyping, interpretation of the respective results and adequate genetic counseling of the patient and his/her family members thus require a deep knowledge of CYP21A2 genetics [27,28,29,30,31].
Quality requirements for diagnostic molecular genetic laboratories
-
Diagnostic CYP21A2 genotyping should be performed only by accredited laboratories (ISO 15189 or ISO 17025) or laboratories with implemented laboratory quality management systems (equivalent to ISO 15189) and in accordance with local laws and standards.
-
Most of the methods used for molecular genetic analyses including sequence analysis, MLPA and Southern Blotting are not CE-IVD certified and therefore diagnostic laboratories should fully validate their analytical procedures before implementing the respective method for patient testing. Nevertheless, it is also necessary for CE-IVD-marked reagents to verify their performance before clinical implementation. For laboratories outside of Europe different regulations and requirements may be relevant and necessary.
-
As a minimum, validation should cover an estimate of test accuracy (specificity and sensitivity) and robustness [32]. Test accuracy can be established by analyzing a series of samples of known genotype, and/or by comparison with other laboratories.
-
Written Standard Operating Procedures (SOPs) for the methods used are mandatory.
-
Laboratories should also refer to and be acquainted with the OECD Guidelines for Quality Assurance of Molecular Genetic Testing, 2007, as well as with EMQN reporting and internal quality control guidelines.
-
Annual participation in external quality assessment schemes for CYP21A2 genotyping (EMQN, UKNEQAS, or similar) is essential.
-
A profound knowledge on the molecular background of the disease, the gene structure, and the respective literature is necessary to correctly interprete the results obtained by CYP21A2 genotyping and to provide adequate genetic counseling.
Pre-test requirements and criteria for testing
Test referral, case history, and essential samples
-
Most referrals to the genetic laboratory for CYP21A2-genotyping come from endocrinologists, pediatricians and obstetricians and the respective reasons are listed in Table 1.
Table 1 Reasons for referrals for CYP21A2-genotyping. -
The type of the test required—confirmation of diagnosis in index patient, prenatal diagnosis, identification of carrier, family analysis (compare Fig. 1)—must be clearly defined by the requesting clinician or geneticist in writing. A proper clinical examination and appropriate biochemical work up is desirable so that a brief clinical description and 17-OHP levels as well as family history are available before diagnostic CYP21A2 genotyping is performed.
Fig. 1: Flow chart of CYP21A2 genotyping. 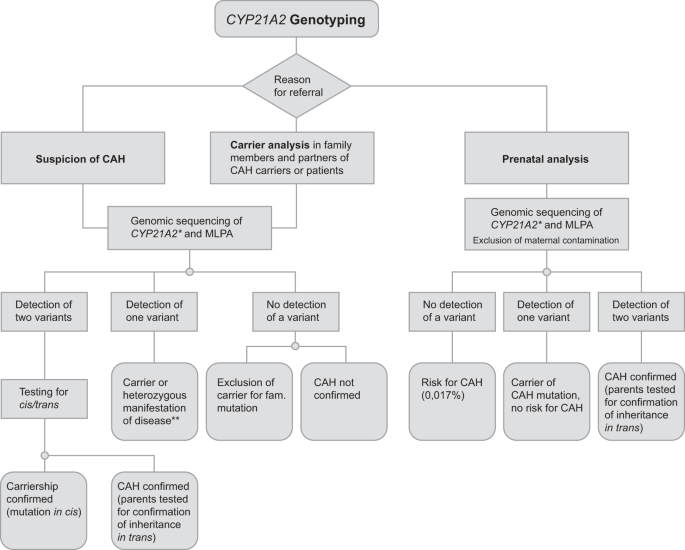
*Best practice genotyping should be PCR-based sequence analysis along with MLPA as addressed in 6.1. of the manuscript. **Second pathogenic variant in trans not detected.
-
It is still controversial whether an ACTH-stimulation test should be performed as the first test for carrier detection in clinically unaffected partners of CAH-patients or suspected carriers. Due to the circumstance that there is an overlap with normal subjects and carriers [33] and that false positive testing has been reported previously [34], 21-OHD genotyping is superior to the stimulation test. However, due to higher costs of the genotyping, the suggested approaches for carrier detection vary and depend on country and region as well as on the respective availability of tetracosactide (active substance of the medicinal product Synacthen®).
-
Since diffuse virilization or elevated levels of different hormones require the exclusion of a hormone producing neoplasm from the clinical side, laboratories could recommend this in parallel to CYP21A2 genotyping.
-
There is broad agreement that parental samples should be requested together with sampling of the index patient (in case he/she is a child) or when reporting (compare Table 2).
Table 2 Analysis of parental samples.

Informed consent and genetic counseling
Policy and procedures concerning initiation of the genotyping, written informed consent, genetic counseling before and after genotyping as well as documentation of genotyping results in different medical records are subject to local practices and national law and have to be handled accordingly. As addressed previously due to the complex genetic background and the wide range of clinical phenotypes CYP21A2 genotyping and genetic counseling should only be offered by experienced laboratories or institutions with a deep understanding thereof.
Significance/scope of CYP21A2 genotyping
Second-tier confirmatory test
Second-tier confirmatory tests of neonatal CAH screening depend on country and/or region and could be both biochemical and molecular genetic approaches. As reported recently, tandem mass spectrometry has proved to be an excellent second-tier test in newborns with positive CAH screening to confirm elevated 17-OHP levels [35,36,37]. On the other hand it is an advantage of the molecular genetic screens [38,39,40,41] that variants affecting 21-hydroxylase function can be detected in DNA extracted from the same dried blood spots that are used for hormonal neonatal screenings. Because more than 90% of disease-causing alleles carry one out of 10 known variants, most 21-OHD cases could be identified by a screening for those 10 most common variants affecting 21-OH function. A relatively easy to handle “strip-test” [40] covering those most common variants as well as a Real-Time-PCR-based assay to detect copy number variations were recently developed, are commercially available and are used in a limited number of institutions in Europe. Of importance, however, the high carrier frequency reported for Europe [21, 42] in a considerable number of samples requires verification of such results by sequencing the whole gene in order to detect other than the most common and known 10 disease causing variants. In contrast to those commercially available test kits, genotyping particularly including sequencing of the whole gene in combination with MLPA or quantitative PCR (for detection of deletions) is more elaborate, concerning both costs and personnel, but would—due to detection of uncommon and new variants as well as duplications and deletions—spare subsequent genotyping necessary for genetic counseling. The latter aspect of detection of uncommon and new variants (not covered by variant specific assays) is particulary important in less well studied populations and in countries with ethnically diverse populations. In general, all negative variant specific sequencing tests should be followed up by expanded testing particularly if there is a positive screening indication or positive family history. It has to be taken into account, however, that now new techniques such as massive parallel sequencing will allow to screen several CAH genes at once in less time with less costs.
Exclusion/confirmation of 21-OHD
Comprehensive CYP21A2 genotyping (not variant specific assays alone) is the best approach to exclude/confirm 21-OHD and 21-OHD-heterozygosity (carrier), because
-
particularly mild clinical symptoms as hyperandrogenism, hirsutism, and acne, or infertility [43,44,45,46,47] show overlap with other diseases (e.g., PCOS),
-
biochemical parameters (e.g., 17-OHP) should have been measured on certain days of the estrous cycle, which in practice turns out to be difficult. Thus, 17-OHP-levels determined in different laboratories (with blood samples from different days of the estrous cycle) show significant differences hampering interpretation.
-
implementation of an ACTH-stimulation test may not be possible in all institutions and depends on availability of Synacthen® (tetracosactide, adrenocorticotropic hormone).
Family planning—genetic counseling
According to the Endocrine Society Clinical Practice Guidelines [3] genetic counseling should be given to parents at birth of a CAH child and to adolescents at the transition to adult care. To provide correct genetic counseling it should be considered that
-
it is only CYP21A2 genotyping that allows the detection and confirmation of the carrier state in parents with a child suffering from 21-OHD.
-
it is of major importance to know whether both parents are carriers or whether a variant affecting 21-OH function in the affected child has occurred de novo.
-
it is indispensable to know the nature of the variants affecting function, which determine the type of CAH from which future children could suffer (salt wasting versus simple virilizing versus nonclassical CAH), since this may influence the parents’ decision for future pregnancies.
-
the nature of the variants affecting 21-OH function is also required for genetic counseling, because they determine whether prenatal diagnosis and prenatal treatment should be considered in further pregnancies.
-
CYP21A2 genotyping is the best method to detect the carrier state in clinically asymptomatic at risk relatives (family analysis) or partners of CAH-patients and of CAH-carriers.
-
in case of in vitro fertilization (IVF): it should be mentioned that it is still a matter of debate whether 21-hydroxylase deficiency genotyping should be considered before IVF and assisted reproductive technology (ART) programs [43, 48, 49].
Prenatal diagnosis (PND) and prenatal therapy (PNT)
Due to the good genotype/phenotype correlation [1, 2, 22, 23] CYP21A2 genotyping of the fetus is predictive for his/her clinical outcome. Therefore, several institutions offer CYP21A2 genotyping in the course of prenatal diagnosis and dexamethasone therapy [50,51,52]. It is, however, important to mention that prenatal therapy is still considered to be an experimental one [3] due to controversial data particularly concerning treatment outcome and maternal as well as fetal safety [53,54,55]. Whereas Miller [53] only recently recommended that “fetal endocrine therapy for congenital adrenal hyperplasia should not be done”, due to the Endocrine Society Clinical Practice Guideline [3] prenatal therapy should be pursued through protocols approved by Institutional Review Boards at centers capable of collecting outcome data, and laboratories should seek for cooperation with such centers in order to provide the best counseling for family planning for such patients and to optimize the therapeutic regimen. Nevertheless in those Guidelines [3] the authors state that prenatal treatment of CAH remains controversial and poses unresolved ethical questions.
In general, most often chorionic villus sampling (CVS) is performed at about 10–12 weeks of gestation (or amniocentesis at 14–16 weeks) followed by determination of sex/karyotype by PCR or cytogenetics (often to exclude other chromosomal abnormalities).
By analysis of free fetal DNA from maternal blood nowadays it is possible to perform fetal sex assignment as early as in the sixth/seventh week of gestation [54,55,56,57]. The latter technique (NIPD; non-invasive prenatal diagnosis) enables sex determination before the start (7th–8th week of gestation) of a potential therapy and helps to minimize the duration of unnecessary dexamethasone treatment in male or unaffected female pregnancies and has become an integral part of prenatal therapy during the last years [52, 58].
NIPD is a rapidly-expanding technique not only for sex determination, but could also be used for detection of CYP21A2 variants using massively parallel sequencing (MPS) of cell-free fetal DNA in maternal plasma. Further development of these techniques will provide new possibilities for the diagnosis of monogenic disorders in utero avoiding the complications of invasive testing by chorionic villus sampling [58, 59].
So far PND has been offered to couples for whom it has previously been proven by diagnostic testing that both partners carry severe disease causing CYP21A2-variants, which would result in severe virilisation of female external genitalia. By genotyping of the parents de novo variants impairing 21-OH function in previous affected children have been ruled out.
In conclusion the requirements for PND are that
-
All steps of PND are performed in accordance with national law and local practice.
-
extensive genetic counseling (including information on risks of dexamethasone therapy for mother and child) by a specialist (medical geneticist, clinical specialist, pediatrician, endocrinologist or equivalent) has been performed,
-
evidence of written informed consent for PND signed by the respective specialist (who did the pre-test genetic counseling) and the parents is provided with the referral, in accordance with local practices and national law,
-
CYP21A2 genotyping is preceded by fetal sex assignment at the earliest time point possible (6th and 7th week of gestation),
-
contamination of the prenatal sample (CVS or amniotic fluid) by maternal tissue is excluded (for example by analysis of polymorphic markers),
-
full gene analysis of CYP21A2 (sequence analysis of all exons and exon/intron boundaries) including copy number detection (MLPA analysis) can be performed, because spontaneous (de novo) variants impairing 21-OH function can occur,
-
absence/presence of CYP21A2 disease causing variants should be confirmed using different techniques (e.g., sequence analysis, MLPA, microsatellite-analysis).
-
It has to be kept in mind that non-paternity is a caveat/limitation for test result interpretation and indication for PND. Thus, if possible, parallel testing of parental samples is recommended.
21-OH genotyping
Nomenclature and gene
The nomenclature of the gene, its localization, transcript and protein is based on specifications by the HUGO Gene Nomenclature Committee (HGNC, genenames.org), Online Mendelian Inheritance in Man (OMIM, omim.org) and Locus Reference Genomic (LRG, lrg-sequence.org). Details are given in Table 3.
General information on gene, disease causing variants, and genotype–phenotype correlation
Gene
The functional CYP21A2 gene and its highly homologous pseudogene CYP21A1P are located in the HLA major histocompatibility complex on chromosome 6p21.33, a highly variable region [60, 61]. Before cloning of the CYP21A genes, prenatal diagnosis was performed by HLA-typing. Moreover, particular forms of 21-OH deficiency, e.g., specific variants or constellations such as the p.(Val281Leu) variant c.841G>T or deletions of the pseudogene are associated with certain HLA haplotypes [62].
Both the functional CYP21A2 gene and the pseudogene form a genetic unit (designated as RCCX module) with several neighboring genes including TNXB. Copy number variations for this RCCX locus have been described, with two modules representing the norm and three or four modules being rare (compare Fig. 2) [63, 64].

The close proximity of CYP21A2 and CYP21A1P as well as copy number variations for this RCCX- locus could result in a deletion of a 30 kb region (including the CYP21A2 gene) due to unequal crossing-over (misalignment of a CYP21A1P pseudogene to the functional CYP21A2 gene) between chromosomes with a monomodular and a standard bimodular form.
Disease causing variants
The most frequent variants impairing 21-OH function are the result of recombination and/or gene conversion events between the functional CYP21A2 gene and its functionally inactive (rendered inactive by variants impairing 21-OH function) pseudogene CYP21A1P [1, 2, 65,66,67]. In a recent review Concolino et al. [68] listed more than 200 CAH causing variants of the CYP21A2 gene. These include the following most common variants affecting 21-OH function representing about 70–75% of CAH alleles listed in Table 4. In that context it has to be considered that uncommon and new variants (not covered by variant specific assays) could occur with higher frequency in less well studied populations and in countries with ethnically diverse populations. Thus, negative variant specific sequencing tests should be followed up by expanded testing particularly if there is a positive screening indication or positive family history.
Variants detected in authors’ departments and so far not published (Table 5) have all been submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar/). A more comprehensive list of known disease-causing and benign variants is given in Table 6.
Large deletions and conversions extending to about 30kb with breakpoints between exons 3 and 8 of CYP21A1P through C4B to the corresponding point in CYP21A2 comprise about 20–30% of CAH alleles [1, 2, 69]. Only a small number of variants affecting 21-OH function (~10%) represent new variants not derived from the pseudogene. CYP21A2 de novo germ-line variantss are estimated to account for about 1–2% of CAH alleles in unexpectedly affected newborns [1, 2, 70]. In case that only one parent of a child (suffering from CAH) carries a disease causing variant, it can be deduced that the child’s second allele has harbored the disease causing variant “de novo”. That means that for children from further pregnancies of that couple there is no significantly increased risk to t should be mentioned that it is still a matter of dbe affected by CAH.
The close proximity of the functional CYP21A2 gene to the pseudogene, as well as copy number variations of the RCCX unit, have been assumed to predispose to de novo gene deletions due to unequal crossing-over between chromosomes with a monomodular and a standard bimodular form (compare Fig. 2) [66, 70], the latter including two complete functional copies of the CYP21A2 gene. It has been suggested that such a bimodular chromosome has no equally-sized homologue to align to during meiosis, resulting in misalignment of a CYP21A1P pseudogene to the functional CYP21A2 gene and causing a deletion of a 30 kb region including the CYP21A2 gene and the 3′ portion of the TNXB gene.
In that context it is of note that in spite of the detection of two CYP21A2 copies, this could be due to a duplicaton of CYP21A2 on one allele, whereas the second allele is lacking a functional CYP21A2 gene, resulting in a CAH-carrier state.
Genotype-phenotype correlation
In general there is a good genotype/phenotype correlation [22, 23] and specific genotypes have been shown to be associated with salt wasting, simple virilizing or non-classical CAH. The genotype/phenotype correlation decreases with diminished severity of the disease [71, 72] or depends on the patient’s background with respect to other genes regulating androgen and oestrogen metabolism. In that context it is of note that Grodnitskaya and Kurtser [73] published only recently that out of 800 women with hyperandrogenism only 1% had non-classical CAH due to 21-OH deficiency. There are also many papers reporting symptomatic patients with clinical signs of hyperandrogenism (hirsutism, precocious puberty, cycle abnormalities in women) and only heterozygous 21-OH function impairing variants and that heterozygotes are more likely to have signs of androgen excess than would genetically-unaffected subjects [74,75,76]. Nevertheless, a recent study [77] showed that out of 205 patients with hyperandrogenemia manifested in adolescence or adulthood the majority (n = 105) were not carriers of 21-OH function impairing variants. Moreover, due to our experience, almost all (heterozygous) parents of classical CAH-patients were clinically asymptomatic or unaware of CAH-related symptoms and were thus identified as 21-OHD-carriers only in the course of family analysis of a 21-OHD index patient. At present it is unclear which conditions lead to “mild” clinical symptoms in heterozygous carriers. As addressed previously [77], benign variants of CYP21A2 (as p.(Asn493Ser) c.1478G>A) could influence the phenotype. Other defects (at a genetic or environmental level) in adrenal and ovarian steroid metabolism and/or metabolic disturbances (e.g., hyperinsulinism, obesity) should be investigated as possible causes for the hyperandrogenism. In these patients, detection of disease causing CYP21A2 variants is only important in the background of family planning.
Another reason for genotype-phenotype variability is the so-called leakiness (e.g., c.290-13C>G) or a different molecular “background” of the respective variant (e.g., p.(Pro30Leu)—small versus large gene conversion – or the circumstance that 3′-and 5′-UTR (untranslated region) are often not analyzed in the course of routine CYP21A2 genotyping [25, 26, 69, 78,79,80,81,82]. This has to be kept in mind, when interpreting and reporting results of 21-OH genotyping.
Methodological approaches—analytical methods
Best practice 21-OHD genotyping
Due to the complex spectrum of disease causing variants (large gene conversions with multiple mutations in cis, deletions and duplications of variable size [83,84,85,86,87,88,89], more than one 21-OH function affecting variant per gene), best practice genotyping should be PCR-based sequence analysis along with MLPA, which would detect the majority of types of potential alterations.
If other methodologies are used, which cover only the most common disease causing variants and/or cannot detect large deletions/duplications the limitations of the employed methods have to be clearly stated in the respective genetic reports.
Nevertheless, we have to be aware, that all methods mentioned so far have limitations and none of those techniques are able to identify 100% of possible variants due to the complexity of the CYP21A2 locus.
In at-risk relatives despite known familial CYP21A2 variants a full screening for CYP21A2 variants (by sequence and MLPA analysis) is preferable, rather than exclusively analyzing the known 21-OH function impairing variant detected in the index patient. Due to a high CYP21A2-carrier rate (as addressed previously in these guidelines [21, 42]) family members may carry other CYP21A2 variants than detected in the index patient and would then escape detection.
Approved/preferred methodologies
PCR-based sequence analysis
Usually PCR-based Sanger sequencing is performed to detect disease causing or clinically relevant single nucleotide substitutions, small deletions and insertions. In brief, one or more fragments covering all 10 exons and the respective exon/intron boundaries are specifically amplified by selective PCR primers differentiating the functional CYP21A2 gene from the CYP21A1P pseudogene followed by Sanger sequencing. So far, only the minority of laboratories sequence parts of the 5′ regulatory and of the 3′ untranslated region.
Of note, PCR based approaches detecting sequence variants, the common 30kb deletion as well as fusion events can also be used for comprehensive CYP21A2 genotyping [89].
Primer design
Since exonic and intronic nucleotide sequences of the pseudogene and the functional gene exhibit 98% sequence homology [61] and are characterized by a high number of SNPs, the design of PCR- and sequencing primers is challenging. A shortlist of different primers used for PCR-based sequencing is provided as Supplementary material.
In principle, allelic dropout has to be considered as for every other gene, since SNPs could lie in primer regions and hamper primer binding. Sequencing overlapping fragments with different primers can help to detect allelic drop out and hence minimize the probability of missing disease causing variants.
Taq-polymerase
The use of proof reading Taq-polymerases is strongly recommended to avoid allelic drop out, as previously reported for the c.290-13C>G locus [90]. Laboratory experience suggests that this phenomenon does not particularly relate to this intronic locus but could be a problem also for other loci.
Promoter and 3′ UTR
Whereas exons and intron/exon boundaries are usually covered by PCR-based sequencing, the majority of the laboratories do not analyze the promoter and the 3′ untranslated region in the course of routine CYP21A2 genotyping. As reported previously [79,80,81,82] promoter variants could result in classic as well as non-classic CAH and/or could modify the phenotype in patients with p.(Pro30Leu) variants [69, 78]. However, due to that lack of information on patients’ data the clinical relevance of promoter sequence variants is unclear and interpretation therefore remains difficult.
In that context it is of note that MLPA covers the c.-113G>A variant in the promoter region with the respective probe for exon 1, but the knowledge on a deletion/conversion in exon 1 (as detected by MLPA) cannot replace for the sequencing of at least a part of the promoter region.
It was reported [26] only recently that the variant c.*13G>A correlates with a mild form of CAH. This variant should therefore be included in routine CYP21A2 genotyping.
Multiplex ligation-dependent probe amplification (MLPA)
A commercially available CYP21A2-MLPA kit is widely used, as this method [29, 91, 92] has the advantage that it is easy to set up and that results should be comparable between laboratories worldwide, although the automatic computer-based plotting is individually done by each lab using different analysis software products. Substitutions of MLPA probes and software updates by the manufacturer require re-evaluation and re-validation of that method (including the used software) regarding IVD (in vitro diagnostic) policies, since the assay is not CE-certified and probe specificity and exons covered by the test may change. Therefore, we strongly encourage the documentation of the MLPA-lot used in the genetic report.
Variant-specific rapid screening strategies
Different methods allowing rapid detection of the most common variants causing CAH are performed by certain laboratories and/or certain countries in the course of stepwise analysis of 21-OH deficiency. Such methods, reviewed in more detail by others [1, 93], include allele-specific oligonucleotide hybridization, amplification-created restriction site and single-stranded conformational analysis, allele-specific oligonucleotide PCR, oligonucleotide arrays, ligase chain reaction or PCR-based minisequencing as well as a commercially available CE-IVD certified Strip assay [40], detecting the most common 21-OH function impairing variants in a time- and cost-effective manner. The latter assay can be used as an independent confirmation for already detected variants by other assays and strategies. Of note, recent EMQN-CAH- schemes revealed that several laboratories using that method did not have sufficient knowledge and experience in interpreting CYP21A2 constellations, as they reported wrong results using that assay. In addition, it was observed by recent EMQN CAH-schemes that disease causing variants as c.898C>T p.(Leu300Phe) were missed by such “CAH-variant-specific” methods. Detection of one disease-causing variant does not exclude the presence of further ones not covered by the respective assay. This has to be mentioned in the respective genetic reports and should also be considered in genetic counseling.
Next generation sequencing
Massive parallel sequencing is expanding quickly and represents a promising tool for future molecular diagnostic approaches. Although already used for 21-OHD genotyping by some laboratories, due to the limited experience a recommendation cannot be given at this point of time. In the course of the 2017 EMQN-CAH scheme two laboratories used NGS and one failed to detect the very common c.515T>A p.(Ile172Asn) variant (in the heterozygous state) in one case, but detected it in the second (in homozygous form). On the basis of the submitted reports of the recent CAH-scheme the assessors strongly encourage to include more information on bioinformatics tools as well as on enrichment kit, etc. in order to get more knowledge on the NGS technique for CYP21A2 analysis. Preliminary data (unpublished observation by the authors) demonstrate difficulties in discriminating localization of variants regarding homologous genes and pseudogenes.
Southern blot analysis (SBA) and quantitative PCR
Originally SBA was used to detect large gene deletions and conversions [1, 2, 93], employing at least two restriction enzymes (Taq I and Bgl II), in order to differentiate large gene deletions from conversions.
Since Southern blot analysis is time-consuming and requires high concentrations and amounts of DNA, in the majority of diagnostic laboratories SBA has been replaced by MLPA or semiquantitative and quantitative real-time PCR methods. Whereas the latter have previously been developed almost exclusively as in house products in different laboratories, in the meantime an IVD-certified kit (ViennaLab) for quantitation of CYP21A copies is commercially available.
Caveats and pitfalls
Controls
Positive controls (with known variants in the respective analyzed exon/intron) should be used for validation of all analyses to ensure that the technique used is appropriate. For predictive testing a close relative carrying the disease causing variant could be analyzed in parallel as internal control.
Duplicated CYP21A2 genes
The wide use of MLPA led to the findings that three or even four functional CYP21A2 genes are present in a certain number of individuals. To avoid false interpretation of positive genotyping it is of utmost importance to be aware of the high frequency of duplicated CYP21A2 genes in association with p.(Gln318*) [28, 42, 94, 95]. In case of detection of a p.(Gln318*) variant, we strongly recommend MLPA analysis in order to clarify the CYP21A2 gene copy number. If three or more CYP21A2 copies are present, PCR with specific primers and/or family analysis has to be performed in order to allocate the disease causing variant to the respective single or duplicated CYP21A2 gene [42]. If two CYP21A2-genes are detected, the p.(Gln318*) bearing allele represents a CAH-allele. If p.(Gln318*) is on an allele carrying a duplicated CYP21A2, it does not represent a CAH allele, but rather a functional normal one.
Currently, the presence of three functional CYP21A2 genes distributed on two alleles are considered to exhibit complete normal function, although proof from the literature is lacking. The observation that the disease causing variant p.(Gln318*) associated with two CYP21A2 gene copies on the same allele or chromosome is more prevalent in the healthy general population than in CAH-patients is in line with the above-mentioned assumption [42, 95].
Of note, the number of functional genes do not only determine the carrier status of a person, but the presence of three or more functional genes could hamper the detection of one mutated allele out of three or four in comparison to one mutated sequence out of two.
In case of duplicated CYP21A2 alleles the risk of no correct assignment of the mutated allele and hence wrong distribution of CYP21A2 genes during meiosis cannot be excluded.
In that context it is of note that in spite of the detection of two CYP21A2 copies, this could be due to a duplicaton of CYP21A2 on one allele, whereas the second allele is lacking a functional CYP21A2 gene, resulting in a CAH-carrier state.
The above mentioned constellations have to be documented and addressed in the respective genetic reports to provide correct genetic counseling.
Pitfalls and false negative results
Pitfalls and false negative results can occur during CYP21A2 analysis and are due to
-
mix up of samples during venipuncture, during DNA isolation or subsequent testing.
-
sequence variations in primer binding regions which could give rise to non-amplification of a normal or disease causing variant carrying allele (“allelic dropout”) [90]; Therefore not amplified CYP21A2 genes are not sequenced and the variant on the other allele appears homozygous instead of heterozygous.
-
homozygosity/hemizygosity for a disease causing variant which has to be verified by a second method (e.g., MLPA)
-
duplication of the functional CYP21A2 gene can mask a deletion on the second allele and can complicate determination of carrier status [28, 69, 94, 95], as outlined above.
-
under certain circumstances hybrid genes can be co-amplified and detected disease causing variants could erroneously be assigned to the functional gene.
-
contamination of fetal with maternal material in prenatal diagnosis.
-
disease causing variant lies in a gene other than the one tested (including double heterozygosity for different genetic entities, e.g., one disease causing variant in CYP21A2 and one in CYP11B1).
Interpretation and reporting
Reporting format and relevant information
While every laboratory has its own reporting format, general guidance on reporting is available from the European Molecular Genetics Quality Network (http://www.emqn.org) on the EMQN website and relevant links including Eurogentest (a project founded in order to harmonize genetic testing across Europe -www.eurogentest.org), the Association for Clinical Genomic Science (www.acgs.uk.com) or the Swiss Society of Medical Genetics (http://sgmg.ch). Some degree of standardization is of pivotal importance for the referring clinicians and for consistency between different laboratories.
The respective recommendations concerning a genetic report and the required informations are:
-
Clear and concise report
-
Individual report issued
-
-
Patient-related information
-
Patient’s name and date of birth, sex, ethnic background
-
Reason for referral
-
Name, address of referring clinician/institution
-
-
Specimen-related information
-
Type of specimen (EDTA-blood, DNA, chorionic villi)
-
Date of referral/arrival noted
-
Laboratory reference/code
-
-
Methods-related information
-
Testing method and respective limitations (error probability) must be included.
-
Clinical sensitivity for full CYP21A2 screening is >95%, analytical sensitivity for sequencing and MLPA is assumed >98%.
-
Both the clinical and analytical accuracy of the assay should be empirically established by each laboratory as part of validation/verification and actually achieved values should be included in the methods.
-
HGVS nomenclature (http://www.hgvs.org) should be used for all variants, and citing the reference sequence on the report—LRG_829t1 (http://www.lrg-sequence.org), NM_000500.9 - is pivotal.
-
Benign variants should not be reported (but can be transmitted upon request).
-
-
Disease-related information
-
OMIM number of the disease and mode of inheritance should be given (www.ncbi.nlm.nih.gov).
-
Detected variants should be related to the different phenotypes (classical CAH, salt wasting, simply virilising, non-classical CAH) [24, 96] and potential risk for prenatal virilisation in females should be addressed.
-
-
Patient and family-related information
-
Availability of carrier screening for relatives and partners should be addressed.
-
Necessity of family analysis should be addressed if applicable (allocation of different disease causing variants to two or more CYP21A2 copies).
-
Risk for children to carry 21-OHD-alleles should be addressed and given.
-
-
In case CAH-index patient is a child.
-
Parents should be genotyped in order to exclude a de novo 21-OH function impairing variant in the affected child and to correctly calculate the risk for the couple’s further children to suffer from CAH (particularly address virilisation of external genitalia in females).
-
Necessity/opportunity of genetic counseling of the patient and her/his partner at a later time point for future children should be mentioned.
-
-
Laboratory related information
-
Name, address of laboratory and its head.
-
Report dated and signed by two suitably qualified persons.
-
-
Recommendations
-
Recommend retesting of an independent blood sample particularly in the case of carrier testing (family planning)
-
Recommend genetic counseling for the index patient
-
Genotype related informations
The CYP21A2 gene harbors benign variants affecting the numbering of nucleotides and codons. Thus, differences in nomenclature existing in the literature and resulting from different reference sequences may cause confusion and make comparison of reports from different laboratories difficult for referring clinicians. Therefore a common reference sequence and standardized nomenclature are desirable. The authors of this manuscript recommend LRG_829t1 (based on NM_000500.9) as a reference sequence.
HGVS nomenclature should be used for all variants including single nucleotide variants, short del/ins and large deletions; for complex variants (insertion–deletions) descriptive explanations can be given in addition.
-
Definitions—explanations
-
In general, the terms “mutation” and “polymorphism” should no longer be used. The term variant is preferred with an explanation of whether the found variants are disease-causing, benign (and so not usually reported) or of uncertain significance.
-
SNP and polymorphisms imply a population allelic frequency of above 1%, but give no indication on pathogenicity.
-
Classification of variants
Variant curation and interpretation is complex and has to be performed in reference to the established guidelines from ACMG (American College of Medical Genetics and Genomics) and ACGS (ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020). In these guidelines only some important aspects can be addressed: Common variants already well known in the literature and being listed in databases e.g., as ClinVar and already classified as “disease causing or pathogenic” do not need further investigation. All other variants, as well as not so common but already included variants in databases, need always a systematic (re-) evaluation for pathogenicity. This can be done by use of so-called search tools as e.g., ClinGen Pathogenicity Calculator (http://calculator.clinicalgenome.org/site/cg-calculator) [97] which interprets the variant on the base of ACMG guidelines (American College of Medical Genetics and Genomics) [98, 99]. The classification is performed in five classes [100]. The systematic interpretation of a variant requires at least
-
the check for frequency in the respective and other populations (dbSNP, 1000 Genomes, ExAC),
-
the search in different databases (for example: Pubmed, HGMD, HGVS, Pharmvar—previously CYP21-alleles, etc.),
-
to perform in silico analysis (for example: MutationTaster, SIFT, PolyPhen-2, SpliceSiteFinder, INNSPLICE, MaxEntScan, etc.),
-
the check of co-segregation with the disease, although this is difficult in recessive disease, and
-
to perform in vitro expression experiments (is not a recommendation, but would bring information about novel variants which would be of general interest).
References
White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–91.
Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776–88.
Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2010;95:4133–60.
Bachelot A, Grouthier V, Courtillot C, Dulon J, Touraine P. 2017 Management of endocrine disease: congenital adrenal hyperplasia due to 21-hydroxylase deficiency: update on the management of adult patients and prenatal treatment. Eur J Endocrinol. 2017;176:R167–81.
Honour JW. 17-Hydroxyprogesterone in children, adolescents and adults. Ann Clin Biochem. 2014;51:424–40. (Pt 4)
Parsa AA, New MI. Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. 2017;165:2–11.
Miller WL. Molecular biology of steroid hormone synthesis. Endocr Rev. 1988;9:295–318.
Therell BL. Newborn screening for congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 2001;30:15–30.
Grosse SD, Van Vliet G. How many deaths can be prevented by newborn screening for congenital adrenal hyperplasia? Horm Res. 2007;67:284–91.
Webb EA, Krone N. Current and novel approaches to children and young people with congenital adrenal hyperplasia and adrenal insufficiency. Best Pract Res Clin Endocrinol Metab. 2015;29:449–68.
Falhammar H, Frisen L, Linden Hirschberg A, Norrby C, Almqvist C, Nordenskjöld A, et al. Increased cardiovascular and metabolic morbidity in patients with 21-hydroxylase deficiency: a Swedisch population-based national cohort study. J Clin Endocrinol Metab. 2015;100:3520–8.
Kim MS, Ryabets-Lienhard A, Dao-Tran A, Mittelman SD, Gilsanz V, Schrager SM, et al. Increased abdominal adiposity in adolescents and young adults with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2015;100:E1153–9.
Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. 2015;50:32–50.
Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxlase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1982;55:817–27.
Kanhere M, Fuqua J, Rink R, Houk C, Mauger D, Lee PA. Psychosexual development and quality of life outcomes in females with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. 2015;2015:21.
Yau M, Vogiatzi M, Lewkowitz-Shpuntoff A, Nimkarn S, Lin-Su K. Health-related quality of life in children with congenital adrenal hyperplasia. Horm Res Paediatr. 2015;84:165–71.
Falhammar H, Wedell A, Nordenström A. Biochemical and genetic diagnosis of 21-hydroxylase deficiency. Endocrine. 2015;50:306–14.
Turcu AF, Auchus RJ. Novel treatment strategies in congenital adrenal hyperplasia. Curr Opin Endocrinol Diabetes Obes. 2016;23:225–32.
Therell BL Jr, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–90.
Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 1985;37:650–67.
Baumgartner- Parzer SM, Nowotny P, Waldhäusl W, Vierhapper H. Carrier frequency of Congenital Adrenal Hyperplasia (21-OH-deficiency) in a middle European population. J Clin Endocrinol Metab. 2004;90:775–8.
Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficieny? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab. 2000;85:1059–65.
Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxlase deficiency. J Clin Investig. 1992;90:584–95.
New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci USA. 2013;110:2611–6.
Araújo RS, Mendonca BB, Barbosa AS, Lin CJ, Marcondes AM, Billerbeck AEC, et al. Microconversion between CYP21A2 and CYP21A1P promoter regions causes the nonclassical form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92/10:4028–34.
Menabo S, Balsamo A, Baldazzi L, Barbaro M, Nicoletti A, Conti V, et al. A sequence variation in 3‵UTR of CYP21A2 gene correlates with a mild form of congenital adrenal hyperplasia. J Endocrinol Invest. 2012;35:298–305.
Day DJ, Speiser PW, Schultz E, Bettendorf M, Fitness J, Barany F, et al. Identification of non-amplifying CYP21-genes when using PCR-based diagnosis of 21-hydroxylase deficiency in congenital adrenal hyperplasia (CAH) affected pedigrees. Hum Mol Genet. 1996;5:2039–48.
Ezquieta B, Beneyto M, Munoz-Pacheco R, Barrio R, Oyarzabal M, Lechuga JL, et al. Gene duplications in 21-hydroxylase deficiency: the importance of accurate molecular diagnosis in carrier detection and prenatal diagnosis. Prenat Diagn. 2006;26:1172–8.
Concolino P, Mello E, Minucci A, Zuppi C, Capoluongo E. Multiplex ligation-dependent probe amplification analysis is useful for diagnosing congenital adrenal hyperplasia but requires a deep knowledge of CYP21A2 genetics. Clin Chem. 2011;57:1079–80.
Tsai LP, Cheng CF, Chuang SH, Lee HH. Analysis of the CYP21A1P pseudogene: indication of mutational diversity and CYP21A2-like and duplicated CYP21A2 genes. Anal Biochem. 2011;413:133–41.
Cantürk C, Baade U, Salazar R, Storm N, Pörtner R, Höppner W. Sequence analysis of CYP21A1P in a German population to aid in the molecular biological diagnosis of congenital adrenal hyperplasia. Clin Chem. 2011;57/3:511–7.
Mattocks CJ, Morris MA, Matthijs G, Swinnen E, Corveleyn A, Dequeker E, et al. A standardized framework for the validation and verification of clinical molecular genetic tests. EuroGentest Valid Group. Eur J Hum Genet. 2010;18:1276–88.
New M, Lorenzen F, Lerner AJ, Kohn B, Oberfield SE, Pollack MS, et al. Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab. 1983;57:320–6.
Ambroziak U, Kępczyńska-Nyk A, Kuryłowicz A, Małunowicz EM, Wójcicka A, Miśkiewicz P, et al. The diagnosis of nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency, based on serum basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol. 2016;84:23–9.
Rauh M, Gröschl M, Rascher W, Dörr HG. Automated, fast and sensitive quantification of 17α-hydroxy-progesterone, androstenedione and testosterone by tandem mass spectrometry with on-line extraction. Steroids. 2006;71:450–8.
Janzen N, Sander S, Terhardt M, Peter M, Sander J. Fast and direct quantification of adrenal steroids by tandem mass spectrometry in serum and dried blood spots. J Chromatogr B Anal Technol Biomed Life Sci. 2008;861:117–22.
Seo JY, Park HD, Kim JW, Oh HJ, Yang JS, Chang YS, et al. Steroid profilinig for congenital adrenal hyperplasia by tandem mass spectrometry as a second-tier test reduces follow-up burdens in a tertiary care hospital: a retrospective and prospective evaluation. J Perinat Med. 2014;42/1:121–7.
Fitness J, Dixit N, Webster D, Torresani T, Pergolizzi R, Speiser PW, et al. Genotyping of CYP21, linked chromosome 6p markers, and a sex-specific gene in neonatal screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1999;84:960–6.
Kösel S, Burggraf S, Fingerhut R, Dörr HG, Roscher AA, Olgemöller B. Rapid second-tier molecular genetic analysis for congenital adrenal hyperplasia attributable to steroid 21-hydroylase deficiency. Clin Chem. 2005;51:298–304.
Nemeth S, Riedl S, Kriegshäuser G, Baumgartner-Parzer S, Concolino P, Neocleous V, et al. Reverse-hybridization assay for rapid detection of common CYP21A2 mutations in dried blood spots from newborns with elevated 17-OH progesterone. Clin Chim Acta. 2012;414:211–4.
Silveira EL, Elnecave RH, dos Santos EP, Moura V, Pinto EM, von der Linden Nader I, et al. Molecular analysis of CYP21A2 can optimize the follow-up of positive results in newborn screening for congenital adrenal hyperplasia. Clin Genet. 2009;76:503–10.
Parajes S, Quinteiro C, Dominguez F, Loidi L. High frequency of copy number variations and sequence variants at CYP21A2 locus: implication for the genetic diagnosis of 21-hydroxylase deficiency. Plos ONE. 2008;3:5.e2138.
Trakakis E, Dracopoulou-Vabouli M, Dacou-Voutetakis C, Basios G, Chrelias C, Kassanos D. Infertility reversed by glucocorticoids and full-term pregnancy in a couple with previously undiagnosed nonclassic congenital adrenal hyperplasia. Fertil Steril. 2011;96/4:1048–50.
Krone N, Wachter I, Stefanidou M, Roscher AA, Schwarz HP. Mothers with congenital adrenal hyperplasia and their children: outcome of pregnancy, birth and childhood. Clin Endocrinol. 2001;55:523–9.
Bidet M, Bellanne-Chantelot C, Galand-Portier M-B, Golmard J-L, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95/3:1182–90.
Wedell A, Thilén A, Ritzén EM, Stengler B, Luthman H. Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: Implications for genetic diagnosis and association with disease manifestation. J Clin Endocrinol Metab. 1994;78:1145–52.
Unluhizarci K, Kula M, Dundar M, Tanriverdi F, Israel S, Colak R, et al. The revalence of non-classic adrenal hyperplasia among Turkish women with hyperandrogenism. Gynecol Endocrinol. 2010;26:139–43.
Ezquieta B, Alonso M, Alvarez E, Arnao DR, Rodriguez A, Siguero PL. Should 21-hydroxylase deficiency genotyping be considered in assisted preproductive technology programs? Fertil Steril. 2007;88:1437.e5.
Baumgartner-Parzer S, Vytiska-Binsdorfer E, Kleinle S, Heinze G, Vierhapper H. Unexplained infertility: increased risk for 21-hydroxylase-deficiency in parents and offspring? Eur J Obstet Gynecol Reprod Biol. 2014;182:258–9.
Yau M, Khattab A, New MI. Prenatal diagnosis of congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 2016;45:267–81.
Speiser PW. Prenatal and neonatal diagnosis and treatment of Congenital Adrenal Hyperplasia. Horm Res. 2007;68(Suppl 5):90–2.
Tardy-Guidollet V, Menassa R, Costa J-M, David M, Bouvattier-Morel C, Baumann C, et al. New management strategy of pregnancies at risk of Congenital Adrenal Hyperplasia using fetal sex determination in maternal serum: French cohort of 258 cases (2002–11). J Clin Endocrinol Metab. 2014;99:1180–8.
Miller WL. Fetal endocrine therapy for congenital adrenal hyperplasia should not be done. Best Pract Res Clin Endocrinol Metab. 2015;29:469–83.
Simpson JL, Rechitsky S. Prenatal genetic testing and treatment for congenital adrenal hyperplasia. Fertil Steril 2019;111:21–3.
Bartha JL, Finning K, Soothill PW. Fetal sex determination from maternal blood at 6 weeks of gestation when at risk for 21-hydroxylase deficiency. Obstet Gynecol. 2003;101:1135–6.
Rijnders RJ, van der Schoot CE, Bossers B, de Vroede MA, Christiaens GC. Fetal sex determination from maternal plasma in pregnancies at risk for congenital adrenal hyperplasia. Obstet Gynecol. 2001;98:374–8.
Tardy V. Management of pregnancies at risk of 21-hydroxylase or 11-hydroxylase deficiency using fetal sex determination in maternal serum proposed in France since 2002: how to limit prenatal exposure to dexamethasone. Horm Res. 2009;70(Suppl 3):348.
Allen S, Young E, Gerrish A. Noninvasive prenatal diagnosis of monogenic disorders. In: Noninvasive Prenatal Testing (NIPT) applied genomics in prenatal screening and diagnosis. 2018; Editors: Lieve P-C and Klein H-G. https://doi.org/10.1016/B978-0-12-814189-2.00009-8.
New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KCA, et al. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014;99:E1022–30.
White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA. 1986;83:5111–5.
Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc Natl Acad Sci USA. 1986;83:2841–5.
Pollack MS, Levine LS, O’Neill GJ, Pang S, Lorenzen F, Kohn B, et al. HLA linkage and B14, DR1, BfS haplotype association with the genes for late onset and cryptic 21-hydroxylase deficiency. Am J Hum Genet. 1981;33:540–50.
Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, et al. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in Caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med. 2000;191:2183–96.
Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular variations of the human major histocompatibility complex class iii genes for serine/threonine kinase rp, complement component c4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX Module). A mechanism for gene deletions and disease associations. J Biol Chem. 1999;274:12147–56.
Higashi Y, Tanae A, Inoue H, Hiromasa T, Fujii-Kuriyama Y. Aberrant splicing and missense mutations cause steroid 21-hydroxylase (P450 (C21)) deficiency in humans: possible gene conversion products. Proc Natl Acad Sci USA. 1988;85:7486–90.
Sinnott P, Collier S, Costigan C, Dyer PA, Harris R, Strachan T. Genesis by meiotic unequal crossover of a de novo deletion that contributes to steroid hydroxylase deficiency. Proc Natl Acad Sci USA. 1990;87:2107–11.
Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2 mutation update: comprehensive analysis of databases and published genetic variants. Hum Mutat. 2018;39:5–22.
Concolino P, Costella A. Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency: a comprehensive focus on 233 pathogenic variants of CYP21A2 gene. Mol Diagn Ther. 2018;22:261–80.
Baumgartner- Parzer SM, Schulze E, Waldhäusl W, Pauschenwein S, Rondot S, Nowotny P, et al. Mutational spectrum of the steroid 21- hydroxylase gene in Austria: Identification of a novel missense mutation. J Clin Endocrinol Metab. 2001;86:4771–5.
Baumgartner-Parzer SM, Nowotny P, Waldhäusl W, Vierhapper H. A rare duplicated 21-OH haplotype and a de novo mutation—a family analysis. J Clin Endocrinol Metab. 2003;88:2794–6.
Wilson RC, Mercado AB, Cheng KC, New MI. Steroid 21-hydroxylase deficiency: genotype may not predict phenotype. J Clin Endocrinol Metab. 1995;80:2322–9.
Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Phenotypic profiling of parents with cryptic nonclassic congenital adrenal hyperplasia: findings in 145 unrelated families. Eur J Endocrinol. 2011;164:977–84.
Grodnitskaya E, Kurtser M. The prevalence of non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Russian women with hyperandrogenism. Hum Fertil. 2017;3:1–6.
Paris F, Tardy V, Chalancon A, Picot MC, Morel Y, Sultan C. Premature pubarche in Mediterranean girls: high prevalence of heterozygous CYP21 mutation carriers. Gynecol Endocrinol. 2010;26:319–24.
Kulle AE, Riepe FG, Hedderich J, Sippell WG, Schmitz J, Niermeyer L, et al. LC-MS/MS based determination of basal- and ACTH-stimulated plasma concentrations of 11 steroid hormones: implications for detecting heterozygote CYP21A2 mutation carriers. Eur J Endocrinol. 2015;173:517–24.
Ghizzoni L, Cappa M, Vottero A, Ubertini G, Carta D, Di Iorgi N, et al. Relationship of CYP21A2 genotype and serum 17-hydroxyprogesterone and cortisol levels in a large cohort of Italian children with premature pubarche. Eur J Endocrinol. 2011;165:307–14.
Skordis N, Shammas C, Phedonos AA, Kyriakou A, Toumba M, Neocleous V, et al. Genetic defects of the CYP21A2 gene in girls with premature adrenarche. J Endocrinol Investig. 2015;38:535–9.
LʼÀllemand D, Tardy V, Grüters A, Schnabel D, Krude H, Morel Y. How a patient homozygous for a 30-kb deletion of the C4-CYP 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2000;85:4562–7.
Zhang HJ, Yang J, Zhang M, Zhang W, Liut J, Wang W, et al. Variations in the promoter of CYP21A2 gene identified in a Chinese patient with simple virilizing form of 21-hydroxlase deficiency. Clin Endocrinol. 2009;70:201–7.
Araújo RS, Billerbeck AEC, Madureira G, Mendonca BB, Bachega TASS. Substitutions in the CYP21A2 promoter explain the simple virilizing form of 21-hydroxylase deficiency in patients harbouring a P30L mutation. Clin Endocrinol. 2005;62:132–6.
Chang SF, Chung B. Difference in transcriptional activity of two homologous CYP21A genes. Mol Endocrinol. 1995;9/10:1330–6.
Chin KK, Chang SF. The -104G nucleotide of the human CYP21 gene is important for CYP21 transcription activity and protein interaction. Nucl Acids Res. 1998;26/8:1959–64.
Concolino P, Mello E, Minucci A, Giardina E, Zuppi C, Toscano V, et al. A new CYP21A1P/CYP21A2 chimeric gene identified in an Italian woman suffering from classical congenital adrenal hyperplasia form. BMC Med Genet. 2009;10:72–7.
Tusié-Luna MT, White PC. Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Proc Natl Acad Sci USA. 1995;92:10796–800.
Lee HH. The chimeric CYP21P/CYP21 gene and 21-hydroxylase deficiency. J Hum Genet. 2004;49:65–72.
Koppens PF, Smeets JH, deWijs IJ, Degenhart JH. Mapping of a de novo uneual crossover causing a deletion of the steroid 21-hydroxylase (CYP1A2) gene and a non-functional hybrid tenascin-X (TNXB) gene. J Med Genet. 2003;40:e53.
Morel Y, Murena M, Nicolino M, Forest MG. Molecular genetics of the congenital adrenal hyperplasia due to 21-hydroxylase deficiency. In: Saez JM, Brownie AC, Capponi A, Chambaz EM, editors. Cellular and molecular biology of the adrenal cortex. 1992;222:123–36.
Wedell A, Luthman H. Steroid 21-hydroxylase (450c21): a new allele and spread of mutations through the pseudogene. Hum Genet. 1993;91:236–40.
Greene CN, Cordovado SK, Turner DP, Keong LM, Shulman D, Mueller PW. Novel method to characterize CYP21A2 in Florida patients with congenital adrenal hyperplasia and commercially available cell lines. Mol Genet Metab Rep. 2014;1:312–23.
Schulze E, Bettendorf M, Maser-Gluth C, Decker M, Schwabe U. Allele-dropout using pcr-based diagnosis for the splicing mutation in intron-2 of the CYP21B-gene: successful amplification with a TAQ/PWO-polymerase mixture. Endocr Res. 1998;24:637–41.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplificiation. Nucleic Acids Res. 2002;30:e57.
Concolino P, Mello E, Toscano V, Ameglio F, Zuppi C, Capoluongo E. Multiplex ligation-dependent probe amplification (MLPA) assay for the detection of CYP21A2 gene deletions/duplications in Congenital Adrenal Hyperplasia: first technical report. Clin Chim Acta. 2009;402:164–70.
Riepe FG, Sippell W. Recent advances in diagnosis, treatment, and outcome of congenital adrenal hyperplasia due to 21-hydroxylase deficiciency. Rev Endocr Metab Disord. 2007;8:349–63.
Wedell A, Stengler B, Luthman H. Characterization of mutations on the rare duplicated C4/CYP21 haplotype in steroid 21-hydroxylase deficiency. Hum Genet. 1994;94:50–4.
Kleinle S, Lang R, Fischer GF, Vierhapper H, Waldhauser F, Födinger M, et al. Duplications of the functional CYP21A2 gene are primarely restricted to Q318X alleles: evidence for a founder effect. J Clin Endocrinol Metab. 2009;94:3954–8.
Gurgov S, Bernabé KJ, Stites J, Cunniff CM, Lin-Su K, Felsen D, et al. Linking the degree of virilization in females with congenital adrenal hyperplasia to genotype. Ann NY Acad Sci. 2017;1402:56–63.
Patel RY, Shah N, Jackson AR, Ghosh R, Pawliczek P, Paithankar S, et al. ClinGen Pathogenicity Calculator: a configurable system for assessing pathogenicity of genetic variants. Genome Med. 2017;9:3.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10:294–300.
Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–91.
Tardy VT, Morel Y. Gene symbol: CYP21A2. Hum Genet. 2007;121/2:292–3.
Toraman B, Ökten A, Kalay E, Karagüzel G, Dinçer T, Açıkgöz EG, et al. Investigation of CYP21A2 mutations in Turkish patients with 21-hydroxylase deficiency and a novel founder mutation. Gene. 2013;513/1:202–8.
Kirac D, Guney AI, Akcay T, Guran T, Ulucan K, Turan S, et al. The frequency and the effects of 21-hydroxylase gene defects in congenital adrenal hyperplasia patients. Ann Hum Genet. 2014;78/6:399–409.
Usui T, Nishisho K, Kaji M, Ikuno N, Yorifuji T, Yasuda T, et al. Three novel mutations in Japanese patients with 21-hydroxylase deficiency. Horm Res. 2004;61/3:126–32.
Baradaran-Heravi A, Vakili R, Robins T, Carlsson J, Ghaemi N, Aʼrabi A, et al. Three novel CYP21A2 mutations and their protein modelling in patients with classical 21-hydroxylase deficiency from northeastern Iran. Clin Endocrinol (Oxf). 2007;67/3:335–41.
Rodrigues NR, Dunham I, Yu CY, Carroll MC, Porter RR, Campbell RD. Molecular characterization of the HLA-linked steroid 21-hydroxylase B gene from an individual with congenital adrenal hyperplasia. EMBO J. 1987;6/6:1653–61.
De Paula Michelatto D, Karlsson L, Lusa AL, Silva CD, Östberg LJ, Persson B, et al. Functional and Structural Consequences of Nine CYP21A2 Mutations Ranging from Very Mild to Severe Effects. Int J Endocrinol. 2016;2016:4209670.
Dolzan V, Stopar-Obreza W, Zerjav-Tansek M, Breskvar K, Krzisnik C, Battelino T. Mutational spectrum of congenital adrenal hyperplasia in Slovenian patients: a novel Ala15Thr mutation and Pro30Leu within a larger gene conversion associated with a severe form of the disease. Europ J Endocrinol. 2003;149/2:137–44.
Kharrat M, Tardy V, MʼRad R, Maazoul F, Jemaa LB, Refaï M, et al. Molecular genetic analysis of Tunisian patients with a classic form of 21-hydroxylase deficiency: identification of four novel mutations and high prevalence of Q318X mutation. Clin Endocrinol Metab. 2004;89/1:368–74.
Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Tardy V, Bilaud L, Laborde K, et al. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab. 2009;94/5:1570–8.
Ezquieta B, Oyarzábal M, Jariego CM, Varela JM, Chueca M. A novel frameshift mutation in the first exon of the 21-OH gene found in homozygosity in an apparently nonconsanguineous family. Horm Res. 1999;51/3:135–41.
Lajic S, Wedell A. An intron 1 splice mutation and a nonsense mutation (W23X) in CYP21 causing severe congenital adrenal hyperplasia. Hum Genet. 1996;98/2:182–4.
Di Pasquale L, Indovina S, Wasniewska M, Mirabelli S, Porcelli P, Rulli I, et al. Novel nonsense mutation (W22X) in CYP21A2 gene causing salt-wasting congenital adrenal hyperplasia in a compound heterozygous girl. J Endocrinol Invest. 2007;30/9:806–7.
Lau IF, Soardi FC, Lemos-Marini SH, Guerra G Jr, Baptista MT, De Mello MP. H28+C insertion in the CYP21 gene: a novel frameshift mutation in a Brazilian patient with the classical form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2001;86/12:5877–80.
Lajic S, Nikoshkov A, Holst M, Wedell A. Effects of missense mutations and deletions on membrane anchoring and enzyme function of human steroid 21-hydroxylase (P450c21). Biochem Biophys Res Commun. 1999;257/2:384–90.
Tusie-Luna MT, Speiser PW, Dumic M, New MI, White PC. A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol Endocrinol. 1991;5/5:685–92.
Vrzalová Z, Hrubá Z, Stʼahlová Hrabincová E, Pouchlá S, Votava F, Kolousková S, et al. Identification of CYP21A2 mutant alleles in Czech patients with 21-hydroxylase deficiency. Int J Mol Med. 2010;26/4:595–603.
Milacic I, Barac M, Milenkovic T, Ugrin M, Klaassen K, Skakic A, et al. Molecular genetic study of congenital adrenal hyperplasia in Serbia: novel p.Leu129Pro and p.Ser165Pro CYP21A2 gene mutations. J Endocrinol Invest. 2015;38/11:1199–210.
Wang R, Yu Y, Ye J, Han L, Qiu W, Zhang H, et al. 21-hydroxylase deficiency-induced congenital adrenal hyperplasia in 230 Chinese patients: Genotype-phenotype correlation and identification of nine novel mutations. Steroids. 2016;108:47–55.
Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, Van Ryzin C, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96/1:E161–72.
Barbaro M, Soardi FC, Östberg LJ, Persson B, de Mello MP, Wedell A, et al. In vitro functional studies of rare CYP21A2 mutations and establishment of an activity gradient for nonclassic mutations improve phenotype predictions in congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2015;82/1:37–44.
Tardy V. Gene symbol: CYP21A2. Disease: steroid 21-hydroxylase deficiency. Hum Genet. 2006;119/3:363.
Marino R, Ramirez P, Galeano J, Perez Garrido N, Rocco C, Ciaccio M, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients: genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2011;75/4:427–35.
Zeng X, Witchel SF, Dobrowolski SF, Moulder PV, Jarvik JW, Telmer CA. Detection and assignment of CYP21 mutations using peptide mass signature genotyping. Mol Genet Metab. 2004;82/1:38–47.
Brønstad I, Breivik L, Methlie P, Wolff AS, Bratland E, Nermoen I, et al. Functional studies of novel CYP21A2 mutations detected in Norwegian patients with congenital adrenal hyperplasia. Endocr Connect. 2014;3/2:67–74.
Loidi L, Quinteiro C, Parajes S, Barreiro J, Lestón DG, Cabezas-Agrícola JM, et al. High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf). 2006;64/3:330–6.
Tardy VT, Morel Y. Gene symbol: CYP21A2. Hum Genet. 2007;121/2:293–4.
Concolino P, Minucci A, Mello E, Zuppi C, Capoluongo E. A new CYP21A2 nonsense mutation causing severe 21-hydroxylase deficiency. Clin Chem Lab Med. 2009;47/7:824–5.
Soardi FC, Barbaro M, Lau IF, Lemos-Marini SH, Baptista MT, Guerra-Junior G, et al. Inhibition of CYP21A2 enzyme activity caused by novel missense mutations identified in Brazilian and Scandinavian patients. J Clin Endocrinol Metab. 2008;93/6:2416–20.
Menassa R, Tardy V, Despert F, Bouvattier-Morel C, Brossier JP, Cartigny M, et al. p.H62L, a rare mutation of the CYP21 gene identified in two forms of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2008;93/5:1901–8.
Ohlsson G, Müller J, Skakkebaek NE, Schwartz M. Steroid 21-hydroxylase deficiency: mutational spectrum in Denmark, three novel mutations, and in vitro expression analysis. Hum Mutat. 1999;13/6:482–6.
Dubey S, Idicula-Thomas S, Anwaruddin M, Saravanan C, Varma RR, Maitra A. A novel 9-bp insertion detected in steroid 21-hydroxylase gene (CYP21A2): prediction of its structural and functional implications by computational methods. J Biomed Sci. 2009;16:3.
Nunez BS, Lobato MN, White PC, Meseguer A. Functional analysis of four CYP21 mutations from spanish patients with congenital adrenal hyperplasia. Biochem Biophys Res Commun. 1999;262/3:635–7.
Krone N, Riepe FG, Grötzinger J, Partsch CJ, Sippell WG. Functional characterization of two novel point mutations in the CYP21 gene causing simple virilizing forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2005;90/1:445–54.
Balraj P, Lim PG, Sidek H, Wu LL, Khoo AS. Mutational characterization of congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Malaysia. J Endocrinol Invest. 2013;36/6:366–74.
Lee HH, Chang SF. Multiple transcripts of the CYP21 gene are generated by the mutation of the splicing donor site in intron 2 from GT to AT in 21-hydroxylase deficiency. J Endocrinol. 2001;171(Dec):397–402.
Friães A, Rêgo AT, Aragüés JM, Moura LF, Mirante A, Mascarenhas MR, et al. CYP21A2 mutations in Portuguese patients with congenital adrenal hyperplasia: identification of two novel mutations and characterization of four different partial gene conversions. Mol Genet Metab. 2006;88/1:58–65.
Rubtsov PM, Igudin EL, Pichugina MIu, Spirin PV, Prasolov VS, Tul´pakov AN. Characterization of new splicing mutation in steroid 21-hydroxylase gene. Bioorg Khim. 2011;37/6:815–20.
Billerbeck AE, Mendonca BB, Pinto EM, Madureira G, Arnhold IJ, Bachega TA. Three novel mutations in CYP21 gene in Brazilian patients with the classical form of 21-hydroxylase deficiency due to a founder effect. J Clin Endocrinol Metab. 2002;87/9:4313–7.
Krone N, Roscher AA, Schwarz HP, Braun A. Comprehensive analytical strategy for mutation screening in 21-hydroxylase deficiency. Clin Chem. 1998;44/10:2075–82.
Wedell A, Ritzén EM, Haglund-Stengler B, Luthman H. Steroid 21-hydroxylase deficiency: three additional mutated alleles and establishment of phenotype-genotype relationships of common mutations. Proc Natl Acad Sci U S A. 1992;89/15:7232–6.
Bruque CD, Delea M, Fernández CS, Orza JV, Taboas M, Buzzalino N, et al. Structure-based activity prediction of CYP21A2 stability variants: A survey of available gene variations. Sci Rep. 2016;6:39082.
White PC, Vitek A, Dupont B, New MI. Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 1988;85/12:4436–40.
Haider S, Islam B, DʼAtri V, Sgobba M, Poojari C, Sun L, et al. Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc Natl Acad Sci USA. 2013;110/7:2605–10.
Concolino P, Vendittelli F, Mello E, Carelli Alinovi C, Minucci A, Carrozza C, et al. Two novel CYP21A2 missense mutations in Italian patients with 21-hydroxylase deficiency: Identification and functional characterisation. IUBMB Life. 2009;61/3:229–35.
Riepe FG, Hiort O, Grötzinger J, Sippell WG, Krone N, Holterhus PM. Functional and structural consequences of a novel point mutation in the CYP21A2 gene causing congenital adrenal hyperplasia: potential relevance of helix C for P450 oxidoreductase-21-hydroxylase interaction. J Clin Endocrinol Metab. 2008;93/7:2891–5.
Massimi A, Malaponti M, Federici L, Vinciguerra D, Manca Bitti ML, Vottero A, et al. Functional and structural analysis of four novel mutaions of CYP21A2 gene in Italian patients with 21-hydroxylase deficiency. Horm Metab Res. 2014;46/7:515–20.
Krone N, Rose IT, Willis DS, Hodson J, Wild SH, Doherty EJ, et al. Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital adreal Hyperplasia Adult Study Executive (CaHASE) cohort. J Clin Endocrinol Metab. 2013;98/2:E346–54.
Taboas M, Gómez Acuña L, Scaia MF, Bruque CD, Buzzalino N, Stivel M, et al. Functional studies of p.R132C, p.R149C, p.M283V, p.E431K and a novel c.652-2A>G mutations of the CYP21A2 gene. PLoS ONE. 2014;9/3:e92181.
Minutolo C, Nadra AD, Fernández C, Taboas M, Buzzalino N, Casali B, et al. Structure-based analysis of five novel disease-causing mutations in 21-hydroxylase-deficient patients. PLoS ONE. 2011;6/1:e15899.
Barbaro M, Soardi FC, de Mello MP, Wedell A, Lajic S. Functional studies of CYP21A2 mutants complement structural and clinical predictions of disease serverity in CAH. Clin Endocrinol (Oxf). 2012;76/5:766–8.
Nermoen I, Brønstad I, Fougner KJ, Svartberg J, Øksnes M, Husebye ES, et al. Genetic, anthropometric and metabolic features of adult Norwegian patients with 21-hydroxylase deficiency. Eur J Endocrinol. 2012;167/4:507–16.
Raisingani M, Contreras MF, Prasad K, Pappas JG, Kluge ML, Shah B, et al. Unusual phenotype of congenital adrenal hyperplasia (CAH) with a novel mutation of the CYP21A2 gene. J Pediatr Endocrinol Metab. 2016;29/7:867–71.
Chu X, Ding H, Cui G, Xu Y, Wang DW, He Y. Functional consequences of a novel point mutation in the CYP21A2 gene identified in a Chinese Han patient with nonclassic 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 2014;80/6:927–8.
Janner M, Pandey AV, Mullis PE, Flück CE. Clinical and biochemical description of a novel CYP21A2 gene mutation 962_963insA using a new 3D model for the P450c21 protein. Eur J Endocrinol. 2006;155/1:143–51.
Hong G, Park HD, Choi R, Jin DK, Kim JH, Ki CS, et al. CYP21A2 mutation in Korean patients with congenital adrenal hyperplasia using complementary methods: sequencing after long-range PCR and restriction fragment length polymorphism analysis with multiple ligation-dependent probe amplification assay. Ann Lab Med. 2015;35/5:535–9.
Robins T, Bellanne-Chantelot C, Barbaro M, Cabrol S, Wedell A, Lajic S. Characterization of novel missense mutations in CYP21 causing congenital adrenal hyperplasia. J Mol Med (Berl). 2007;85/3:247–55.
Tardy V, Menassa R, Sulmont V, Lienhardt-Roussie A, Lecointre C, Brauner R, et al. Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patiens affected with 21-hydroxylase deficiency and in one carrier. J Clin Endocrinol Metab. 2010;95/3:1288–1300.
Grischuk Y, Rubtsov P, Riepe FG, Grötzinger J, Beljelarskaia S, Prassolov V, et al. Four novel missense mutations in the CPY21A2 gene detected in Russian patients suffering from the classical form of congenital adrenal hyperplasia: identification, functional characterization, and structural analysis. J Clin Endocrinol Metab. 2006;91/12:4976–80.
Witchel SF, Smith R, Suda-Hartman M. Identification of CYP21 mutations, one novel, by single strand conformational polymorphism (SSCP) analysis. Mutations in brief no. 218. Online. Hum Mutat. 1999;13/2:172.
Barbaro M, Baldazzi L, Balsamo A, Lajic S, Robins T, Barp L, et al. Functional studies of two novel and two rare mutations in the 21-hydroxylase gene. J Mol Med (Berl). 2006;84/6:521–8.
Amor M, Parker KL, Globerman H, New MI, White PC. Mutation in the CYP21B gene (Ile-172–Asn) causes steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 1988;85/5:1600–4.
Concolino P, Rizza R, Costella A, Carrozza C, Zuppi C, Capoluongo E. CYP21A2 intronic variants causing 21-hydroxylase deficiency. Metabolism. 2017;71:46–51.
Stikkelbroeck NM, Hoefsloot LH, de Wijs IJ, Otten BJ, Hermus AR, Sistermans EA. CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J Clin Endocrinol Metab. 2003;88/8:3852–9.
Khajuria R, Walia R, Bhansali A, Prasad R. The spectrum of CYP21A2 mutations in Congenital Adrenal Hyperplasia in an Indian cohort. Clin Chim Acta. 2017;464:189–94.
Higashi Y, Fujii-Kuriyama Y. Functional analysis of mutant P450(C21) genes in COS cell expression system. Methods Enzymol. 1991;206:166–73.
Concolino P, Mello E, Patrosso MC, Penco S, Zuppi C, Capoluongo E. p.H282N and p.Y191H: 2 novel CPY21A2 mutations in Italian congenital adrenal hyperplasia patients. Metabolism. 2012;61/4:519–24.
Nikoshkov A, Lajic S, Vlamis-Gardikas A, Tranebjaerg L, Holst M, Wedell A, et al. Naturally occuring mutants of human steroid 21-hydroxylase (P450c21) pinpoint residues important for enzyme activity and stability. J Biol Chem. 1998;273/11:6163–5.
Niceta M, Bono M, Fabiano C, Pojero F, Niceta F, Sammarco P, et al. A large view of CYP21 locus among Sicilians and other populations: identification of a novel CYP21A2 variant in Sicily. J Endocrinol Invest. 2011;34/11:847–54.
Speiser PW, New MI, White PC. Molecular genetic analysis of nonclassic steriod 21-hydroxylase deficiency associated with HLA-B14,DR1. N Engl J Med. 1988;319/1:19–23.
Girgis R, Ajamian F, Metcalfe P. A previously undescribed mutation detected by sequence analysis of CYP21A2 gene in an infant with salt wasting congenital adrenal hyperplasia. J Pediatr Endocrinol Metab. 2013;26/7-8:765–6.
Concolino P, Santonocito C, Minucci A, Carrozza C, Zuppi C, Capoluongo E, et al. Novel human pathological mutations. Gene Symbol: CYP21A2. Disease: Non-classic 21-hydroxylase deficiency. Hum Genet. 2007;122/5:559.
Krone N, Riepe FG, Partsch CJ, Vorhoff W, Brämswig J, Sippell WG. Three novel point mutations of the CYP21 gene detected in classical forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Exp Clin Endocrinol Diabetes. 2006;114/3:111–7.
Ezquieta B, Cueva E, Oyarzábal M, Oliver A, Varela JM, Jariego C. Gene conversion (655G splicing mutation) and the founder effect (Gln318Stop) contribute to the most frequent severe point mutations in congenital adrenal hyperplasia (21-hydroxylase deficiency) in the Spanish population. Clin Genet. 2002;62/2:181–8.
Falhammar H, Thorén M, Hagenfeldt K. A 31-year-old woman with infertility and polycystic ovaries diagnosed with non-classic congenital adrenal hyperplasia due to anovel CYP21 mutation. J Endocrinol Invest. 2008;31/2:176–80.
Robins T, Barbaro M, Lajic S, Wedell A. Not all amino acid substitutions of the common cluster E6 mutation in CYP21 cause congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2005;90/4:2148–53.
Concolino P, Mello E, Zuppi C, Toscano V, Capoluongo E. CYP21A2 p.E238 deletion as result of multiple microconversion events: a genetic study on an Italian congenital adrenal hyperplasia (CAH) family. Diagn Mol Pathol. 2013;22/1:48–51.
Concolino P, Mello E, Zuppi C, Capoluongo E. Molecular diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency: an update of new CYP21A2 mutations. Clin Chem Lab Med. 2010;48/8:1057–62.
Loke KY, Lee YS, Lee WW, Poh LK. Molecular analysis of CYP-21 mutations for congenital adrenal hyperplasia in Singapore. Horm Res. 2001;55/4:179–84.
Bleicken C, Loidi L, Dhir V, Parajes C, Quinteiro C, Dominguez F, et al. Functional characterization of three CYP21A2 sequence variants (p.A265V, p.W302S, p.D322G) employing a yeast co-expression system. Hum Mutat. 2009;30/2:E443–50.
Asanuma A, Ohura T, Ogawa E, Sato S, Igarashi Y, Matsubara Y, et al. Molecular analysis of Japanese patients with steroid 21-hydroxylase deficiency. J Hum Genet. 1999;44/5:312–7.
Wu DA, Chung BC. Mutations of P450c21 (steriod 21-hydroxylase) at Cys428, Val281, and Ser268 result in complete, partial or no loss of enzymatic activity, respectively. J Clin Invest. 1991;88/2:519–23.
Barbat B, Bogyo A, Raux-Demay MC, Kuttenn F, Boué J, Simon-Bouy B, et al. Screening of CYP21 gene mutations in 129 French patients affected by steroid 21-hydroxylase deficiency. Hum Mutat. 1995;5/2:126–30.
Speiser PW, New MI, White PC. Molecular genetic analysis of nonclassic steriod 21-hydroxylase deficiency associated with HLA-B14,DR1. N Engl J Med. 1988;319/1:19–23.
Ezquieta B, Cueva E, Varela J, Oliver A, Fernández J, Jariego C. Non-classical 21-hydroxylase deficiency in children: association of adrenocorticotropic hormone-stimulated 17-hydroxyprogesterone with the risk of compound heterozygosity with severe mutations. Acta Paediatr. 2002;91/8:892–8.
de Carvalho DF, Miranda MC, Gomes LG, Madureira G, Marcondes JA, Billerbeck AE, et al. Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur J Endocrinol. 2016;175/2:107–16.
Bleicken C, Loidi L, Dhir V, Parajes S, Quinteiro C, Dominguez F, et al. Functional characterization of three CYP21A2 sequence variants (p.A265V, p.W302S, p.D322G) employing a yeast co-expression system. Hum Mutat. 2009;30(Feb):E443–50. https://doi.org/10.1002/humu.20926.
Levo A, Partanen J. Novel nonsense mutation (W302X) in the steroid 21-hydroxylase gene of a Finnish patient with the salt-wasting form of congenital adrenal hyperplasia. Hum Mutat. 1997;9/4:363–5.
Lajić S, Clauin S, Robins T, Vexiau P, Blanché H, Bellanne-Chantelot C, et al. Novel mutations in CYP21 detected in individuals with hyperandrogenism. J Clin Endocrinol Metab. 2002;87/6:2824–9.
Wedell A, Luthman H. Steroid 21-hydroxylase deficiency: two additional mutations in salt-wasting disease and rapid screening of disease-causing mutations. Hum Mol Genet. 1993;2/5:499–504.
Ordoñez-Sánchez ML, Ramírez-Jiménez S, López-Gutierrez AU, Riba L, Gamboa-Cardiel S, Cerrillo-Hinojosa M, et al. Molecular genetic analysis of patients carrying steroid 21-hydroxylase deficiency in the Mexican population: identification of possible new mutations and high prevalence of apparent germ-line mutations. Hum Genet. 1998;102/2:170–7.
Dolzan V, Sólyom J, Fekete G, Kovács J, Rakosnikova V, Votava F, et al. Mutational spectrum of steroid 21-hydroxylase and the genotype-phenotype association in Middle European patients with congenital adrenal hyperplasia. Eur J Endocrinol. 2005;153/1:99–106.
Lee HH, Chao HT, Lee YJ, Shu SG, Chao MC, Kuo JM, et al. Identification of four novel mutations in the CYP21 gene in congenital adrenal hyperplasia in the Chinese. Hum Genet. 1998;103/3:304–10.
Deneux C, Tardy V, Dib A, Mornet E, Billaud L, Charron D, et al. Phenotype-genotype correlation in 56 women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2001;86/1:207–13.
Bojunga J, Welsch C, Antes I, Albrecht M, Lengauer T, Zeuzem S. Structural and functional analysis of a novel mutation of CP21B in a heterozygote carrier of 21-hydroxylase deficiency. Hum Genet. 2005;117/6:558–64.
Globerman H, Amor M, Parker KL, New MI, White PC. Nonsense mutation causing steroid 21-hydroxylase deficiency. J Clin Invest. 1988;82/1:139–44.
Bernal González C, Fernández Salas C, Martínez S, Ezquieta Zubicaray B. [Premature androgenetic alopecia in adult male with nonclassic 21-OH deficiency. A novel nonsense CYP21A2 mutation (Y336X) in 2 affected siblings]. Med Clin (Barc). 2006;127/16:617–21.
Helmberg A, Tusie-Luna MT, Tabarelli M, Kofler R, White PC. R339H and P453S: CYP21 mutations associated with nonclassic steroid 21-hydroxylase deficiency that are not apparent gene conversions. Mol Endocrinol. 1992;6/8:1318–22.
Balsamo A, Cicognani A, Baldazzi L, Barbaro M, Baronio F, Gennari M, et al. CYP21 genotype, adult height, and pubertal development in 55 patients treated for 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2003;88/12:5680–8.
Krone N, Riepe FG, Grötzinger J, Partsch CJ, Brämswig J, Sippell WG. The residue E351 is essential for the activity of human 21-hydroxylase: evidence from a naturally occuring novel point mutation compared with artificial mutants generated by single amino acid substitutions. J Mol Med (Berl). 2005;83/7:561–8.
Abid F, Tardy V, Gaouzi A, El Hessni A, Morel Y, Chabraoui L. CYP21A2 gene mutation analysis in Moroccan patients with classic form of 21-hydroxylase deficiency: high regional prevalence of p.Q318X mutation and identification of a novel p.L353R mutation. Clin Chem Lab Med. 2008;46/12:1707–13.
Dubey S, Tardy V, Chowdhury MR, Gupta N, Jain V, Deka D, et al. Prenatal diagnosis of steroid 21-hydroxylase-deficient congenital adrenal hyperplasia: Experience from a tertiary care centre in India. Indian J Med Res. 2017;145/2:194–202.
Chiou SH, Hu MC, Chung BC. A missense mutation at Ile172–Asn or Arg356–Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. 1990;265/6:3549–52.
Levo A, Partanen J. Mutation-haplotype analysis of steroid 21-hydroxylase (CYP21) deficiency in Finland. Implications for the population history of defective alleles. Hum Genet. 1997;99/4:488–97.
Lajic S, Levo A, Nikoshkov A, Lundberg Y, Partanen J, Wedell A. A cluster of missense mutations at Arg356 of human steroid 21-hydroxylase may impair redox partner interaction. Hum Genet. 1997;99/6:704–9.
Menabò S, Balsamo A, Baldazzi L, Barbaro M, Nicoletti A, Conti V, et al. A sequence variation in 3ˈUTR of CYP21A2 gene correlates with a mild form of congenital adrenal hyperplasia. J Endocrinol Invest. 2012;35/3:298–305.
Levo A, Partanen J. Novel mutations in the human CYP21 gene. Prenat Diagn. 2001;21/10:885–9.
Kirby-Keyser L, Porter CC, Donohoue PA. E380D: a novel point mutation of CYP21 in an HLA-homozygous patient with salt-losing congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Mutat. 1997;9/2:181–2.
Wasniewska M, Mirabelli S, Baldazzi L, Salzano G, Messina MF, Lombardo F, et al. Novel mutation of CYP21A2 gene (N387K) affecting a non-conserved amino acid residue in exon 9. J Endocrinol Invest. 2009;32/7:633.
Baradaran-Heravi A, Vakili R, Robins T, Carlsson J, Ghaemi N, Aʼrabi A, et al. Three novel CYP21A2 mutations and their protein modelling in patients with classical 21-hydroxylase deficiency from northeastern Iran. Clin Endocrinol (Oxf). 2007;67/3:335–41.
Katsumata N, Shinagawa T, Horikawa R, Fujikura K. Novel intronic CYP21A2 mutation in a Japanese patient with classic salt-wasting steroid 21-hydroxylase deficiency. Metabolism. 2010;59/11:1628–32.
Yu Y, Wang J, Huang X, Wang Y, Yang P, Li J, et al. Molecular characterization of 25 Chinese pedigrees with 21-hydroxylase deficiency. Genet Test Mol Biomarkers. 2011;15(Mar):137–42. https://doi.org/10.1089/gtmb.2010.0131.
Billerbeck AE, Bachega TA, Frazetto ET, Nishi MY, Goldberg AC, Marin ML, et al. A novel missense mutation, GLY424SER, in Brazilian patients with 21-hydroxylase deficiency. J Clin Enocrinol Metab. 1999;84/8:2870–2.
Baumgartner-Parzer SM, Schulze E, Waldhäusl W, Pauschenwein S, Rondot S, Nowotny P, et al. Mutational spectrum of the steroid 21-hyxdroxylase gene in Austria: identification of a novel missense mutation. J Clin Endocrinol Metab. 2001;86/10:4771–5.
Dain L, Minutolo C, Buzzalino N, Belli S, Oneto A, Charreau E, et al. Gene symbol: CYP21A2. Disease: adrenal hyperplasia, CYP 21. Hum Genet. 2006;119/3:363.
Carvalho B, Pereira M, Marques CJ, Carvalho D, Leão M, Oliveira JP, et al. Comprehensive genetic analysis and structural characterization of CYP21A2 mutations in CAH patients. Exp Clin Endocrinol Diabetes. 2012;120/9:535–9.
Owerbach D, Sherman L, Ballard AL, Azziz R. Pro-453 to Ser mutation in CYP21 is associated with nonclassic steroid 21-hydroxylase deficiency. Mol Endocrinol. 1992;6/8:1211–5.
Wang H, Jiang L, Wang PP, Zhou HB, Wang JL, Song LL. [Mutation analysis in families with 21-hydroxylase deficiency]. Zhonghua Yi Xue Chuan Xue Za Zhi. 2007;24/6:681–4.
Ordoñez-Sánchez ML, Ramírez-Jiménez S, López-Gutierrez AU, Riba L, Gamboa-Cardiel S, Cerrillo-Hinojosa M, et al. Molecular genetic analysis of patients carrying steroid 21-hydroxylase deficiency in the Mexican population: identification of possible new mutations and high prevalence of apparent germ-line mutations. Hum Genet. 1998;102/2:170–7.
Di Pasquale G, Wasniewska M, Caruso M, Salzano G, Coco M, Lombardo F, et al. Salt wasting phenotype in a compound heterocygous girl with P482S mutation associated with a novel mutation of CYP21 gene (Q481P). J Endocrinol Invest. 2005;28/11:1038–9.
Barbaro M, Lajic S, Baldazzi L, Balsamo A, Pirazzoli P, Cicognani A, et al. Functional analysis of two recurrent amino acid substitutions in the CYP21 gene from Italian patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2004;89/5:2402–7.
Wedell A, Luthman H. Steroid 21-hydroxylase (P450c21): a new allele and spread of mutations through the pseudogene. Hum Genet. 1993;91/3:236–40.
White PC, Tusie-Luna MT, New MI, Speiser PW. Mutations in steroid 21-hydroxylase (CYP21). Hum Mutat. 1994;3/4:373–8.
Neocleous V, Fanis P, Toumba M, Phedonos AAP, Picolos M, Andreou E, et al. Variations in the 3ʼUTR of the CYP21A2 Gene in Heterozygous Females with Hyperandrogenaemia. Int J Endocrinol. 2017; https://doi.org/10.1155/2017/8984365.
Falhammar H, Nyström HF, Ekström U, Granberg S, Wedell A, Thorén M. Fertility, sexuality and testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia. Eur J Endocrinol. 2012;166:441–9.
Yu MK, Jung MK, Kim KE, Kwon AR, Chae HW, Kim DH, et al. Clinical manifestations of testicular adrenal rest tumor in males with congenital adrenal hyperplasia. Ann Pediatr Endocrinol Metab. 2015;20:155–61.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–61.
Acknowledgements
We wish to thank Dr Wim Wuyts for organizing the Best Practice Meeting in Antwerp in 2009 and all the participants of that EMQN-CAH-Best-Practice-Meeting (Christine Bellanne-Chantelot, Berta Carvalho, Edward Edkins, Rob Elles, João Gonçalves, Lies Hoefsloot, Mirjana Kocova, Ivan McGown, Yves Morel, Nicole Motton, Vassos Neocleous, Josep Oriola, Gill Rumsby, Egbert Schulze, Nicos Skordis, Veronique Tardy, Chu-Lee Thakkar) as well as all participants of the EMQN-CAH schemes for their input. We gratefully acknowledge the help of Stefan Nemeth who did the illustration of figures and we thank Daniel Wedemeyer for preparing the list of CYP21A2 variants. Funding for the best practice meeting was provided by the European Molecular Genetics Quality Network (http://www.emqn.org) and EuroGentest.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baumgartner-Parzer, S., Witsch-Baumgartner, M. & Hoeppner, W. EMQN best practice guidelines for molecular genetic testing and reporting of 21-hydroxylase deficiency. Eur J Hum Genet 28, 1341–1367 (2020). https://doi.org/10.1038/s41431-020-0653-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0653-5
This article is cited by
-
Genetic diagnosis of endocrine disorders in Cyprus through the Cyprus Institute of Neurology and Genetics: an ENDO-ERN Reference Center
Orphanet Journal of Rare Diseases (2024)
-
Analysis of the CYP21A2 gene pathogenic variants in CAH patients from Surgut using next-generation sequencing (NGS)
Egyptian Journal of Medical Human Genetics (2024)
-
High carrier frequency of CYP21A2 gene mutations in Southern India – underscoring the need for genetic testing in Congenital Adrenal Hyperplasia
Endocrine (2024)
-
Identification and functional characterization of compound heterozygous CYP11B1 gene mutations
Endocrine (2024)
-
Salt-wasting congenital adrenal hyperplasia phenotype as a result of the TNXA/TNXB chimera 1 (CAH-X CH-1) and the pathogenic IVS2-13A/C > G in CYP21A2 gene
Hormones (2023)
