
Abstract
Next generation sequencing provides an important opportunity for improved diagnosis in epilepsy. To date, the majority of diagnostic genetic testing is conducted in the paediatric arena, while the utility of such testing is less well understood in adults with epilepsy. We conducted whole exome sequencing (WES) and copy number variant analyses in an Irish cohort of 101 people with epilepsy and co-morbid intellectual disability to compare the diagnostic yield of genomic testing between adult and paediatric patients. Variant interpretation followed American College of Medical Genetics and Genomics (ACMG) guidelines. We demonstrate that WES, in combination with array-comparative genomic hybridisation, provides a diagnostic rate of 27% in unrelated adult epilepsy patients and 42% in unrelated paediatric patients. We observe a 2.7% rate of ACMG-defined incidental findings. Our findings indicate that WES has similar utility in both adult and paediatric cohorts and is appropriate for diagnostic testing in both epilepsy patient groups.
Similar content being viewed by others

Introduction
Epilepsy is a common neurological disorder affecting around 50–60 million people worldwide [1]. An estimated 70–80% of patients with epilepsy are thought to have an underlying genetic cause or predisposition, a proportion of which are monogenic which can be detected by whole exome sequencing (WES) or array-comparative genomic hybridisation (array-CGH) [2] and in this context, next-generation sequencing (NGS) offers great potential for genetic diagnostics in the clinic. To date, reports from large-scale WES projects in epilepsy have focused mainly on cohorts with severe epilepsies of infancy and childhood, particularly the epileptic encephalopathies [3,4,5]. These studies have reported diagnostic, monogenic causes in circa 27% of cases, identifiable via exome sequencing [5,6,7,8,9]. In contrast, less is known about the diagnostic yield of WES in adult epilepsy populations, and it is unknown if adult patients with epileptic encephalopathy who survive into adulthood have a different genetic aetiology to a paediatric patient cohort. Furthermore, it can be difficult to diagnose childhood onset epilepsy syndromes in adult patients due to the continuing development of the patient’s disease phenotype.
There have been only limited efforts to establish the diagnostic yield of WES in adults with epilepsy and co-morbid learning disability specifically. Previous studies, in mixed-age populations with multiple seizure types, have reported a diagnostic yield of 33–38% [9,10,11]. Thevenon et al. used WES to study a cohort of 43 patients with severe epilepsy (epileptic encephalopathy) and/or non-syndromic intellectual disability, ranging in age (at time of testing) from <5 to 55 years [10]. The authors reported an overall diagnostic yield of 32.5% [10]. Schouwenaars-Snoeijen et al. reported a diagnostic yield of 28.3% in adult patients with epilepsy and intellectual disability [9]. Helbig et al. performed WES in people with seizures and intellectual disability and/or developmental delay, referred for diagnostic WES at a commercial laboratory [11]. Of the recruited 314 patients, 38 were adults. An overall diagnostic rate of 38.2% (112/293) was reported in the sequenced patients with seizures [11]. A majority of the studies mentioned above did not implement American College of Medical Genetics and Genomics (ACMG) guidelines [12] when estimating the diagnostic single nucleotide variant (SNV) yield from NGS in their cohorts [5, 7, 8, 10], which likely will have resulted in higher yields than if applying the ACMG criteria.
Similar to SNVs, the diagnostic yield of copy number variants (CNVs) is better understood in paediatric epilepsy, relative to adult patients with epilepsy [13,14,15,16,17]. Olson and colleagues for example, conducted microarray testing for CNVs in paediatric patients with seizures and/or epilepsy, reporting a diagnostic rate of 5% (40/323) using stringent variant classification guidelines [13, 18]. Analysing a cohort of adults with paediatric-onset epilepsy and intellectual disability, Borlot et al. reported a diagnostic rate of 16.1% (23/143) when applying ACMG guidelines [14]. Striano et al. used microarray-based analyses to study 279 adult patients with unexplained epilepsy (29% with ID), reporting a diagnostic rate of 9.3% [19].
Here, we describe the ACMG-guided diagnostic yield of genomic testing (WES and array-CGH), and associated spectrum of variants, present in a predominantly adult cohort with unexplained refractory epilepsy and intellectual disability. This study was conducted to inform the clinical utility of genomic testing in adult epilepsy patients compared with a similar cohort of children tested during the same study period.
Subjects and methods
Patient cohort
Adult and paediatric patients were recruited via tertiary referral clinical centres throughout Ireland, specifically; Beaumont Hospital, Cork University Hospital, Galway University Hospital, Our Lady’s Children’s Hospital Crumlin, St. James’ Hospital, and the Daughters of Charity (St. Vincent De Paul). Inclusion criteria for adult patients were: a diagnosis of epilepsy with co-morbid ID, no clear cause and DNA samples available from both parents. Inclusion criteria for paediatric patients were: a diagnosis of epilepsy with co-morbid ID, no clear cause, a history of epileptic encephalopathy and DNA samples available from both parents.
Previous genetic testing with a targeted gene panel or WES, was an exclusion criteria for both adult and paediatric arms of the study. However, patients were included if they had previously undergone single gene test(s) (e.g., SCN1A and/or SLC2A1 etc).
Information on the patient’s medical and family history (defined as a first-degree relative with epilepsy), and phenotype was collected by the primary research team, via face-to-face interview and by manual review of patients’ medical charts.
Next-generation sequencing
DNA from the proband and both parents were extracted in a research setting, from peripheral blood leucocytes (Qiagen,QIAcube), saliva or buccal swabs. A second DNA sample from the proband was extracted in an accredited laboratory setting for confirmatory DNA testing by an accredited service provider, as required. Whole-exome capture was conducted in a research setting using Roche NimbleGen SeqCap EZ V3 as per the manufacturer’s instructions using 500 ng of input gDNA. Sequencing was performed on an Illumina NextSeq platform. Targeted panel sequencing was conducted using a custom Roche NimbleGen SeqCap panel (compiled on the basis of literature review, this panel was established at the start of the study and remained fixed) (Table S1) as per manufacturer’s instructions using 500 ng of input DNA and sequenced using an Illumina MiSeq. Array-CGH was performed on a subset of index cases and selected parental samples as required using Agilent SureTag labelling kit and 44 K Human Genome array-CGH Microarray Kit.
Bioinformatic analyses
Exome data were aligned to the GRCh37/hg19 reference genome [20] and processed using a Burrows-Wheeler Aligner and Picard [21]. Variants were identified using the Genome Analysis ToolKit (GATK) best practices protocol and annotated using ANNOVAR. Exomes with a minimum of 85% of target bases covered at >10× were included for analysis. Relatedness between proband and parents was confirmed using identity by descent testing in PLINK [22]. Bioinformatic pipelines were hosted on a dedicated Microsoft Azure server. Following analysis using the in-house pipeline, unsolved cases were analysed using a separate pipeline via SapientiaTM v1.9, a commercially available diagnostic decision support platform by Congenica Ltd (Hinxton, UK) (www.congenica.com).
Variant classification
We selected, for discussion at a multidisciplinary team (MDT) meeting, ‘qualifying variants’ from WES that satisfied the following criteria; (1) minor allele frequency (MAF) < 1% (for a recessive model) or MAF < 0.1% (for a dominant model) in gnomAD control database [23], (2) functional (exonic/splicing variant), predicted damaging by at least two of three prediction software tools (PolyPhen [24], SIFT [25] and/or MutationTaster [26]) and (3) in a gene either known to cause epilepsy or intellectual disability (as per the Online Mendelian Inheritance in Man (OMIM) website, accessed most recently in January 2019 [27]) or included in the ACMG recommended list of clinically actionable incidental findings [28]. Candidate genes from across the exome were assessed for a seizure or intellectual disability phenotype using OMIM for each case, and variants assessed were not limited to a specific gene panel. We considered ‘qualifying’ CNVs if the event was (1) large (>1 Mb), (2) spanned a gene listed in OMIM as morbid in neurological disease and (3) was not commonly seen in control populations (<3 similar sized variants in non-disease populations in the Database of Genomic Variation [29]). The genomic results from this study were uploaded to an electronic patient record via a custom designed ‘genomics module’ [30]. This module facilitated integration of clinical and genomics data, discussion of each patient’s genomics results at an MDT meeting and sharing of clinically important results with referring clinicians throughout the Irish public healthcare system [30]. The inheritance pattern of each qualifying variant was considered during discussion at MDT. Where there were multiple affected first-degree relatives, we tested the relevant variant for segregation in additional family members. Variants which did not match the expected segregation pattern for pathogenicity were considered as benign or VUS. The MDT meetings had input from a clinical geneticist (MG), geneticist/bioinformaticians (KB, GLC), adult and paediatric neurologists (ND, NA, HE, DC, CD, MK, JL, DW, SB) and an epilepsy advanced nurse practitioner (MW).
Clinical interpretation and variant validation
Qualifying variants were classified in terms of pathogenicity according to ACMG guidelines [12, 18, 31]. Additional details on local interpretation of the ACMG guidelines are provided in the Supplementary Information (Table S2). Cascade testing was considered when requested by family members. Candidate variants were confirmed on an independent DNA sample (extracted in an accredited setting) using Sanger sequencing, array-CGH or Multiplex Ligation-dependent Probe Amplification as appropriate. Confirmation testing was conducted by an accredited service provider (CeGaT GmbH, Germany). The accredited service provider is also an independent clinical genetics interpretation of variant pathogenicity. Once the presence and pathogenicity of the candidate variant were confirmed by the accredited lab, results were returned to the patient and/or carers via the clinical geneticist. All variants discussed in this manuscript have been submitted to ClinVar (see Table 2) and requests for access to raw sequence data can be made via direct contact with the corresponding author.
Incidental findings
Adult patients were informed on the concept of incidental findings [28] during the consenting process and provided with the option whether or not to receive clinically actionable incidental findings, as previously defined by the ACMG [28, 32]. This list of clinically actionable genes can cause disorders which have preventative measures and/or treatments available. In the case of ID, the patients’ legal guardian/parent provided consent on behalf of the patient. We did not analyse for incidental findings in all paediatric patients, as per requirements of local ethics committees.
Mosaicism detection
Deep sequencing (coverage > 100×) of selected patients and their parents was considered in cases of suspected mosaicism (multiple affected siblings with apparent de novo inheritance) using a targeted Roche SeqCap EZ custom sequencing panel (Table S1).
Results
Patient cohort
A cohort of 101 adult and paediatric patients (representing 96 families) with epilepsy and co-morbid ID and their parents underwent WES, of which 80 patients also had array-CGH testing. Of the sequenced patients, 74/101 (73%) were adult (age ≥ 18 years) and 27/101 (27%) were paediatric (age < 18 years). All patients had recurrent seizures. Five affected sibling pairs were recruited (four adult, one paediatric pair). Patient characteristics are provided in Table 1.
Next generation sequencing results
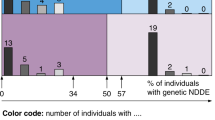
A mean coverage of 44× was obtained from WES (minimum 11.31× and maximum 112.6×). Index samples (n = 16) with coverage of <85% at 10× were re-sequenced using a custom Roche NimbleGen targeted epilepsy sequencing panel to ensure that relevant genes were covered. After identification of qualifying variants, discussion at the MDT meeting and confirmation via the accredited laboratory, 30 ACMG-classified pathogenic/likely pathogenic variants (in disease-causing genotype state) were detected in 33/101 (33%) patients and 30/96 (31%) of families. Three variants were identified in multiple, related patients (Table 2). Diagnostic variants included SNVs in 23 established epilepsy genes as well as two CNVs in established epilepsy loci.Footnote 1 Of the 28 SNVs we identified as diagnostic, 23/28 (82%) were novel variants and absent from the literature and/or relevant public databases (See Table 2). In total, 82% (23/28) of unique SNVs were de novo, one was a result of parental mosaicism, two were recessive, one was X-linked, and one was inherited from a partially affected parent. Of the SNVs, 8/28 (28.6%) were stop-gain, 3/28 (10.7%) were frameshifts and 17/28 (60.7%) were non-synonymous (See Table 2). A diagnostic rate of 30% (22/74) was obtained in adult patients and 41% (11/27) in paediatric patients. In unrelated individuals (excluding siblings), a diagnostic rate of 27% (19/70) was obtained in adult patients and 42% (11/26) in paediatric patients. We detected three diagnostic SNVs in CHD2; all in patients with seizure onset between 3 and 4 years of age, mild ID at the time of seizure onset and with features of autistic spectrum disorder. Two potentially disease-causing, ACMG ‘likely pathogenic’/‘pathogenic’ deletions were identified from array-CGH (See Table 2). Deep sequencing (coverage > 100×) of selected patients (n = 3 probands) and their parents was conducted in cases of suspected mosaicism. In one family, confirmation of mosaicism provided additional evidence of pathogenicity.
Previous evidence has suggested a correlation between paternal age >35 and epilepsy in offspring [33, 34]. In this study, the average paternal age was 33 years and the average maternal age was 30 at the time of birth. The average paternal age in trios with an identified pathogenic de novo variant was 33 years, and not significantly different to average paternal age in trios without a pathogenic de novo variant (32 years), (p value from two-tailed student’s t test: 0.57). The percentage of patients with a molecular diagnosis and age of seizure onset ≤3 years was 43% (10/23) in the adult cohort and 70% (7/10) in the paediatric cohort. This difference was not significant (Fisher’s Exact test p value: 0.197). All variants identified are shown in Table 2. Patient phenotype information is listed in Table 3. A total of 26 of the diagnostic variants were identified from the in-house GATK based bioinformatics pipeline, two by array-CGH and two using Sapientia 1.9.
Parental mosaicism
Three cases from two families were tested for parental mosaicism, and mosaicism was confirmed in one family. A shared variant in MBD5 was detected in two affected full siblings. The variant was not identified in either parent from WES data or Sanger sequencing. Deep sequencing using our gene panel identified mosaicism in the mother, explaining the segregation pattern. MBD5 is associated with autosomal dominant developmental delay with ID, autism, and multiple seizure types [35]. A third sibling with ID without seizures (a different phenotype from the proband) also carried the variant, identified through segregation testing. The pedigree chart is shown in Fig. 1.

a ( ) Probands: Epilepsy, history of febrile seizures, mild/moderate intellectual disability (ID), ±Asperger syndrome. (
) Probands: Epilepsy, history of febrile seizures, mild/moderate intellectual disability (ID), ±Asperger syndrome. ( ) Sister of probands: Moderate ID, behavioural problems, no epilepsy. (
) Sister of probands: Moderate ID, behavioural problems, no epilepsy. ( ) Epilepsy, no ID. Variant of Interest: MBD5 p.(D242fs) ± = Heterozygote for MBD5 p. (D242fs). (±) = Mosaic heterozygote for MBD5 p.(D242fs). −/− = Tested negative for MBD5 p. (D242fs). b II5 Unaffected father of proband. II6 Unaffected mother of probands. II7 Developed epilepsy at 52 years. III3 Proband (Patient 37): Epilepsy, mild intellectual disability (ID), Aspeger syndrome, history of febrile seizures. Tetralogy of Fallot at birth. III4 Unaffected twin sister. III6 Moderate ID, complex mood disorder, challenging behaviour. III7 Proband (Patient 39): Moderate ID, epilepsy, history of febrile seizures. III10 Few seizures as teenager. No seizures now.
) Epilepsy, no ID. Variant of Interest: MBD5 p.(D242fs) ± = Heterozygote for MBD5 p. (D242fs). (±) = Mosaic heterozygote for MBD5 p.(D242fs). −/− = Tested negative for MBD5 p. (D242fs). b II5 Unaffected father of proband. II6 Unaffected mother of probands. II7 Developed epilepsy at 52 years. III3 Proband (Patient 37): Epilepsy, mild intellectual disability (ID), Aspeger syndrome, history of febrile seizures. Tetralogy of Fallot at birth. III4 Unaffected twin sister. III6 Moderate ID, complex mood disorder, challenging behaviour. III7 Proband (Patient 39): Moderate ID, epilepsy, history of febrile seizures. III10 Few seizures as teenager. No seizures now.
Incidental findings
Four potential incidental findings were identified across KCNH2, KCNQ1, MYH11 and VHL from WES. Although only two of these satisfied the ACMG guidelines (Table 2) (yield of ACMG-classified incidental findings: 2.7% (2/74)), we proceeded with confirmatory testing in all four at the accredited provider to get a second opinion, since these were all previously reported as disease-causing (ClinVar database [36]). Two of these four variants were deemed pathogenic by the accredited service provider. Following consultation with our clinical geneticist, it was deemed important for clinical care to refer three patients (patients 6, 65 and 108) for appropriate clinical testing as a direct result of these incidental findings. The genes KCNH2 and KCNQ1 are associated with long-QT syndrome as well as mild seizure phenotypes.
Impact on patient care
Of the 33 patients provided with a molecular diagnosis, four findings may have a clear impact on epilepsy treatments, three SCN1A variants (two adults, one child) and one TSC1 variant (adult). The patient with the TSC1 variant may be suitable for treatment with evorolimus, an mTOR inhibitor originally developed as a cancer drug shown to improve seizures in patients with tuberous sclerosis [37, 38]. In three patients with SCN1A variants, clinicians may choose to avoid sodium channel blockers commonly used to treat other forms of epilepsy as these medications can worsen seizures in patients with SCN1A variants [39, 40]. In other diagnosed patients there may be n-of-1 trials or small cases series of other therapies or management approaches which may be relevant to the phenotype, and the provision of support groups or links to communities to explore other therapeutic avenues and clinical research.
Discussion
Our findings reveal an overall diagnostic rate for WES and array-CGH of 31% in a combined adult and paediatric cohort of unrelated patients with medically refractory epilepsy and co-morbid ID. Our findings are in line with those from comparable studies which report a diagnostic yield in mixed age cohorts of 33–38% [9,10,11]. The rate of molecular diagnosis was nominally higher in the paediatric cohort (41%) than the adult cohort (30%) but the difference was not statistically significant (Fisher’s Exact test; p = 0.209). When restricted to unrelated probands, this difference remains non-significant (Fisher’s Exact test; p value 0.12). Slight differences in inclusion criteria for adult and paediatric patients were used as there is no clear hallmark to distinguish between epileptic encephalopathy and intellectual disability plus epilepsy in adult epilepsy patients, some of whom may have had epilepsy since early childhood. The set of genes identified in adult versus paediatric patients were comparable, and the frequency of core epileptic encephalopathy genes (as defined by Heyne et al. [41]) was not significantly higher in the paediatric cohort (p value from Fisher’s Exact test: 0.27). Our diagnostic yields are comparable to previous reports in both paediatric and mixed-age cohorts [3,4,5,6,7, 10, 11]. The implementation of dual-bioinformatics pipelines (in-house and via Sapientia v1.9) allowed the identification of additional variants, likely due to differences in variant-quality scoring threshold differences. A large proportion of the ACMG-classified ‘likely pathogenic’/‘pathogenic’ variants were de novo (82%). This is consistent with previous studies emphasising the role of de novo variants in patients with epileptic encephalopathies [6, 42]. We encountered difficulties in the implementation of ACMG guidelines for both SNVs and CNVs [12, 18, 43]. For example, the ability to assign additional evidence of pathogenicity in cases where the patient inherited a candidate variant on the X chromosome from an unaffected mother would be a valuable addition to the current guidelines. To illustrate, in patient 36 (male) a qualifying variant on the X chromosome in OPHN1, inherited from the (unaffected) mother was not classified as pathogenic by ACMG guidelines. There is no category of evidence which can be attributed to this X-linked variant inherited from an unaffected mother (and therefore unable to cause disease in that parent) although this is pertinent information. The MDT team deemed this variant disease-causing on the basis of a close phenotypic match, but it did not satisfy ACMG criteria (Table 2). It is probable that some disease-causing variants were missed due to low WES coverage or insufficient array-CGH probes in key regions.
Whole exome sequencing for molecular diagnosis in adult epilepsy patients
The utility of WES for molecular diagnosis of patients with epilepsy and ID is established in paediatric cohorts, but the effectiveness of this approach in adult patients is less understood, with published work reporting on relatively small cohort sizes and mixed age patient populations. We demonstrate a molecular diagnostic rate of ~30% in adult patient groups with childhood-onset disease, and ID. This is due to a number of factors including the absence of genetic knowledge when these children were younger, and an under-appreciation and poor availability of appropriate genetic testing as these individuals grew into adulthood. The benefits of genomic testing to patients and their families are well established [44, 45]. A molecular diagnosis can provide clarification of diagnosis bringing a conclusion to the ‘diagnostic odyssey', enhancing cost-effectiveness from alternative fruitless investigations [46] and inform prognosis. In some cases, alternative treatment options may become apparent following genetic diagnosis, for example Dravet syndrome patients with SCN1A variants should generally avoid carbamazepine related drugs, phenytoin and lamotrigine [47]. The differential effects of a potential precision medicine initiated in childhood versus adulthood are unknown. While interventions in childhood may improve prognosis and outcomes, interventions in adulthood may positively affect seizure control and have a considerable impact on quality of life. Genomics approaches, such as this one, are particularly relevant as we enter an era of personalised medicine which may allow for better seizure control with fewer side effects of therapy in both children and adults while increasing value from limited healthcare resources. Genomic testing can have valuable implications in terms of family planning for the patient’s parents and siblings and remove any sense of blame within families, including the adult cohort diagnosed in this study [48, 49]. Therefore, we suggest that all adult patients with epilepsy and ID of unclear cause should undergo diagnostic WES and array-CGH testing.
Incidental findings
The rate of incidental findings for adult patients consented to receive these results (2.7%) is slightly higher than reported by the ACMG (1%) [32], although it is the same as that reported by Ding et al. who predicted a rate of 2.7% based on mathematical modelling and ACMG guidelines [50]. Two of our incidental findings were in genes associated with long-QT syndrome (KCNQ1 and KCNH2), but also known to infer risk of seizures [39, 40]. We speculate that any inflation in the rate of incidental findings in our cohort may indicate that the variants in genes associated with mild seizure phenotypes may, in part, contribute to the phenotype of interest. However, we have concluded that due to the patients’ severe seizure profile and intellectual disabilities, these variants are unlikely to explain the entire epilepsy phenotype in these individuals.
Conclusion
This study illustrates how WES provides diagnostic and prognostic value for neurologists in adult, as well as paediatric settings. Although extremely helpful for the standardisation of variant interpretation, current ACMG guidelines require continuous development to incorporate growing evidence for pathogenicity. The next challenges are to educate health care professionals in the evolving advantages of genomic medicine, and to inform state and private funders of the clinical and cost benefit of the integration of diagnostic exome sequencing into healthcare systems for epilepsy patients with complex phenotypes [51].
Supplementary information is available at European Journal of Human Genetics’ website.
Notes
One of these CNVs (patient 84) awaits confirmation and segregation testing from accredited laboratory.
References
Covanis A, Guekht A, Li S, Secco M, Shakir R, Perucca E. From global campaign to global commitment: The World Health Assembly’s Resolution on epilepsy. Epilepsia. 2015;56:1651–7.
Myers CT, Mefford HC. Advancing epilepsy genetics in the genomic era. Genome Med BioMed Cent. 2015;7:91.
Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21.
Pagnamenta AT, Murakami Y, Taylor JM, Anzilotti C, Howard MF, Miller V, et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet. 2017;25:669–79.
Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, Van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–14.
Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. 2017;101:664–85.
Ortega-Moreno L, Giráldez BG, Soto-Insuga V, Pozo RL Del, Rodrigo-Moreno M, Alarcón-Morcillo C. et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS ONE. 2017;12:e0188978.
Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SDJ, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia. 2016;57:e12–7.
Snoeijen-Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia. 2019;60:155–64.
Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. 2016;89:700–7.
Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18:898–905.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Olson H, Shen Y, Avallone J, Sheidley BR, Pinsky R, Bergin AM, et al. Copy number variation plays an important role in clinical epilepsy. Ann Neurol. 2014;75:943–58.
Borlot F, Regan BM, Bassett AS, Stavropoulos DJ, Andrade DM. Prevalence of pathogenic copy number variation in adults with pediatric-onset epilepsy and intellectual disability. JAMA Neurol. 2017;74:1301–11.
Mefford HC. Copy number matters in epilepsy. Epilepsy Curr. 2015;15:180–2.
Galizia EC, Srikantha M, Palmer R, Waters JJ, Lench N, Ogilvie CM, et al. Array comparative genomic hybridization: results from an adult population with drug-resistant epilepsy and co-morbidities. Eur J Med Genet. 2012;55:342–8.
Coppola A, Cellini E, Stamberger H, Saarentaus E, Cetica V, Lal D, et al. Diagnostic implications of genetic copy number variation in epilepsy plus. Epilepsia. 2019;60:689–706.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–5.
Striano P, Coppola A, Paravidino R, Malacarne M, Gimelli S, Robbiano A, et al. Clinical significance of rare copy number variations in epilepsy: a case-control survey using microarray-based comparative genomic hybridization. Arch Neurol. 2012;69:322–30.
Church DM, Schneider VA, Graves T, Auger K, Cunningham F, Bouk N, et al. Modernizing reference genome assemblies. PLoS Biol. 2011;9:e1001091.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(Jul):1754–60.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore M. Online Mendelian Inheritance in Man, OMIM®. https://omim.org/.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55.
MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–92.
Delanty N, White M, Benson K, McCormack M, Heavin S, Comerford E, et al. Development of a genomics module within an epilepsy-specific electronic health record: toward genomic medicine in epilepsy care. Epilepsia. 2019;epi.16278.
Palmer EE, Schofield D, Shrestha R, Kandula T, Macintosh R, Lawson JA, et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: evidence of clinical utility and cost effectiveness. Mol Genet Genomic Med. 2018;6:186–99.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
Saha S, Barnett AG, Foldi C, Burne TH, Eyles DW, Buka SL. et al. Advanced paternal age is associated with impaired neurocognitive outcomes during infancy and childhood. PLoS Med. 2009;6:e1000040.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–30.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5.
Samueli S, Abraham K, Dressler A, Gröppel G, Mühlebner-Fahrngruber A, Scholl T, et al. Efficacy and safety of Everolimus in children with TSC—associated epilepsy—Pilot data from an open single-center prospective study. Orphanet J Rare Dis. 2016;11:145.
French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388:2153–63.
EpiPM Consortium. A roadmap for precision medicine in the epilepsies. Lancet Neurol. 2015;14:1219–28.
Nabbout R, Chemaly N, Chipaux M, Barcia G, Bouis C, Dubouch C, et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis. 2013;8:176.
Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. 2018;50:1048–53.
EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project and EC, Balling R, Barisic N, Baulac S, Caglayan H, Craiu D, et al. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2014;95:360–70.
Smith LA, Douglas J, Braxton AA, Kramer K. Reporting incidental findings in clinical whole exome sequencing: incorporation of the 2013 ACMG recommendations into current practices of genetic counseling. J Genet Couns. 2015;24:654–62.
Berkovic SF. Genetics of epilepsy in clinical practice. Epilepsy Curr. 2015;15:192–6.
Mefford HC. Clinical genetic testing in epilepsy. Epilepsy Curr. 2015;15:197–201.
Forman EB, Gorman KM, Conroy J, Arthur N, Grant C, Ennis S, et al. Cost of exome sequencing in epileptic encephalopathy: is it “worth it”? Arch Dis Child. 2018;103:304.
Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012;135:2329–36.
Poduri A. When should genetic testing be performed in epilepsy patients? Epilepsy Curr. 2017;17:16–22.
Poduri A, Sheidley BR, Shostak S, Ottman R. Genetic testing in the epilepsies-developments and dilemmas. Nat Rev Neurol. 2014;10:293–9.
Ding L-E, Burnett L, Chesher D. The impact of reporting incidental findings from exome and whole-genome sequencing: predicted frequencies based on modeling. Genet Med. 2015;17:197–204.
Delanty N, Goldstein DB. Neuron NeuroView diagnostic exome sequencing: a new paradigm in neurology. 2013;80:841–3.
Acknowledgements
This publication has emanated from research supported in part by eHealth Ireland, Health Service Executive (HSE), Grant Number 13/CDA/2223 and a research grant from Science Foundation Ireland (SFI) under Grant Number 16/RC/3948 and co-funded under the European Regional Development Fund and by FutureNeuro industry partners. We thank Prof. Brendan Loftus and Catherine Moss of University College Dublin for access to sequencing platforms. We also thank the patients, their families, carers and clinical staff involved in the care of these patients for making this research possible.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
NJL, ASR and CAS are employed by Congenica Ltd.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Benson, K.A., White, M., Allen, N.M. et al. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur J Hum Genet 28, 1066–1077 (2020). https://doi.org/10.1038/s41431-020-0610-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0610-3
This article is cited by
-
Genetische Diagnostik der Epilepsien: Empfehlung der Kommission Epilepsie und Genetik der Deutschen Gesellschaft für Epileptologie (DGfE)
Clinical Epileptology (2023)
-
The association between early childhood onset epilepsy and attention-deficit hyperactivity disorder (ADHD) in 3237 children and adolescents with Autism Spectrum Disorder (ASD): a historical longitudinal cohort data linkage study
European Child & Adolescent Psychiatry (2023)
-
Trio-based exome sequencing reveals a high rate of the de novo variants in intellectual disability
European Journal of Human Genetics (2022)
-
Diagnostic Considerations in the Epilepsies—Testing Strategies, Test Type Advantages, and Limitations
Neurotherapeutics (2021)
-
Next-generation gene panel testing in adolescents and adults in a medical neuropsychiatric genetics clinic
neurogenetics (2021)
