
Abstract
The Pamirs, among the world’s highest mountains in Central Asia, are one of homelands with the most extreme high altitude for several ethnic groups. The settlement history of modern humans on the Pamirs remains still opaque. Herein, we have sequenced the mitochondrial DNA (mtDNA) genomes of 382 individuals belonging to eight populations from the Pamirs and the surrounding lowlands in Central Asia. We construct the Central Asian (including both highlanders and lowlanders) mtDNA haplogroup tree at the highest resolution. All the matrilineal components are assigned into the defined mtDNA haplogroups in East and West Eurasians. No basal lineages that directly emanate from the Eurasian founder macrohaplogroups M, N, and R are found. Our data support the origin of Central Asian being the result of East–West Eurasian admixture. The coalescence ages for more than 93% mtDNA lineages in Central Asians are dated after the last glacial maximum (LGM). The post-LGM and/or later dispersals/admixtures play dominant roles in shaping the maternal gene pool of Central Asians. More importantly, our analyses reveal the mtDNA heterogeneity in the Pamir highlanders, not only between the Turkic Kyrgyz and the Indo-European Tajik groups, but also among three highland Tajiks. No evidence supports positive selection or relaxation of selective constraints in the mtDNAs of highlanders as compared to that of lowlanders. Our results suggest a complex history for the peopling of Pamirs by multiple waves of migrations from various genetic resources during different time scales.
Similar content being viewed by others


Introduction
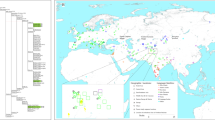
The settlements of high-altitude regions are great achievements for various human populations in their dispersal history [1]. In Central Asia, the Pamirs are a mountain knot (i.e., the Pamirian Knot) in the heartland of Asia at the junction of the Himalayas with the Hindu Kush, Tien Shan, Karakoram, and Kunlun ranges (Fig. 1). Due to having high average elevation (2500–4500 m), the Pamirs was first referred to as “Roof of the World” in 1838 [2]. The harsh conditions (e.g., hypoxia, strong ultraviolet radiation, and cold temperature) associated with high altitude makes the Pamirs as one of the most challenging environments for human colonization [3, 4]. The modern Pamir highlanders mainly refer to two linguistic groups. The Indo-European speakers, highland Tajiks, are proposed to be indigenous to the Pamirs [4]. The Turkic-speaking Kyrgyz people are recent immigrants. Their settlement in Pamirs can be traced back to the Turko-Mongol expansions of the medieval period. Some tribes were driven into the Pamirs during the war against the Dzungar Khanate around 1680–1740s AD [5].

The early peopling of the Pamirs was around 10 kya (thousand years ago), as revealed by limited archeological sites and rock paintings [6]. Around 4000 years before present, the migrations of Andronovo Culture reached the Pamirs. And then, the Scythians (also known as Saka and Sai), the speakers of Indo-Iranian languages, began to dominate this plateau [7]. It indicates a wave of eastward expansion of the Indo-European language family [7]. Although the Scythians are evident in archeological and historical records [6], their population history remains unclear. Nowadays, the Pamirs are the homelands to the highland Tajiks [4]. Both linguistic and physical anthropological evidence suggest the highland Tajiks likely being the descendants of ancient Scythians [7, 8]. Thus, to investigate the gene pool of modern highland, Tajiks may provide us an opportunity to retrace the origin of early highlanders as well as their settlement history in the Pamirs.
During the past two decades, the study of mitochondrial DNA (mtDNA) has extensively improved our understanding of genetic diversity and population history of Central Asians [9,10,11]. Several efforts have been done to retrieving the genetic information from ancient mtDNA sequences [12, 13]. However, due to various kinds of difficulty in sample collection, only limited samples of highland Tajiks were referred recently [14]. Meanwhile, most studies relied upon partial mtDNA sequences (i.e., D-loop and certain coding regions). The limited information hampers further dissection of mtDNA lineages into fine resolution. The recurrent variants occurring in the D-loop can make the haplogroup assignment ambiguous [15]. In previous studies, researchers just selected particular Central Asian individuals of interest for complete mtDNA genome sequencing (e.g., ref. [16]). Such biased sampling and limited sequence data are unsuitable for many demographic analyses [17]. To date, there were few reports focusing on the mtDNA genomes of Central Asians. Lacking of a well-resolved phylogeny of Central Asian mtDNA lineages impedes our knowledge of human evolution and demographic history in this region.
In this study, we focus on the highlanders living in the Pamirs, including three highland Tajik populations and two highland Kyrgyz populations (Table 1). For comparisons, we also investigated three lowland populations neighboring to the Pamirs. We used the next-generation sequencing (NGS) technique to generate the unbiased, population-based data including a total of 382 mtDNA genomes. Our results not only help further understand the mtDNA phylogeny in Central Asia, but also shed novel light on the peopling of the Pamirs.
Materials and methods
Sampling and data collection
We collected the peripheral blood samples of 382 unrelated adult individuals, which can be attributed to eight populations based on geographic location and ethnic information (Table 1 and Fig. 1). Written informed consents were obtained from all individuals. The study protocol was approved by the Internal Review Board of Kunming Institute of Zoology, Chinese Academy of Sciences, Xinjiang Medical University, and E.N. Pavlovsky Institute of Zoology and Parasitology, Academy of Sciences of Republic of Tajikistan. The published population data from East Asia, South Asia, West Asia [18], and Caucasus [17] were collected for comparison (Table 1).
DNA extraction, mitochondrial genome amplification, and sequencing
Genomic DNA was extracted from the blood samples by using the phenol-chloroform method. The complete mtDNA genome was amplified by the long-range PCR protocol to generate two overlapping amplicons of ~8.5 kb [19]. We constructed the pooled libraries according to the instruction provided by Ion Torrent (Life Technologies). The pooled libraries were amplified by emulsion PCR and sequenced in the Ion 316TM Chip with the Ion PGM™ 400 Sequencing Kit. The raw data have been deposited into Genome Sequence Archive (http://bigd.big.ac.cn/gsa; project ID: PRJCA000486; accession numbers SAMC014618-SAMC014999; Table S1).
NGS data analyses
In the initial quality control, Torrent Suite removes the low-quality base to make sure the average Q value is more than 15 within 30 bp window on the 3′ end of reads. We used the revised Cambridge Reference Sequence (rCRS; GenBank: NC_012920) [20] as the reference. The reads mapping and variants calling were carried out with Ion Torrent Software Suite Plugin Variant Caller v5.0.2.1 as recommended by the manufacturer. For all the scored variants in vcf files, we further manually checked the corresponding bam files with Integrative Genomics Viewer [21] to confirm the status. We developed a python script to transfer the confirmed vcf file (vcf format) into the consensus sequence (fasta format) with the cutoff value of heteroplasmy as 0.2. The sequences of fasta format are available at GenBank under accession numbers MF522841-MF523222.
Analyses of mtDNA genome sequences
By using HaploGrep 2 [22], we scored the variants out for each of sequences relative to rCRS, and then assigned the mtDNA sequences into specific haplogroups. The results were further confirmed by using MitoTool [23]. The transition at 16519, the C-length variants in regions 16180–16193 and 303–315, and AC indels at 515–522 were disregarded in subsequent analyses. The nomenclatures were referred to PhyloTree (http://phylotree.org/; Build 17) [24]. The mtDNA haplogroup trees were reconstructed manually.
The indexes for nucleotide and haplotype diversity, and Tajima’s D, Fu’s Fs, and AMOVA tests were computed with ARLEQUIN 3.5.1.3 [25]. We compared the numbers of synonymous (S) and nonsynonymous (N) substitutions in the mtDNA protein-coding regions from the Pamir highlanders and the surrounding lowlanders to estimate the potential selective constraints as described before [26]. The 3639 sequences after a stringent quality-control procedure [26] were retrieved as the general populations for comparisons.
We estimated the average sequence divergence of the haplotypes to their most recent common ancestor by the rho statistic [27]. A heuristic estimate of the standard error (sigma) was calculated as described before [28]. The rho ± sigma was converted into the coalescent age for certain haplogroup by using the calibration rates for mtDNA coding region (np 577–16024) variants [29] and only synonymous substitutions [30], respectively. We also employed BEAST v1.8 [31] to date the nodes based on 382 coding region sequences with the fixed rate as 1.404 × 10−8 substitutions per site per year [29]. The strict clock model and coalescent Bayesian skyline were adopted. We used jModelTest 2.1.3 [32] to select GTR + I + G substitution model as well as estimate the related parameters. MCMC was run for 100,000,000 generations and sampled every 2000 generations. The burn-in for trees was set as 10%.
The Bayesian skyline plots (BSPs) for each of populations were conducted by using BEAST v1.8 [31] based on coding region sequences. The TrN + I + G substitution model as well as the related parameters were estimated with jModelTest 2.1.3 [32]. We chose strict clock model with the rate 1.404 × 10−8 substitutions per site per year [29]. Each MCMC was run for 300,000,000 generations and sampled every 5000 generations. The effective sample size values for the parameters of interest were over 300. The results were visualized with Tracer v1.5 (http://tree.bio.ed.ac.uk/software/tracer/).
The approximate Bayesian computation (ABC) analyses were done by using DIYABC 2.1 [33]. We used Beijing Han Chinese and Persian populations (Table 1) to represent the modern East and West Eurasians, respectively. It should be noted that the two populations cannot be treated as the parental or ancestral proxies for Central Asians. We only considered Kyrgyz and Tajik populations. We carried out the pairwise sample/population comparison in terms of using four summary statistics: number of segregating sites in the pooled sample, mean of within-sample pairwise differences, mean of between-sample pairwise differences, and FST between two samples, as suggested before [17]. The number of simulated data sets in computation is 200,000. The scenario prior combination pre-evaluating and model/scenario checking are done by using PCA based on the Euclidian distance between observed and simulated summary statistics.
Results
Sequencing of mtDNA
After filtering the unmapped reads and reads with poor quality, the average mapped reads are 26,492 per individual (minimum 4346; maximum 76,814) (Table S1). The average depth is 300× per individual, ranging from 51× to 964×. The mtDNA genome is well covered for each of the individuals. Due to some technological defects of NGS, certain positions had higher false insertion—deletion rates [34] and then identified as phantom variants. We detect a total of 53 positions with potential errors and then corrected them (Table S2 and see “Materials and methods” for details).
MtDNA genetic diversity
We excluded the West Pamir Kyrgyz consisting of only three individuals (Table 1) in the subsequent analyses at the population level. The genetic diversity indexes in seven Central Asian populations are at a similar level, with the values observed in populations from East Asia (Beijing Han Chinese and Sherpa), South Asia (Punjabi), West Asia (Persian and Qashqai), and Caucasus (Armenian, Azeri, and Georgian) (Table 2). In Central Asians, the relatively low nucleotide diversity and haplotype diversity values are observed in lowland Tajik (0.00168) and Wakhi Tajik (0.979), respectively. The values of Tajima’s D and Fu’s Fs are negative in all populations, indicating that the high frequencies of rare or private variants in the mitochondrial genomes. It indicates that those populations underwent recent expansions [35], although some negative Fu’s Fs values are not significant statistically.
MtDNA haplogroup profiles
In terms of PhyloTree (http://www.phylotree.org/; Build 17) [24], we classify all 382 mtDNA sequences into 73 specific (sub-)haplogroups within the non-African macrohaplogroups M, N, and R (Table S3). We then incorporate all sequences to build the haplogroup tree for Central Asians (Table S4). No basal lineages directly split from macrohaplogroups M, N, and R are defined. Most (sub-)haplogroups in Central Asians are derived from the characterized haplogroups in East and West Eurasians. Some South Asian autochthonous haplogroups, such as M30, M33a, M35, M3, and M5, are also found (Table S3). In the Turkic group, the most dominant haplogroup is D4, accounting 13.2% in East Pamir Kyrgyz, 20.4% in lowland Kyrgyz, and 18.5% in lowland Uygur. In the Indo-European group, haplogroup H is prevailing in Sarikoli Tajik (22.1%), Pamiri Tajik (48.0%), Wakhi Tajik (37.9%), and lowland Tajik (32.1%). Within haplogroups, our data characterize a series of new subhaplogroups. For instance, three sequences (ST143, WT199, and WT217) cluster as a subhaplogroup of haplogroup H (Table S4). Interestingly, subhaplogroup U5a2a1, which was detected in one 700-year-old individual excavated from the eastern Pamirs [16], exists in four modern individuals of four populations (Table S4).
In terms of the haplogroup frequency profiles (Table S3), we carry out the analysis of molecular variance (AMOVA) to discern the genetic differentiations among groups. The division between the Turkic (East Pamir Kyrgyz, lowland Kyrgyz, and lowland Uygur) and Indo-European (lowland Tajik, Pamiri Tajik, Sarikoli Tajik, and Wakhi Tajik) speakers contributes 1.86% of total variation, and it is significant in AMOVA (P = 0.03128 ± 0.00588). We further compare the highlanders and lowlanders within the Turkic and Indo-European groups, respectively. The differentiation is not significant in both East Pamir Kyrgyz vs. lowland Kyrgyz and lowland Uygur (P = 0.33040 ± 0.01275) and Pamiri Tajik and Sarikoli Tajik and Wakhi Tajik vs. lowland Tajik (P = 0.33040 ± 0.01275) in AMOVA, suggesting a small differentiation of mtDNA haplogroup profiles between the highlanders and lowlanders.
The coalescent time of mtDNA lineages
The Central Asian mtDNA genomes cluster with the published sequences into 252 (sub-)haplogroups (Table S5). Among of them, the numbers of sequences are less than six in 85 subhaplogroups. We adopted the rho statistic [27] to calculate the coalescent ages for mtDNA lineages from Central Asians based on the haplogroup tree (Table S4). When just considering the mean values, the most ancient coalescent ages for Central Asians (including both highlanders and lowlanders) are observed in haplogroup U (Fig. 2 and Table S5), including U2b2 (~32–30 kya), U2c1b (~39–32 kya), and U3b (~32–30 kya). In the Pamir highlanders, the most ancient lineages of subhaplogroups U2b2 and U3b occur in East Pamir Kyrgyz (1/68; 1.4%) and Sarikoli Tajik (1/86, 1.1%), respectively (Table S4). The coalescence ages of lineages of haplogroups B4c1a, D4m, D4v, F2e, M30 (WT292), M33a3b, M7c1, T2g, and ×2p fall within the LGM. Those sporadic lineages make contributions of 2.9% (11/382) for the maternal gene pool. Both in the Pamir highlanders and the surrounding lowlanders, most (>93%) lineages coalesced after the LGM. More than 70% lineages coalesced within 10 kya. Notably, some subhaplogroups contain the small number of sequences, or show not in the ideally star-like structure. Those dating results are with huge standard errors (Table S5). The Bayesian dating for the 382 mtDNA genomes shows that more than 95% sequences coalesced after the LGM (Fig. S1), which is largely in agreement with the results of rho statistic (Fig. 2).

. The trees are scaled with the rho statistic based on coding region variations (Table S5). Colors indicate geographic affiliation of sequences (Table S4). The percentage indicates the proportion in both Pamir highlanders and surrounding lowlanders. The percentage in parentheses indicates the proportion in highlanders only. Haplogroups referring to highlanders, lowlanders, and both are labeled as rectangle, oval, and rhombus, respectively. Haplogroups including sequences from Indo-European and Turkic speakers are framed and underlined, respectively. WT292 and LT320 can be assigned as M30* and H*, respectively. Their ages are retrieved from the previous study [30]. a Schematic trees of macrohaplogroup M and haplogroups N9, A, B, and R9. Most lineages originate from East Asia; and some (i.e., M3, M5, M30, and M65) are from South Asia. b Schematic trees of haplogroups N1, W, X, R0, R2, JT, and U. These linages are prevailing in West Eurasia and South Asia
Genetic relationship of the Pamir highlanders with other populations
We employ PCA based on the distribution frequency of 23 haplogroups (Table S6) to display the relationships of the Pamir highlanders with other Eurasian populations (Fig. 3). The first principal component (PC1) mainly reflects a separation between East and West Eurasian. The East Pamir Kyrgyz, lowland Kyrgyz, and lowland Uygur populations are close to Beijing Han Chinese. The lowland Tajik, Pamiri Tajik, Sarikoli Tajik, and Wakhi Tajik cluster with the populations from Iran (Persian and Qashqai) and Caucasus (Armenian, Azeri, and Georgian). The PC2 further distinguishes the genetic differentiation within the Indo-European Tajiks. It separates Sarikoli Tajik and Pamiri Tajik from lowland Tajik and Wakhi Tajik. In the east pole, the Turkic speakers (East Pamir Kyrgyz, lowland Kyrgyz, and lowland Uygur) are close to each other.

. PC map of populations based on mtDNA haplogroup frequencies (Table S5). The populations generated in this study are in bold
Inference of maternal effective population size changes
We conduct Bayesian skyline plot (BSP) to infer historical variations in maternal effective population size based on mtDNA coding region sequences (Fig. 4). The earliest population increase took place 45–50 kya, which was most likely to be attributed to the common background of ancient demographics in various Asian populations [36]. From 40 to 20 kya, population growth lasts in lowland Uygur, whereas constant size is observed in other six populations. After the LGM (26.5–19 kya) [37] but before the Holocene, the population growths occur in East Pamir Kyrgyz, lowland Kyrgyz, lowland Tajik, Pamiri Tajik, and Sarikoli Tajik populations but not in lowland Uygur and Wakhi Tajik populations. During the last 2 kya, a decline of population size is seen in all seven Central Asian populations as well as in the Iranian population. The recent declines in BSPs are in conflicts with the expansions revealed by the negative values of Tajima’s D and Fu’s Fs (Table 2). One possible explanation for this discrepancy is that the false signals of population decline can be detected in BSP with biologically plausible combinations of population structure and sampling strategy [38].

. The bold black line represents the median population size estimate from Bayesian posterior distribution. The gray lines delimit the 95% highest posterior density (HPD) boundaries, accounting for uncertainty in the reconstructed phylogeny and substitution model parameters. All the lines were drawn within the lower 95% HPD boundary of the maximum time to the coalescent age
Approximate Bayesian computation analyses
To address the origin and admixture history of the Pamir highlanders, we perform ABC to test different scenarios. We propose the scenario of three populations: West Eurasian—Pamir highlander—East Eurasian (Fig. 5a). The admixture model indicates genetic admixture playing plays a fundamental role in the origin of Pamir highlander. Whereas the split model suggests that the Pamir highlander is an isolated population receiving minor or few gene flows from West and East Eurasians. In the more complex scenario off our populations: West Eurasian—Ancient Central Asian—Pamir highlander—East Eurasian, we propose the Ancient Central Asian as the unsampled ancestor population. And our aim is to test the admixture-admixture and split-admixture models for the origin of Pamir highlander (Fig. 5b).

. a The scenario of three populations. The split and admixture models for Kyrgyz (East Pamir Kyrgyz and lowland Kyrgyz) and Tajik (lowland Tajik, Pamiri Tajik, Sarikoli Tajik, and Wakhi Tajik) are included. b The scenario of four populations. The split-admixture and admixture-admixture models for Kyrgyz and Tajik are included. c The priors are list in the table. The generation time is 20 years [50]. The individual locus mutation rate ranges 1.00 × 10−9 to 5.00 × 10−6 substitutions per site per generation, following a gamma distribution (Shape = 2)
The pre-evaluation based on PCA shows the plots of observed data sets are overlapping with those of scenario prior combinations (Fig. S2), suggesting our proposed scenarios and priors are compatible with the observed data sets. In the scenarios of three populations, the data of East Pamir Kyrgyz, lowland Kyrgyz, and Sarikoli Tajik fit the admixture model well, while lowland Tajik, Pamiri Tajik, and Wakhi Tajik prefer the split model (Fig. S3). When considering the more complex scenarios of four populations, with the exception of lowland Kyrgyz fitting, the admixture-admixture model, the rest five populations prefer the split-admixture model. The model checking based on PCA suggests the selected models with posterior parameters estimated by ABC fit the observed data sets better as compared with those models with prior parameters (Fig. S4). The posterior parameters of interests are shown in Table 3. The parameter estimation for the same population varies in different scenarios. For instance, the split of Pamiri Tajik from West Eurasian is dated 405 and 1190 generations according to the scenarios of three and four populations, respectively. It should be noted that, in the scenario of four populations, the recent admixture time (both mean and median values) is older than the ancient split time in lowland Tajik. It implies the current model is unable to distinguish the ancient split and recent admixture. Or both demographic events occurred simultaneously, so the time scales were overlapping. The most ancient split time is observed in Wakhi Tajik (1410–1510 generations, mean value) around 28–30 kya (a generation time of 20 years), which is before the LGM. In the Pamir highlanders, the admixture time (not the split time) is estimated within 15 ky, that is after the LGM. The gene flow from East Eurasian is stronger in East Pamir Kyrgyz (35.3–55.8%) than Pamiri Tajik (17.6%), Sarikoli Tajik (19.7–21.8%), and Wakhi Tajik (16.4%).
Detection of selection
The number of nonsynonymous substitution (N), the number of synonymous substitution (S), and the ratio N/S in the Pamir highlanders, the lowlanders, and the general population [26] are shown Fig. S5. The N/S ratio in the highlanders (0.430) is higher than that in the lowlanders (0.381), but lower than that in the general population (0.495). The ratio differences between the highlanders and the lowlanders/the general population are not significant (P > 0.05; Table S3). The distribution of N/S ratio in 13 protein-coding genes indicates no significant differences between the highlanders and lowlanders with an exception of COX2 gene (Table S3). In COX2 gene, the N/S ratio in the highlanders (2/27) is significantly lower (P = 0.013) than that in the lowlanders (82/213). According to the genetic affinity revealed by PCA (Fig. 3), we further compared the highland Tajiks and highland Kyrgyz with their corresponding lowlanders (Table S7). Only the N/S ratio of CYTB gene in Pamiri Tajik (17/12) is significantly higher (P = 0.016) than that (33/69) in the corresponding lowlanders. Nevertheless, the P value is higher than 0.05 after the Bonferroni correction.
Discussion
Central Asia, located in the Eurasian heartland, is a hotspot for studying human evolutionary history. The paleoanthropological and archeological evidence suggests that Central Asia was occupied by ancient hominin since the early Pleistocene (800–900 ka) [39]. It was also inhabited by Neanderthals [40]. No archaic mtDNA lineages with origins from the other archaic hominids in Central Asia, such as Denisovans [41] and Neanderthals [40], are detected (Fig. 1 and Tables S3 and S4). Given that some gene flows from archaic hominids are revealed in various populations around Central Asia (e.g., ref. [42]), the lack of mtDNA introgression can be explained by the small effective size of mtDNA (i.e., one quarter that of the autosomes), which may be subjected to severe bottleneck effect/genetic drift. There is a possibility that the current absence of archaic hominid types may be caused by insufficient sampling. It is also compatible with the model of strong reproductive isolation between modern humans and archaic hominids [43].
We first provide a detailed mtDNA haplogroup tree at the highest molecular resolution for Central Asian populations (Table S4). As compared with previous studies mainly based on mtDNA D-loop sequences and certain coding regions [9,10,11], it can provide deeper insights into the origins of Central Asians. Especially in terms of the mtDNA genomes, we can date the Central Asian lineages into a fine scale. We find no basal lineages of macrohaplogroups M, N, and R in our data. The mtDNA lineages in Central Asians are classified into a subset of those typical of the East and the West Eurasians rather than ancient and/or unique lineages. The estimated ages for all lineages are within 40 kya (Fig. 2 and Table S5), i.e., after the initial arrival of modern human in Central Asia during 50–40 kya [6]. It supports the genetic admixture hypothesis (e.g., refs. [9, 10]) rather than the Central Asian heartland hypothesis which proposed that Central Asia was a heartland and reservoir for the genetic diversity of Eurasia and America [44]. Also, our results do not support the scenario that people carrying mtDNA N lineages crossed Asia via a northern route and then migrated to Australia [45]. Most (i.e., 93–97%) lineages coalesced after the LGM (Fig. 2 and Fig. S1). As a result, the post-LGM and/or later dispersals/admixtures play dominant roles in shaping the maternal gene pool of Central Asians.
According to the population mtDNA genomes, we are able to conduct extensive analyses to uncover the population history for Pamir highlanders. The genetic differentiation between the Tajik and Kyrgyz groups revealed in PCA (Fig. 3) reflects their different linguistic sources. Both East Pamir Kyrgyz and low Kyrgyz populations have close relationships in PCA and similar patterns in BSPs, suggesting the genetic differentiation is small. It is in agreement that the migrations of Kyrgyz to the Pamirs are recent [5]. More interestingly, we detect substantial genetic differentiation among three highland Tajik populations. The PCA shows Wakhi Tajik from the Wakhan Corridor, the southern edge of Pamirs, separating from Pamiri Tajik and Sarikoli Tajik (Fig. 3). Wakhi Tajik underwent a different demographic history in which no obvious female effective size increasing happened within 50 kya (Fig. 4). It should be noted that, the split time of Wakhi Tajik is before the LGM (Table 3), which is largely in disagreement with the dating of mtDNA lineages (Fig. 2 and Table S5). In addition to the setting of scenarios and models, the split time estimation relies on the selection of the representative West Eurasian. The lineages of haplogroup W3 account for 12.1% in Wakhi Tajik (Table S3), but are absent in Persian. So the coalescent time for haplogroup W lineages of Wakhi Tajik and Persian would be the age of haplogroup W, i.e., before the LGM [30]. As a result, the split time estimation is overestimated. And it cannot be simply explained as the time for the origin of Wakhi Tajik or the initial settlement of the Pamirs. As for Pamiri Tajik and Sarikoli Tajik from West and East Pamirs, respectively, the ABC analyses detect stronger gene flow from East Eurasian to Sarikoli Tajik than that to Pamiri Tajik and Wakhi Tajik (Table 3). Thus, the mtDNA heterogeneity in Pamir highlanders probably mirrors the complex history for peopling of Pamirs from the matrilineal genetic perspective.
Furthermore, Jeong et al. proposed that genetic admixture mediated selective signals for high-altitude adaptation in Tibetan Plateau [46]. Some studies showed positive selection on mtDNA lineages in Tibetans [47] and Sherpas [48]. Our results indicate that the Pamir highlanders are admixed populations. It gives us an opportunity to test whether admixture can mediate selection on mtDNAs in the Pamir highlanders or not. The comparisons of the N/S ratio between the highlanders and lowlanders/general population (Fig. S5 and Table S7) provide no evidence to supporting positive selection or relaxation of selective constraints in the highlanders. The patterns are different from those observed in Tibetans and Sherpas from Tibetan Plateau. It is expected that, future analyses based on extensive samplings (e.g., ref. [49]), genome-wide genetic markers (e.g., ref. [46]), even together with ancient DNAs (e.g., ref. [49]), will provide more details about the admixture history in the Pamirs.
References
Simonson TS. Altitude adaptation: a glimpse through various lenses. High Alt Med Biol. 2015;16:125–37.
Keay J. When men and mountains meet: the explorers of the Western Himalayas 1820–1875. 1st ed. Karachi: Oxford University Press; 1994.
Aldashev A, Naeije R, (eds). Problems of high altitude medicine and biology. Dordrecht: Springer; 2007.
Liu M. Migration and adaptation: the ethnography of the Tajik in the Pamirs. Beijing: Social Sciences Academic Press; 2014.
Dani AH, Masson VM. History of civilizations of Central Asia: development in contrast: from the sixteenth to the mid-nineteenth century. UNESCO; 2003.
Baumer C. The history of Central Asia: the age of the Steppe Warriors. I.B. Tauris; 2012.
Kuzʹmina EE, Mallory JP. The origin of the Indo-Iranians. Brill; 2007.
Han K. The racial anthropological study of the ancient Xinjiang inhabitants. Urumqi: Xinjiang People’s Publishing House; 1994.
Comas D, Calafell F, Mateu E, et al. Trading genes along the silk road: mtDNA sequences and the origin of central Asian populations. Am J Hum Genet. 1998;63:1824–38.
Comas D, Plaza S, Wells RS, et al. Admixture, migrations, and dispersals in Central Asia: evidence from maternal DNA lineages. Eur J Hum Genet. 2004;12:495–504.
Heyer E, Balaresque P, Jobling MA, et al. Genetic diversity and the emergence of ethnic groups in Central Asia. BMC Genet. 2009;10:49.
Ricaut FX, Keyser-Tracqui C, Cammaert L, Crubézy E, Ludes B. Genetic analysis and ethnic affinities from two Scytho-Siberian skeletons. Am J Phys Anthropol 2004;123:351–60.
Li C, Li H, Cui Y, et al. Evidence that a West-East admixed population lived in the Tarim Basin as early as the early Bronze Age. BMC Biol 2010;8:15.
Ovchinnikov IV, Malek MJ, Drees K, Kholina OI. Mitochondrial DNA variation in Tajiks living in Tajikistan. Leg Med. 2014;16:390–95.
Duggan AT, Stoneking M. A highly unstable recent mutation in human mtDNA. Am J Hum Genet. 2013;92:279–84.
Ning C, Gao S, Deng B, et al. Ancient mitochondrial genome reveals trace of prehistoric migration in the east Pamir by pastoralists. J Hum Genet. 2016;61:103–8.
Schönberg A, Theunert C, Li M, Stoneking M, Nasidze I. High-throughput sequencing of complete human mtDNA genomes from the Caucasus and West Asia: high diversity and demographic inferences. Eur J Hum Genet. 2011;19:988–94.
Derenko M, Malyarchuk B, Bahmanimehr A, et al. Complete mitochondrial DNA diversity in Iranians. PLoS ONE. 2013;8:e80673.
Fendt L, Zimmermann B, Daniaux M, Parson W. Sequencing strategy for the whole mitochondrial genome resulting in high quality sequences. BMC Genomics. 2009;10:139.
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147–47.
Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Weissensteiner H, Pacher D, Kloss-Brandstätter A, et al. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016;44:W58–63.
Fan L, Yao YG. An update to MitoTool: using a new scoring system for faster mtDNA haplogroup determination. Mitochondrion. 2013;13:360–63.
van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30:E386–94.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–67.
Liu J, Wang LD, Sun YB, et al. Deciphering the signature of selective constraints on cancerous mitochondrial genome. Mol Biol Evol. 2012;29:1255–61.
Forster P, Harding R, Torroni A, Bandelt HJ. Origin and evolution of native American mtDNA variation: a reappraisal. Am J Hum Genet. 1996;59:935–45.
Saillard J, Forster P, Lynnerup N, Bandelt HJ, Norby S. mtDNA variation among Greenland Eskimos: the edge of the Beringian expansion. Am J Hum Genet. 2000;67:718–26.
Perego UA, Achilli A, Angerhofer N, et al. Distinctive Paleo-Indian migration routes from Beringia marked by two rare mtDNA haplogroups. Curr Biol. 2009;19:1–8.
Soares P, Ermini L, Thomson N, et al. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am J Hum Genet. 2009;84:740–59.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Cornuet JM, Pudlo P, Veyssier J, et al. DIYABCv2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics. 2014;30:1187–89.
Seo SB, Zeng X, King JL, et al. Underlying data for sequencing the mitochondrial genome with the massively parallel sequencing platform Ion Torrent PGM. BMC Genomics. 2015;16:S4.
Avise JC. Phylogeography: the history and formation of species.. Cambridge: Harvard University Press; 2000.
Atkinson QD, Gray RD, Drummond AJ. mtDNA variation predicts population size in humans and reveals a major southern Asian chapter in human prehistory. Mol Biol Evol. 2008;25:468–74.
Clark PU, Dyke AS, Shakun JD, et al. The last glacial maximum. Science. 2009;325:710–14.
Heller R, Chikhi L, Siegismund HR. The confounding effect of population structure on Bayesian skyline plot inferences of demographic history. PLoS ONE. 2013;8:e62992.
Glantz MM. The history of hominin occupation of central Asia in review. In: Norton CJ, Braun DR, editors. Asian paleoanthropology: from Africa to China and beyond. Dordrecht: Springer; 2010.
Krause J, Orlando L, Serre D, et al. Neanderthals in Central Asia and Siberia. Nature. 2007;449:902–4.
Sawyer S, Renaud G, Viola B, et al. Nuclear and mitochondrial DNA sequences from two Denisovan individuals. Proc Natl Acad Sci USA. 2015;112:15696–700.
Ding Q, Hu Y, Xu S, Wang J, Jin L. Neanderthal introgression at chromosome 3p21.31 was under positive natural selection in East Asians. Mol Biol Evol. 2014;31:683–95.
Currat M, Excoffier L. Strong reproductive isolation between humans and Neanderthals inferred from observed patterns of introgression. Proc Natl Acad Sci USA. 2011;108:15129–34.
Wells RS, Yuldasheva N, Ruzibakiev R, et al. The Eurasian Heartland: a continental perspective on Y-chromosome diversity. Proc Natl Acad Sci USA. 2001;98:10244–49.
Fregel R, Cabrera V, Larruga JM, Abu-Amero KK, González AM. Carriers of mitochondrial DNA macrohaplogroup N lineages reached Australia around 50,000 years ago following a northern Asian route. PLoS ONE. 2015;10:e0129839.
Jeong C, Alkorta-Aranburu G, Basnyat B, et al. Admixture facilitates genetic adaptations to high altitude in Tibet. Nat Commun. 2014;5:3281.
Gu M, Dong X, Shi L, Lin K, Huang X, Chu J. Differences in mtDNA whole sequence between Tibetan and Han populations suggesting adaptive selection to high altitude. Gene. 2012;496:37–44.
Kang L, Zheng HX, Chen F, et al. mtDNA lineage expansions in Sherpa population suggest adaptive evolution in Tibetan highlands. Mol Biol Evol. 2013;30:2579–87.
Olivieri A, Sidore C, Achilli A, et al. Mitogenome diversity in Sardinians: a genetic window onto an island’s past. Mol Biol Evol. 2017;34:1230–39.
Hey J. On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol. 2005;3:e193.
Acknowledgments
We are grateful to all volunteers and samplers. We thank Ni-Ni Shi, Quan-Kuan Shen, Ya-Jiang Wu, Mu-Yang Wang, Yao-Ming Li, and Feng Xu for the technical assistance. This study was supported by grants from National Natural Science Foundation of China (31301026), Science & Technology Department of Xinjiang Uygur Autonomous Region (201491188), Research Center for Ecology and Environment of Central Asia, Chinese Academy of Sciences, and Bureau of Science and Technology of Yunnan Province. M.-S.P. thanks the support from the Youth Innovation Promotion Association, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Min-Sheng Peng, Weifang Xu, Jiao-Jiao Song, and Xing Chen contributed equally to this work.
Electronic supplementary material
41431_2017_28_MOESM3_ESM.pdf
Figure S3. Posterior probabilities of scenarios for target populations based on the linear discriminant analysis on summary statistics
41431_2017_28_MOESM5_ESM.pdf
Figure S5. The N/S ratio values of mitochondrial genomes from the Pamir highlanders, the lowlanders, and the general population
Rights and permissions
About this article

Cite this article
Peng, MS., Xu, W., Song, JJ. et al. Mitochondrial genomes uncover the maternal history of the Pamir populations. Eur J Hum Genet 26, 124–136 (2018). https://doi.org/10.1038/s41431-017-0028-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0028-8
This article is cited by
-
Mitogenomics of the Koryaks and Evens of the northern coast of the Sea of Okhotsk
Journal of Human Genetics (2023)
-
Dual origins of the Northwest Chinese Kyrgyz: the admixture of Bronze age Siberian and Medieval Niru’un Mongolian Y chromosomes
Journal of Human Genetics (2022)
-
Genetic structure and forensic characteristics of the Kyrgyz population from Kizilsu Kirghiz autonomous prefecture based on autosomal DIPs
International Journal of Legal Medicine (2022)
-
Mitochondrial DNA genomes revealed different patterns of high-altitude adaptation in high-altitude Tajiks compared with Tibetans and Sherpas
Scientific Reports (2020)
-
Novel insight into the genetic basis of high-altitude pulmonary hypertension in Kyrgyz highlanders
European Journal of Human Genetics (2019)
