
Abstract
Congenital myasthenic syndromes (CMS) are a clinically and genetically heterogeneous group of disorders caused by mutations which lead to impaired neuromuscular transmission. SLC25A1 encodes a mitochondrial citrate carrier, associated mainly with the severe neurometabolic disease combined D-2- and L-2-hydroxyglutaric aciduria (D/L-2-HGA). We previously reported a single family with a homozygous missense variant in SLC25A1 with a phenotype restricted to relatively mild CMS with intellectual disability, but to date no additional cases of this CMS subtype had been reported. Here, we performed whole exome sequencing (WES) in three additional and unrelated families presenting with CMS and mild intellectual disability to identify the underlying causative gene. The WES analysis revealed the presence of a homozygous c.740G>A; p.(Arg247Gln) missense SLC25A1 variant, the same SLC25A1 variant as identified in the original family with this phenotype. Electron microscopy of muscle from two cases revealed enlarged and accumulated mitochondria. Haplotype analysis performed in two unrelated families suggested that this variant is a result of recurrent mutation and not a founder effect. This suggests that p.(Arg247Gln) is associated with a relatively mild CMS phenotype with subtle mitochondrial abnormalities, while other variants in this gene cause more severe neurometabolic disease. In conclusion, the p.(Arg247Gln) SLC25A1 variant should be considered in patients presenting with a presynaptic CMS phenotype, particularly with accompanying intellectual disability.
Similar content being viewed by others


Introduction
Congenital myasthenic syndromes (CMS) are a heterogeneous group of disorders caused by mutations leading to impairment of neuromuscular transmission. Most often, these disorders arise from mutations affecting postsynaptic components of the neuromuscular junction (NMJ). CMS due to mutations affecting the motor nerve terminal comprise a rarer subset; they are however, increasingly recognised, and the majority of the recently discovered CMS genes result in presynaptic NMJ defects [1].
SLC25A1 is a mitochondrial citrate carrier which mediates the exchange of citrate/isocitrate with cytosolic malate [2]. Variants in the SLC25A1 gene are associated with severe neurometabolic disease [3,4,5]. We previously identified a homozygous c.740G>A; p.(Arg247Gln) missense variant in SLC25A1 in a British sib-pair presenting with a mild form of CMS with intellectual disability [6]. Here we report three additional unrelated CMS families carrying the same missense variant in SLC25A1, presenting with a similar phenotype, confirming the genotype–phenotype association.
Materials and methods
Patients
Patients were identified and fully investigated with standard clinical, electrophysiological and myopathological examinations at specialised neurology and neuromuscular clinics, namely Department of Neurology, National Institute of Mental Health and Neuroscience, India, Department of Neuropediatrics and Muscle Disorders, Freiburg University, Germany and the NHS Highly Specialised Mitochondrial disease service, Newcastle Upon Tyne hospitals NHS Foundation Trust, UK. Written consent was obtained for all participants to publish their clinical photographs. DNA samples were submitted to the Newcastle MRC Centre Biobank for Neuromuscular Diseases for which ethical approval was granted by the NRES Committee North East—Newcastle & North Tyneside 1 (reference 08/H0906/28). We have conducted the research in accordance with the Declaration of Helsinki.
Whole exome analysis (WES) and Haplotype analysis
WES was performed by the Genomics Platform at the Broad Institute of MIT and Harvard, Cambridge, USA. Libraries were created with an Illumina exome capture (38 Mb target) and sequenced with a mean target coverage of >80×. Exome data was then processed at the Centro Nacional de Análisis Genómico (CNAG), Barcelona, Spain, and variant prioritization was carried out on the Genome–Phenome Analysis Platform. Standard filtering criteria were applied, including minor allele frequency of 1% and high to moderate effect on protein structure (i.e. nonsense, splice site, frame shift, in-frame and non-synonymous variants), and using a gene list of 416 genes known to be associated with neuromuscular disease. The identified SLC25A1 variant has been submitted to ClinVar ((https://www.ncbi.nlm.nih.gov/clinvar/) under accession number: SCV000853306.1). Genomic data for patients 1/1, 2/1 and 2/2 were analysed for runs of homozygosity (RoH) using PLINK (http://zzz.bwh.harvard.edu/plink/ibdibs.shtml) and the respective haplotypes were identified manually.
Results
Clinical description
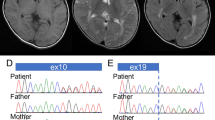
We identified six patients from four families of different ethnicity (British, Indian, Greek and Pakistani) (Table 1 and Fig. 1). Three families were consanguineous. All patients developed a non-progressive proximal weakness in early infancy, which fluctuated with physical activity and was clearly fatigable on examination. Some degree of involvement of extraocular muscles was found in all patients, which ranged from severe bilateral ptosis (Fig. 1c, patient 3/1) to subjective diplopia only (patient 1/2). Respiratory function was normal in all cases. In addition, all patients had mild intellectual disability. No symptoms of autonomic dysfunction were reported by any of the patients. MRI brain was performed in four cases and was normal in all. Serum lactate was normal at rest in all, but in two cases showed increase after exercise. Two patients had functional benefit from acetylcholinesterase (AChE) inhibitor treatment, but in other cases it was not beneficial despite adequate dosing. One case was treated with 3, 4-diaminopyridine and responded well to the treatment, in-keeping with a presynaptic NMJ defect (Table 1).

Representative clinical pictures of patients with a pathogenic SLC25A1 variant. a Symmetrical facial weakness and ophthalmoparesis (patient 2/1). b Ptosis and mild facial weakness (patient 2/2). c Severe ptosis with compensatory over-activation of frontalis and mild facial weakness (patient 3/1)
Repetitive nerve stimulation (RNS) showed decrement in two patients at 3 Hz stimulation, and significantly increased jitter on single fibre electromyography in five patients.
Muscle biopsy was performed in three cases and showed non-specific myopathic features in all. In addition, on electron microscopy two cases showed enlarged and accumulated mitochondria.
Genetic analysis
After WES variant filtering, the same homozygous missense variant hg19 chr22:g.19164098C>T; c.740G>A; p.(Arg247Gln) in SLC25A1 (GenBank NM_005984.5) was identified in all patients. Sequencing data for patients 1/1 (British origin), 2/1 and 2/2 (Indian origin) were analysed to identify possible RoH around the SLC25A1 variant. As expected due to their consanguinity, patients 2/1 and 2/2 shared a large RoH of 1.43 Mb (from chr22:18870865 to 20325552), whereas patient 1/1 had a much smaller RoH of 0.15 Mb (from chr22:19148244 to 19299703). The genotypes within the shared region between the two families differ in at least five homozygous calls (i.e. hg38 chr22: g.19177322A>C; rs3213491, hg38 chr22:g.19199939G>A; rs2236760, hg38 chr22:g.19199940C>G; rs2236761, hg38 chr22:g.19208992G>A; rs1060376 and hg38 chr22:g.19258298A>G; rs3788295), suggesting that they represent two distinct haplotypes.
Discussion
Pathogenic variants in SLC25A1 cause combined D-2- and L-2-hydroxyglutaric aciduria (D/L-2-HGA), a severe metabolic neurodevelopmental disorder with early lethality [3,4,5]. We previously reported that the homozygous p.(Arg247Gln) missense variant caused a CMS phenotype but without systemic manifestations of a mitochondrial disease [6]. Here, we report three additional CMS families of different ethnicity harbouring the same homozygous missense variant. Interestingly, the haplotype of the Indian and British families [6] were distinct, suggesting this variant is not seen as a result of a founder effect but rather to recurrent mutation.
Clinically these families had early onset, fatigable ocular, bulbar and proximal muscle weakness, which are clinical hallmarks of impaired neuromuscular transmission. In addition, cognitive impairment was found in all cases. Several presynaptic CMS genes including SNAP25B [7], MUNC13 [8], MYO9A [9], SLC18A3 [10] and SLC5A7 [11], as well as mutations in DPAGT1 [12], are known to be associated with cognitive and behavioural problems, which may reflect the importance of these genes in central as well as peripheral synapses. Our findings suggest that SLC25A1 and in particular the p.(Arg247Gln) variant should also be considered in CMS cases with intellectual disability.
Ptosis, ophthalmoplegia and fatigable muscle weakness can also occur in mitochondrial disease along with many other severe systemic symptoms [13]. Unlike the typical clinical progression in mitochondrial disorders, the muscle weakness in our cohort was non-progressive. Pathogenic variants in SLC25A1 which cause D/L-2-HGA are typically in amino acid residues which are crucial for protein activity, whereas the variant documented here was shown to result in reduced carrier activity with compensatory increase in protein expression [6, 14]. Consistent with this, all patients were found to have normal urinary 2-HG levels. In addition, muscle biopsy showed subtle mitochondrial abnormalities on electron microscopy, but normal or only mildly reduced mitochondrial oxidative enzyme reactivities in-keeping with a mild mitochondrial defect.
In previous studies, we showed that SLC25A1 deficiency results in a primary presynaptic defect [6]. Mitochondria play important roles in the presynaptic nerve terminal, which is a site of high energy requirement [15]. In particular, mitochondria are involved in the regulation of presynaptic calcium [16]. Lambert Eaton Myasthenic Syndrome (LEMS) is caused by antibodies against the presynaptic voltage gated calcium channel. Another CMS phenotype overlapping with LEMS, which results in impaired presynaptic calcium signalling, has been described recently [17]. This includes increment on RNS after a period of maximum voluntary contraction, and response to 3,4-diaminopyridine (3,4-DAP), a presynaptic potassium channel blocker. However, none of the patients in this cohort exhibited features in-keeping with LEMS on neurophysiological tests. 3,4-DAP resulted in good clinical benefit in one case, but has not been trialled in other cases.
In summary, the p.(Arg247Gln) SLC25A1 variant should be considered in patients presenting with a presynaptic CMS, particularly with accompanying intellectual disability.
Change history
20 November 2019
The original publication had the author name Atcharayam Nalini displaying incorrectly. This has been corrected in the PDF and XML.
References
Engel AG. Congenital myasthenic syndromes in 2018. Curr Neurol Neurosci Rep. 2018;18:46.
Kaplan RS, Mayor JA, Wood DO. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J Biol Chem. 1993;268:13682–90.
Nota B, Struys EA, Pop A, Jansen EE, Fernandez Ojeda MR, Kanhai WA, et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am J Hum Genet. 2013;92:627–31.
Cohen I, Staretz-Chacham O, Wormser O, Perez Y, Saada A, Kadir R, et al. A novel homozygous SLC25A1 mutation with impaired mitochondrial complex V: possible phenotypic expansion. Am J Med Genet A. 2018;176:330–6.
Smith A, McBride S, Marcadier JL, Michaud J, Al-Dirbashi OY, Schwartzentruber J, et al. Severe neonatal presentation of mitochondrial citrate carrier (SLC25A1) deficiency. JIMD Rep. 2016;30:73–9.
Chaouch A, Porcelli V, Cox D, Edvardson S, Scarcia P, De Grassi A, et al. Mutations in the mitochondrial citrate carrier SLC25A1 are associated with impaired neuromuscular transmission. J Neuromuscul Dis. 2014;1:75–90.
Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology. 2014;83:2247–55.
Engel AG, Selcen D, Shen XM, Milone M, Harper CM. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol Genet. 2016;2:e105
O’Connor E, Topf A, Muller JS, Cox D, Evangelista T, Colomer J, et al. Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome. Brain. 2016;139:2143–53.
O’Grady GL, Verschuuren C, Yuen M, Webster R, Menezes M, Fock JM, et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology. 2016;87:1442–8.
Bauche S, O’Regan S, Azuma Y, Laffargue F, McMacken G, Sternberg D, et al. Impaired presynaptic high-affinity choline transporter causes a congenital myasthenic syndrome with episodic apnea. Am J Hum Genet. 2016;99:753–61.
Selcen D, Shen XM, Brengman J, Li Y, Stans AA, Wieben E, et al. DPAGT1 myasthenia and myopathy: genetic, phenotypic, and expression studies. Neurology 2014;82(May):1822–30.
Chinnery PF. Mitochondrial disease in adults: what’s old and what’s new? EMBO Mol Med. 2015;7:1503–12.
Prasun P, Young S, Salomons G, Werneke A, Jiang YH, Struys E, et al. Expanding the clinical spectrum of mitochondrial citrate carrier (SLC25A1) deficiency: facial dysmorphism in siblings with epileptic encephalopathy and combined D,L-2-hydroxyglutaric aciduria. JIMD Rep. 2015;19:111–5.
Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010;2:139.
Zenisek D, Matthews G. The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron. 2000;25:229–37.
Whittaker RG, Herrmann DN, Bansagi B, Hasan BA, Lofra RM, Logigian EL, et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology. 2015;85:1964–71.
Acknowledgements
The authors are grateful to the patients for giving them permission to share their data and photographs. We thank the Broad Institute for performing WES in patients 1/1 and 1/2 as part of the CMG project; the Broad Centre for Mendelian Genomics (UM1 HG008900) is funded by the National Human Genome Research Institute with supplemental funding provided by the National Heart, Lung, and Blood Institute under the Trans-Omics for Precision Medicine (TOPMed) program and the National Eye Institute. Data was analysed using the RD-Connect Genome–Phenome Analysis platform developed under FP7/2007–2013 funded project (grant agreement no. 305444). We thank CNAG members for their technical support.
Funding
The project is supported by Newton fund under the governance of Academy of Medical Sciences. SB is a Newton International postdoctoral fellow (Ref no.NIF003/1002). RH was supported by the Medical Research Council (UK) [MR/N025431/1], the Wellcome Investigator fund [109915/Z/15/Z], the Newton Fund [UK/Turkey, MR/N027302/1], the European Research Council [309548] and the Wellcome Trust Pathfinder Scheme [201064/Z/16/Z]. RM and RWT are supported by the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), the MRC Centre for Neuromuscular Diseases (G0601943), Newcastle University Centre for Ageing and Vitality (supported by the Biotechnology and Biological Sciences Research Council and Medical Research Council (G016354/1)), the UK NIHR Biomedical Research Centre in Age and Age Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation, the MRC/ESPRC Newcastle Molecular Pathology Node, the Lily Foundation and the UK National Health Service Highly Specialised Service for Rare Mitochondrial Disorders. HL is supported by a project grant of the Canadian Institutes of Health Research (CIHR, PJT 162265).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Balaraju, S., Töpf, A., McMacken, G. et al. Congenital myasthenic syndrome with mild intellectual disability caused by a recurrent SLC25A1 variant. Eur J Hum Genet 28, 373–377 (2020). https://doi.org/10.1038/s41431-019-0506-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0506-2
