
Abstract
A distinct neurodevelopmental phenotype characterised mainly by mild motor and language delay and facial dysmorphism, caused by heterozygous de novo or dominant variants in the TLK2 gene has recently been described. All cases reported carried either truncating variants located throughout the gene, or missense changes principally located at the C-terminal end of the protein mostly resulting in haploinsufficiency of TLK2. Through whole exome sequencing, we identified a homozygous missense variant in TLK2 in a patient showing more severe symptoms than those previously described, including cerebellar vermis hypoplasia and West syndrome. Both parents are heterozygous for the variant and clinically unaffected highlighting that recessive variants in TLK2 can also be disease causing and may act through a different pathomechanism.
Similar content being viewed by others


Introduction
The TLK2 gene encodes the tousled-like kinase 2, a nuclear serine/threonine kinase known to be involved in DNA replication and chromatin assembly by phosphorylating chromatin assembly factors, such as ASF1 and regulating histone usage [1]. TLK2 was suggested as a candidate gene for intellectual disability (ID) in a meta-analysis of de novo variants in 2104 exome trios [2], and recently Reijnders et al. [3] associated it with ID within a broader phenotype in a cohort of 40 individuals from 38 families. The cases presented with a distinct neurodevelopmental phenotype characterised by mild motor and language delay, behavioural problems, facial dysmorphism and gastro-intestinal symptoms caused by heterozygous de novo or dominant variants in TLK2. Truncating (i.e. nonsense, frameshift and splice site) variants located throughout the gene, or missense changes principally located at the C-terminal end of the protein were identified amongst the cohort. RNA analysis from patient derived cell lines suggested that the variants resulted in loss-of-function and that the most likely disease mechanism was haploinsufficiency of TLK2. Here, we report a child carrying a homozygous missense variant in TLK2 showing more severe symptoms than those previously described.
Subject and methods
Patient and family members were recruited at the Department of Paediatric Neurology, Izmir, Turkey. Written informed consent was obtained. Whole exome sequencing (WES) was performed by the Genomics Platform at the Broad Institute of MIT and Harvard, Cambridge, USA. Libraries were created with an Illumina exome capture (38 Mb target) and sequenced with a mean target coverage of >80 × . Exome sequencing data were processed and analysed on the RD-Connect Genome-Phenome Analysis Platform (https://platform.rd-connect.eu/genomics). Likely pathogenic variants, affecting the function of the gene, and potentially causing disease, were identified applying standard filtering criteria: minor allele frequency <1%, and high to moderate variant effect predictor (i.e. nonsense, splice site, frameshift, in-frame and non-synonymous variants). Shortlisted variants were interrogated for their predicted in silico deleteriousness and previous known association with human disease. Genomic and phenotypic data have been submitted to RD-Connect under accession numbers E287350, E180542 and E227711 (index case, mother and father, respectively) where they can be accessed under a controlled access agreement (https://platform.rd-connect.eu/datasubmission/). Annotation of the TLK2 gene is according to NM_006852.3.
Clinical description
The patient is a 6-year-old girl and the second child of unaffected consanguineous parents of Turkish origin; her 9-year-old brother is healthy (Fig. 1). She was born at term but had intrauterine growth retardation (birth weight 2400 g). Intrauterine foetal movements were reduced and she had hypotonia and hip dislocation at birth. She presented with epileptic spasms at the age of 6 months. Her EEG showed hypsarrhythmia suggesting West syndrome. She has been seizure-free since 3 years of age. On clinical examination at 6 years of age her psychomotor and speech development were severely delayed, she could sit without support but was unable to walk or speak. She had microcephaly, coarse facial appearance and dysmorphic facial features including telecanthus, upslanting palpebral fissures, thin vermilion upper lip, big mouth and broad nasal bridge (Fig. 2a, b), and a hemangioma on the left parietal scalp. In addition, she had feeding difficulties, dysphagia, peripapillary atrophy on fundoscopic examination and conductive hearing loss in the left ear. Neurological examination detected spastic tetraparesis with increased deep tendon reflexes, head titubation, trunk ataxia and dysmetria. She also presented with constipation and behavioural problems, including aggression and irritability. Her brain MRI at age 2 years showed cerebellar vermis hypoplasia (Dandy-Walker variant) and dilatation of lateral ventricles (Fig. 2c, d). Abdominal MRI detected medullary nephrocalcinosis and hydronephrosis.

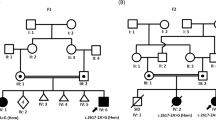
Pedigree and Sanger segregation of the TLK2 variant (hg19 chr17:60599574 A>G). The index case is homozygous for the alternate allele (G/G), whereas her unaffected brother is homozygous wild type (A/A) and his parents are heterozygous (A/G) (indicated by an arrow). No dysmorphic features are observed in the healthy family members, including those who are carriers for the TLK2 variant

Clinical findings. a, b Patient photographs at 6 years of age showing facial dysmorphism: telecanthus and upslanting palpebral fissures, thin vermilion upper lip, big mouth and broad nasal bridge; b Sagittal T2-weighted cranial MR images showing cerebellar vermis hypoplasia (Dandy-Walker variant); c Axial T2-weighted cranial MR images showing cystic dilatation of the cerebellum indicating cerebellar vermis hypoplasia
Results
Trio analysis of the patient and her parents identified a homozygous missense variant in the TLK2 gene (hg19 chr17: g.60599574 A>G; c.163 A>G; p.(Lys55Glu)), which affects a highly conserved amino acid. The variant is predicted to be damaging by all tested in silico tools, is amongst the top 0.2% most deleterious variants (CADD score 27) [4] and is classified as ‘likely pathogenic’ by the ACMG Guidelines [5]. This variant has been detected only once, in a heterozygous European, out of 122,547 individuals in gnomAD and is absent from a cohort of 1182 ethnically-matched Turkish control individuals (TUBITAK MAM-GMBE dataset; http://gmbe.mam.tubitak.gov.tr/en). Sanger sequencing showed that the variant is not present in her healthy brother, and that both parents are heterozygous carriers. On examination they were clinically unaffected and showed no signs of facial dysmorphism or cognitive impairment (Fig. 1).
Discussion
We report a homozygous missense variant in TLK2 in a patient with a neurodevelopmental disorder with severe motor and language delay, West syndrome, pontocerebellar hypoplasia, behavioural problems, facial dysmorphism and gastro-intestinal symptoms. The clinical presentation fits what has been described by Reijnders et al. [3] for heterozygous TLK2 patients and the dysmorphic features were remarkably similar. Our index case, however, presented with more severe symptoms, with profound ID, spastic tetraparesis and a structural brain anomaly. Whilst conductive hearing loss, microcephaly and non-specific brain anomalies were present in a minority of reported cases (<25%), we are not aware of cerebellar vermis hypoplasia, which we observed in the homozygous individual. Although the majority of the patients reported by Reijnders had language and cognitive delay (92% and 74%, respectively), this was mainly mild ID (IQ 50–70). In contrast, our patient presents severe neurodevelopmental delay with no acquired speech (although non-verbal IQ was not formally assessed). Constipation and feeding difficulties were similar to other individuals with heterozygous variants in TLK2. Medullary nephrocalcinosis and hydronephrosis were not observed in the previously published cohort.
All nine missense variants identified in the 38 families described by Reijnders et al. [3] were located either in the catalytic domain or in the coiled-coil motifs of the protein. Our variant is located at the N -terminus in a region where no functional domains are known (Fig. 3). This region, however, is expected to contain a nuclear localisation signal (NLS) as mutants lacking the first 160 amino acids fail to localise to the nucleus [6]. In addition, more than 20 phosphorylation sites, including p.Thr52 and p.Tyr70, have been identified in the N-terminal domain of TLK2, suggesting that this is potentially an important regulatory domain in vivo (https://www.phosphosite.org). In silico tools (cNLS Mapper) predict a monopartite NLS (61RNRKRKAEPY70) starting at position p.Arg61. NLSs consist of stretches of basic amino acids, primarily lysine and arginine, which bind to negative charges in the binding grooves of the transport receptor. Amino acid substitutions of lysine residues within NLSs can completely abolish their cargo nuclear import [7]. Although speculative, it may be hypothesised that the change from a positive (Lys) to a negative (Glu) side chain amino acid at the proximal position p.Lys55 may result in a change in conformation, partially occluding the binding between the NLS-cargo and the import receptor. This would lead to impaired nuclear import and in turn, diminished TLK2 activity.

Schematic representation of the TLK2 missense variants identified. In black, the p.(Lys55Glu) reported here; in grey, the nine variants previously reported by Reijnders et al. [3]. Annotation is according to NM_006852.3
In summary, here we describe a patient with a homozygous TLK2 variant leading to an autosomal recessive severe neurodevelopmental disorder, highlighting that certain TLK2 variants can cause a recessive disease, as the heterozygous parents were healthy. The clinical presentation of our patient shows similarities with the previously reported patients carrying heterozygous TLK2 variants, however the symptoms are more severe and complicated with additional features, such as cerebellar vermis hypoplasia and West syndrome. A reduction of available nuclear TLK2 would be the proposed mechanism, although this requires further functional investigation. We also highlight the importance of searching not only for de novo and dominant, but also recessive variants in TLK2 in patients with neurodevelopmental disease. The identification of additional cases will help to further delineate the recessive TLK2 phenotype.
References
Sillje HH, Nigg EA. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr Biol: CB. 2001;11:1068–73.
Lelieveld SH, Reijnders MR, Pfundt R, Yntema HG, Kamsteeg EJ, de Vries P, et al. Meta-analysis of 2104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–6.
Reijnders MRF, Miller KA, Alvi M, Goos JAC, Lees MM, de Burca A, et al. De novo and inherited loss-of-function variants in TLK2: clinical and genotype-phenotype evaluation of a distinct neurodevelopmental disorder. Am J Hum Genet. 2018;102:1195–203.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D94.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Mortuza GB, Hermida D, Pedersen AK, Segura-Bayona S, Lopez-Mendez B, Redondo P, et al. Molecular basis of Tousled-Like Kinase 2 activation. Nat Commun.
Kalderon D, Richardson WD, Markham AF, Smith AE Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–8.
Acknowledgements
We thank the family for allowing to present their photographs.
Funding
The project is supported by TUBITAK (The Scientific and Technological Research Council of Turkey) Project No. 216S771. RH is a Wellcome Trust Investigator (109915/Z/15/Z), who receives support from the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), Medical Research Council (UK) (MR/N025431/1), the European Research Council (309548), the Wellcome Trust Pathfinder Scheme (201064/Z/16/Z) and the Newton Fund (UK/Turkey, MR/N027302/1). We thank the Broad Institute for carrying out WES. The Broad Centre for Mendelian Genomics (UM1 HG008900) is funded by the National Human Genome Research Institute with supplemental funding provided by the National Heart, Lung, and Blood Institute under the Trans-Omics for Precision Medicine (TOPMed) programme and the National Eye Institute. Data were analysed using the RD-Connect Genome-Phenome Analysis platform developed under FP7/2007-2013 funded project (grant agreement no. 305444).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Töpf, A., Oktay, Y., Balaraju, S. et al. Severe neurodevelopmental disease caused by a homozygous TLK2 variant. Eur J Hum Genet 28, 383–387 (2020). https://doi.org/10.1038/s41431-019-0519-x
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0519-x
This article is cited by
-
Novel insights into PORCN mutations, associated phenotypes and pathophysiological aspects
Orphanet Journal of Rare Diseases (2022)
-
Molecular causes of primary microcephaly and related diseases: a report from the UNIA Workshop
Chromosoma (2020)
