
Abstract
Clinical genomic sequencing can identify pathogenic variants unrelated to the initial clinical question, but of medical relevance to the patients and their families. With ongoing discussions on the utility of disclosing or searching for such variants, it is of crucial importance to obtain unbiased insight in the prevalence of these incidental or secondary findings, in order to better weigh potential risks and benefits. Previous studies have reported a broad range of secondary findings ranging from 1 to 9%, merely attributable to differences in study design, cohorts tested, sequence technology used and genes analyzed. Here, we analyzed WES data of 1640 anonymized healthy Dutch individuals to establish the frequency of medically actionable disease alleles in an outbred population of European descent. Our study shows that 1 in 38 healthy individuals (2.7%) has a (likely) pathogenic variant in one of 59 medically actionable dominant disease genes for which the American College of Medical Genetics and Genomics (ACMG) recommends disclosure. Additionally, we identified 36 individuals (2.2%) to be a carrier of a recessive pathogenic disease allele. Whereas these frequencies of secondary findings are in line with what has been reported in the East-Asian population, the pathogenic variants are differently distributed across the 59 ACMG genes. Our results contribute to the debate on genetic risk factor screening in healthy individuals and the discussion whether the potential benefits of this knowledge and related preventive options, outweigh the risk of the emotional impact of the test result and possible stigmatization.
Similar content being viewed by others


Introduction
Clinical genomic sequencing can identify pathogenic variants unrelated to the initial clinical question, that are of medical relevance to the patient and their families [1]. To promote standardized reporting of these incidental (unintentionally detected in analysis) and/or secondary findings (deliberate analysis of available data), the American College of Medical Genetics and Genomics (ACMG) published a list of 59 medically actionable genes recommended for return of such findings [2]. The potential impact of reporting actionable variants in these genes would be significant and far-reaching as it presents opportunities to prevent disease.
There is an ongoing debate among medical genetic societies worldwide, and the general public, on whether, how, and when, incidental findings and/or secondary findings are to be disclosed or screened for [3]. Simultaneously, discussions on obligatory genetic testing of employees and disclosure of the results to their employers are taking place. Some important arguments in favor of routine screening of genomic data are potential improvement of an individual’s health, contribution to scientific progress and circumventing expensive treatments. Arguments against routine screening include possible harm to a person by complications of (unnecessary) medical interventions, stigmatization, and negative psychosocial impact [4]. Yet, with the decreasing costs for genome sequencing and a growing commercial (direct-to-consumer) market, genetic testing of healthy individuals might eventually be inevitable.
It is of importance to obtain unbiased insight in the potential risks and benefits of opportunistic screening, and to develop adequate education for the general public. To foster such discussions, knowledge on the prevalence of secondary findings in medically actionable genes in the general population is required. Recently, multiple studies have reported frequencies of secondary findings ranging from 1 to 9% in various populations [5,6,7,8,9,10,11,12,13]. This broad range of reported frequencies is largely explained by the cohorts tested (e.g., inclusion of individuals more prone to have a pathogenic variant) in combination with differences in sequence technology (e.g., whole-exome sequencing (WES) of inferior quality), classification of variants, and amount of genes for which pathogenic variants are taken into account. To the best of our knowledge, an unbiased prevalence of secondary findings in healthy individuals of European descent identifiable using clinical WES has not yet been described. Here, we analyzed clinical grade WES data of >1500 healthy individuals to establish the frequency of medically actionable disease alleles in the general Dutch population.
Material and methods
In our tertiary clinical genetic center in the Netherlands, 1640 healthy parents (50% males) received family based WES to allow for the interpretation of de novo mutations as cause of the intellectual disability observed in their child [14]. The parents were predominantly of Caucasian origin and from an outbred, nonconsanguineous population [14]. For the purpose of this exploratory and observational study, parental exome data were anonymized. None of these parents carried a known detrimental allele for intellectual disability.
WES was performed following our routine diagnostic procedures [15]. In essence, DNA was outsourced to BGI (Copenhagen, Denmark) where exomes were captured using Agilent Sureselect v4 and sequenced to a median coverage of 75-fold on an Illumina HiSeq instrument with 101-bp paired-end reads. Sequence reads were aligned to the hg19 using BWA version 0.5.9-r16. Variants were called in-house using GATK unified genotyper (version 3.2–2) and annotated using custom diagnostic annotation pipeline, using Human Genome Variant Society nomenclature [16]. Variant interpretation was limited to high quality variants (GATK quality score ≥ 500) eliminating false-positive calls [17], and to those that occurred in the 59 medically actionable genes [2]. Of note, 97.7% of the coding sequence for these genes was covered ≥20-fold. Variants in these genes were prefiltered for truncating, canonical splice sites, insertion deletion and/or missense variants based on frequency of occurrence in dbSNPv137 (<5%), ExACr0.2 (<1%) and our in-house database (<1%) containing exome data of 12,244 exomes. Remaining variants were classified according to the ACMG guidelines for diagnostic variant interpretation [18]. Variants classified as pathogenic and likely pathogenic, referring to the potential of the variant to cause disease in a specific context, were considered medically actionable, and percentages referred to in our study are based on these classifications.
Results
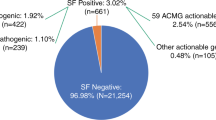
In a cohort of 1640 anonymized healthy individuals, we classified all variants in the 59 ACMG medically actionable genes, including 56 dominant and 3 recessive genes, using the standardized ACMG interpretation and classification variant guidelines [18].
In total, 44 individuals (2.7%) of our cohort had a dominant medically actionable variant, including 33 unique variants, which were detected in 18 out of the 56 dominant actionable genes. Six of 33 variants were detected in more than one individual. Disease alleles in genes for cardiac disease were most frequently observed (24 individuals, 1.5%), with variants in MYBPC3 (NM_000256.3), responsible for hypertrophic cardiomyopathy, most often reported (seven individuals). Pathogenic variants in genes predisposing to hereditary cancer were detected in 11 individuals (0.7%), including five individuals with a pathogenic variant in BRCA1 (NM_007300.3) and three others in BRCA2 (NM_000059.3), both associated with hereditary breast and ovarian cancer. None of the individuals had more than one dominant high-risk disease allele.
In addition to dominant disease alleles, we also identified 36 individuals (2.2%) to be carriers of a high-risk disease allele in two of the three recessive actionable genes (Fig. 1; Supplementary Table S1). Pathogenic variants were observed in MUTYH (NM_001128425.1; 31 individuals) and ATP7B (NM_000053.2; five individuals), known to cause MUTYH Associated Polyposis and Wilson disease, respectively, when present in compound heterozygous or homozygous state. None of the 36 individuals carried homozygous or compound heterozygous recessive high-risk disease alleles. One carrier of a heterozygous recessive high-risk disease allele also had a dominant high-risk disease allele.

Schematic representation of actionable (likely) pathogenic variants identified in 1640 healthy individuals in the 59 ACMG genes. Data is visualized by type of disease (cardiogenetic, oncogenetic, connective tissue, and other). Mode of inheritance is represented in blue for dominant disease genes and orange for recessive disease genes. X-linked genes are indicated by #. All detected (likely) pathogenic variants and their classification according to HGVS recommendations [16] and ACMG-AMP guidelines [18], respectively, are provided in Supplementary Table 1. Abbreviations: HCM hypertrophic cardiomyopathy; DCM dilated cardiomyopathy; ARVC arrhythmogenic right ventricular cardiomyopathy; TSC tuberous sclerosis complex; HBOC hereditary breast and ovarian cancer; n.a. not applicable
Discussion
On March 8 2017, the US House of Representatives approved a Bill that would allow companies to require employees to undergo genetic testing and disclose the results to their employers [19]. As a response, the European Society of Human Genetics provided a statement that strongly argued against obligatory genetic testing as decisions on whether or not to undergo genetic testing must be a voluntary choice of the individual [20]. For both obligatory and voluntary testing of healthy individuals, it is, however, important to know the prevalence of medically actionable disease alleles in an unbiased population. In this study, we set out to determine this frequency by screening for secondary findings in a healthy population of European descent using existing (anonymized) WES data. From our data we conclude that 2.7% of healthy Dutch individuals has a (likely) pathogenic variant in a medically actionable dominant disease allele for which returning of secondary findings is indicated by the ACMG. These individuals are predisposed to develop for instance cancer or cardiomyopathy.
Given the wide range of reported secondary findings, we systematically compared our results to studies published previously from other populations [5,6,7,8,9,10,11,12,13] in order to explain the differences in frequencies observed, focussing on (i) the sequence technology used, (ii) the cohort tested, (iii) the certainty of pathogenicity of variants, as well as (iv) the genes for which pathogenic variants were assessed. This comparison yielded three distinct categories: those studies reporting lower [5, 7, 10, 11, 13], higher [6, 8, 9], or comparable [12] frequencies of secondary findings when compared to our observation of 2.7%.
In comparison to the first studies reporting incidental findings [5, 7, 10, 11], the frequency identified in our cohort is elevated up to twice as high. This could partially be explained by the presence of Dutch founder mutations. From the 33 unique dominant risk alleles detected, two have been reported as founder mutation for the Dutch population (BRCA1 (c.2685_2686del) and BRCA2 (c.9672dup)) [21]. However, both variants were observed in a single individual in our cohort, thus not accounting for the higher frequency observed in our study. We then examined whether the differences are explained by experimental design and data analysis, previous versions of databases for variant filtering and interpretation, absence of ethnicity specific variation information and a shorter list of ACMG medically actionable genes for disclosing secondary findings. However, all variants identified in our study were also present in databases at time of the initial studies, and also, the extension of the ACMG genes with 3 additional genes to 59 as analyzed here, is not sufficient to explain our observed higher frequency. It thus is more likely that the increased sequence coverage in our study allowed more sensitive detection of (likely) pathogenic variants. It is, however, noteworthy that, Thompson et al. [13] also identified a relatively low frequency (1.5%) of secondary findings when based on (likely) pathogenic variants in the 59 ACMG genes, despite using WES at an average sequence depth of 71×, covering 80% of bases at least 20-fold [13, 22]. Whether this coverage is also achieved for the coding sequencing of the 59 genes is, however, not reported [13], but any deviation from this, could potentially explain the differences observed. Overall, since also other recent publications report higher frequencies than the 1–1.5% previously reported, it seems reasonable to conclude that the frequencies initially reported are too low.
Higher prevalence of secondary findings compared to our 2.7% for the Dutch population have also been reported [6, 8, 9]. For instance, Dewey et al. (2016) reported 49 variants in the 59 ACMG genes in 1415 individuals (3.5%). Their cohort, however, consisted of patients of whom some were affected with conditions likely attributable to the disease alleles in the ACMG genes, thus creating bias towards higher frequency. When excluding this bias, their findings are more in line with our frequency. Interestingly, several papers report frequencies of over 5% [8, 9]. Since these studies were conducted at the same time as ours, the difference cannot be explained by the previously mentioned issues like sequence coverage and availability of newer releases of databases for variant filtering. As was also noted by Tang et al. [12], the high frequency reported in these studies is mainly due to improper classification of variants as pathogenic. For instance, the NM_198056.2:c.3575G > A variant in SCN5A reported by Lawrence et al. [9] is present in 6% of the Asian population (including homozygotes) and truncating variants in the RYR1 (NM_000540.2) gene are not causative for malignant hyperthermia susceptibility, as proposed by Jang et al. [8].
Importantly, the frequency of secondary findings observed in our cohort is almost identical to the prevalence of 2.5% published by Tang et al. [12], who tested a cohort of 954 East-Asian individuals using WGS. Interestingly, however, the distribution of pathogenic variants over the genes differs markedly despite the overall the frequency of secondary findings in the cardiogenetic genes and oncogenes being similar; that is, Tang et al. [12] reported 36% of their pathogenic variants in seven of the 59 ACMG genes in which no pathogenic variants were detected in our cohort. Conversely, in our Dutch cohort 48% of the detected pathogenic variants were in nine of the 59 ACMG genes in which no pathogenic variants were detected by Tang et al. Hence, this may indicate that, although the frequency of secondary findings is similar between different ethnicities, different genes contribute to their prevalence.
We also identified 2.2% of the population to be a carrier of a recessive pathogenic disease allele in one of the 59 ACMG genes. Whereas the identification of carriers of recessive disease alleles in the population is not unexpected given our study set up, our unbiased analysis of these alleles in the healthy population elicits discussion on their return. The ACMG recommends to only return bi-allelic pathogenic variants, but one may wonder whether it is not relevant to return carrier status (e.g., for reproductive decisions). Using the carrier frequency determined in our cohort, we can now determine that ~1 in 3000 and ~1 in 100,000 couples are both carrier of a heterozygous disease allele in MUTYH or ATP7B, respectively. For comparison, in March 2017 the Committee on Genetics of the American College of Obstetricians and Gynecologists stated that cystic fibrosis screening, with a carrier frequency of 1 in 30 in the Caucasian population, should be offered to all pregnant women, or ideally before pregnancy [23]. Our data do not only contribute to the discussion on genes that could be selected for preconception carrier screening based on absolute carrier frequency, such as here presented for MUTYH (~1:50), but also reopens the discussion whether or not more prevalent diseases, such as cystic fibrosis (~1:30), should be included in secondary screening programs.
Routine screening of healthy individuals for secondary findings in the 59 ACMG medically actionable dominant disease genes will impact at least 1 in 38 individuals. These individuals have an increased risk for life-threatening disease, and could profit from early monitoring and possible preventive treatment. On the other hand, some of the individuals in whom an incidental findings is identified may spend their lives worrying about a disease that may never manifest itself. This is mostly due to a highly variable disease penetrance for these conditions, ranging from 20 to 100%. Our study design did not allow to link the secondary findings to individuals and their families, but it can be expected that the penetrance is even lower in the absence of a positive family history. In terms of policy decisions about reporting and counseling of individuals in whom an incidental and/or secondary finding is observed, this may lead to a redefinition of what is perceived as a “medically actionable disease allele”. Also, individuals at risk may face difficulties—or may even be unable to—acquire job positions, obtain mortgages, and/or health and/or life insurances. These implications not only affect the individuals in whom the incidental or secondary finding was uncovered, but will also directly impact their blood relatives and extended families. Apart from practical implications, such as the impact on the health care system to screen healthy individuals, it is presently unclear whether the potential benefits of early monitoring and possible preventive treatment outweigh the risks of the emotional impact of the test result and possible stigmatization.
Taken together, we believe that our conclusion that 2.7% of healthy Dutch individuals has a dominant acting disease allele, is expected to be representative for the European population given the current guidelines on variant interpretation and the limited number of genes studied. Yet, with genetic knowledge still advancing, the number of genes and medically actionable variants for which disclosure could be considered will likely continue to expand. In addition, improvements in sequencing technology will likely allow detection of more variants, and simultaneously, increasing clinical interpretation of the noncoding parts of the genome will allow for the detection of more pathogenic variation. Hence, it may be expected that our estimate that 1 in 38 healthy individuals is genetically affected with a dominant high-risk disease is an underrepresentation for the true prevalence of dominant medically actionable disease alleles in the population.
References
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SFv2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55.
Vears DF, Senecal K, Clarke AJ, et al. Points to consider for laboratories reporting results from diagnostic genomic sequencing. Eur J Hum Genet. 2018;26:36–43.
Wouters RHP, Bijlsma RM, Frederix GWJ, et al. Is it our duty to hunt for pathogenic mutations? Trends Mol Med. 2018;24:3–6.
Amendola LM, Dorschner MO, Robertson PD, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015;25:305–15.
Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354:1550–59.
Jamuar SS, Kuan JL, Brett M, et al. Incidentalome from genomic sequencing: a barrier to personalized medicine? EBioMedicine. 2016;5:211–6.
Jang MA, Lee SH, Kim N, Ki CS. Frequency and spectrum of actionable pathogenic secondary findings in 196 Korean exomes. Genet Med. 2015;17:1007–11.
Lawrence L, Sincan M, Markello T, et al. The implications of familial incidental findings from exome sequencing: the NIH Undiagnosed Diseases Program experience. Genet Med. 2014;16:741–50.
Natarajan P, Gold NB, Bick AG, et al. Aggregate penetrance of genomic variants for actionable disorders in European and African Americans. Sci Transl Med. 2016;8:364ra151.
Olfson E, Cottrell CE, Davidson NO, et al. Identification of medically actionable secondary findings in the 1000 genomes. PLoS One. 2015;10:e0135193.
Tang CS, Dattani S, So MT, et al. Actionable secondary findings from whole-genome sequencing of 954 East Asians. Hum Genet. 2018;137:31–37.
Thompson ML, Finnila CR, Bowling KM, et al. Genomic sequencing identifies secondary findings in a cohort of parent study participants. Genet Med. 2018 https://doi.org/10.1038/gim.2018.53. [Epub ahead of print].
de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Haer-Wigman L, van Zelst-Stams WA, Pfundt R, et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur J Hum Genet. 2017;25:591–9.
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–9.
Strom SP, Lee H, Das K, et al. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet Med. 2014;16:510–5.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
USA Congress: H.R. 1313—Preserving Employee Wellness Programs Act. https://www.congress.gov/bill/115th-congress/house-bill/1313/text.
European Society of Human Genetics: The European Society of Human Genetics condemns move to impose obligatory genetic testing for employees in the USA. March 16, 2017. https://www.eshg.org/.
Rebbeck TR, Friebel TM, Friedman E, et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat. 2018;39:593–620.
Bowling KM, Thompson ML, Amaral MD, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9:43.
Committee on G. Committee Opinion No. 691: carrier screening for genetic conditions. Obstet Gynecol. 2017;129:e41–e55.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Haer-Wigman, L., van der Schoot, V., Feenstra, I. et al. 1 in 38 individuals at risk of a dominant medically actionable disease. Eur J Hum Genet 27, 325–330 (2019). https://doi.org/10.1038/s41431-018-0284-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0284-2
This article is cited by
-
Rapid genomic sequencing for genetic disease diagnosis and therapy in intensive care units: a review
npj Genomic Medicine (2024)
-
An spanish study of secondary findings in families affected with mendelian disorders: choices, prevalence and family history
European Journal of Human Genetics (2023)
-
Actionable secondary findings in the 73 ACMG-recommended genes in 1559 Thai exomes
Journal of Human Genetics (2022)
-
Lessons learned from unsolicited findings in clinical exome sequencing of 16,482 individuals
European Journal of Human Genetics (2022)
-
Molecular Diagnosis of Primary Cardiomyopathy in 231 Unrelated Pediatric Cases by Panel-Based Next-Generation Sequencing: A Major Focus on Five Carriers of Biallelic TNNI3 Pathogenic Variants
Molecular Diagnosis & Therapy (2022)
