
Abstract
Extracellular matrix (ECM) bioscaffolds have been widely studied to repair bladder tissue defects; however, insufficient angiogenesis and weak mechanical strength limit their applicability for tissue regeneration. In this work, ECM bioscaffolds were derived from decellularized sheets and then modified with vascular endothelial growth factor (VEGF)-conjugated superparamagnetic iron oxide nanoparticles (Fe3O4 NP–VEGF) to promote angiogenesis. Electrospun silk fibroin (SF) is a natural biocompatible protein with excellent mechanical properties. To further improve the mechanical properties of ECM bioscaffolds, SF was added to the Fe3O4 NP–VEGF-modified ECM bioscaffolds to develop a biomimetic ECM-like proangiogenic scaffold. In vitro characterization with endothelial cells revealed that the proangiogenic scaffold supports cell adhesion and proliferation. Furthermore, the biomimetic proangiogenic scaffold significantly accelerated vascularization in vivo in a rat bladder augmentation model. Importantly, these improvements in vascularization promoted urothelium and smooth muscle regeneration. These findings demonstrate that the developed biomimetic proangiogenic scaffold possesses great potential for bladder regeneration applications.
Similar content being viewed by others


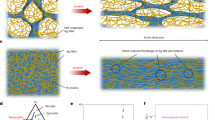
Introduction
Extracellular matrix (ECM) bioscaffolds have been widely used for bladder reconstruction in recent years1,2. Decellularized ECM bioscaffolds contain various cell-secreted growth factors, such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF), and structural proteins, including collagen and fibronectin, all of which play an important role in generating new vascular networks, recruiting regeneration-related cells, and inducing new tissue formation3. Cell sheet-based technologies are a promising approach in the field of regenerative medicine4. Additionally, a cell sheet can preserve various therapeutic cytokines and important adhesive proteins within the ECM. With these advantages, cell sheets may be used as an ECM source5.
Regeneration of the bladder requires early and rapid vascularization of bioscaffolds to supply both adequate nutrients and oxygen. Angiogenic processes are induced by numerous growth factors in ECM bioscaffolds6. Among them, VEGF is a critical regulator of angiogenesis7. However, the level of VEGF in ECM bioscaffolds is not high. To establish a VEGF concentration gradient and attract endothelial sprouts toward regenerated regions, exogenous VEGF can be covalently bound to ECM bioscaffolds by crosslinking agents, thus mimicking the natural protease-mediated release of growth factors from the ECM.
Superparamagnetic iron oxide nanoparticles (Fe3O4 NPs) have been receiving research attention because of their excellent biocompatibility and ability to deliver various biological molecules, such as growth factors, nucleic acids, and small drug molecules8. Moreover, Fe3O4 NPs can easily be modified and effectively bound to scaffolds9. On this basis, ECM bioscaffolds can be functionalized with Fe3O4 NPs binding VEGF, and then angiogenic factors can be released via ECM degradation10. However, proangiogenic ECM bioscaffolds are compromised by poor mechanical properties. Electrospun silk fibroin (SF) has been explored in a variety of tissue engineering and regenerative medicine applications because of numerous advantages, such as suitable mechanical properties, tunable biodegradability, and excellent biocompatibility11. Thus, we propose that electrospun SF can be combined with proangiogenic ECM bioscaffolds to produce ideal structural scaffolds with mechanical superiority and bioactive elements for bladder regeneration.
In this study, we developed a novel construction strategy on the basis of thermally triggered fabrication of cell sheets to improve the angiogenesis of biological bladder grafts. As shown in Fig. 1, decellularized ECM bioscaffolds were created using adipose-derived stem cell (ASC) sheets that were further modified with VEGF via nanoparticles, which were then layered on the SF to construct proangiogenic bladder grafts. The design of the proangiogenic scaffolds may inspire new options for bladder tissue engineering.

A The ECM bioscaffolds were acquired by decellularization of ASC sheets. The Fe3O4 NPs were conjugated with VEGF and then covalently bound to ECM bioscaffolds to form ECM–Fe3O4 NP–VEGF scaffolds. Thereafter, the SF nanofiber biomaterials and ECM–Fe3O4 NP–VEGF scaffold were assembled. B The therapeutic effect of this SF/ASC–ECM–Fe3O4 NP–VEGF scaffold was evaluated in a bladder defect model in SD rats.
Materials and methods
Isolation and characterization of ASCs
Adipose tissues of female Sprague–Dawley (SD) rats were collected to isolate ASCs in accordance with a previously reported protocol12. All animal experiments were approved by the animal welfare ethics committee of Shanghai Sixth People’s Hospital with approval number 2020-0586. Briefly, the adipose tissues were digested with 0.1% collagenase I (Sigma, St. Louis, MO, USA). The cell suspension was then filtered and centrifuged (1500 rpm, 5 min). The isolated ASCs were seeded and cultured in 10-cm dishes supplemented with low-glucose Dulbecco’s modified Eagle’s medium (LG-DMEM) (Gibco, Gaithersburg, MD, USA) and 10% fetal bovine serum (FBS) (Gibco). The ASCs were identified by the trilineage differentiation potential as previously reported13. For adipogenic differentiation, the cultured ASCs were incubated in 10% FBS-containing LG-DMEM supplemented with 1 μM dexamethasone, 10 μg/ml insulin, 0.5 mM 1-methyl-3-isobutylxanthine, and 100 μM indomethacin. After 2 weeks, the cells were stained with oil red O. For osteogenic differentiation, ASCs were cultured in 10% FBS-containing LG-DMEM supplemented with 10 mM b-glycerol phosphate, 0.1 μM dexamethasone, and 50 μM ascorbic acid. After 3 weeks, the cells were stained with Alizarin red. For chondrogenic differentiation, ASCs were induced in a chondrogenic induction medium consisting of LG-DMEM, 0.1 mM dexamethasone, 10 ng/ml transforming growth factor-β, 10 ng/ml BMP-6, and 50 ng/ml IGF-1. After 4 weeks, the cells were identified by Alcian Blue staining.
Construction of the ASC sheet-derived ECM (ASC–ECM)
The ASC sheet was fabricated by a previously described method12. Briefly, second passage ASCs were seeded in a 60-mm, temperature-responsive cell culture dish (Thermo Fisher Scientific, San Jose, CA, USA) at 1 × 105 cells/cm2. When the ASCs reached 90–100% confluence, they were stimulated with 50 μg/mL vitamin C to induce cell sheet formation. The culture medium consisted of low-glucose DMEM, 10% FBS, 50 μg/mL vitamin C, 1% penicillin/streptomycin, and 3.7 g/L sodium bicarbonate (Sigma). After 2 weeks, the cell sheet was collected from the bottom of the temperature-responsive cell culture dish by reducing the culture temperature to 20 °C for 30 min.
The harvested ASC sheets were rinsed with sterile phosphate-buffered solution (PBS) three times and then decellularized with 1% sodium dodecyl sulfate (SDS, Sigma) for 24 h. The treated cell sheets were further washed with deionized water three times to remove the residual reagents. Finally, the ASC–ECM was lyophilized in a vacuum freeze-drier (Virtis Benchtop 6.6, SP Industries, Gardiner, NY, USA) and stored at −20 °C for later use. H&E staining was performed to evaluate whether the decellularization process was successful. The decellularized ASC–ECM was stained using antibodies against collagen I and collagen III, followed by the addition of an appropriate Alexa Fluor secondary antibody. The morphological structures of ASC–ECM samples were observed by scanning electron microscopy (SEM, JSM-7800, Japan).
Preparation of Fe3O4 NPs
The Fe3O4 NPs were synthesized using a previously reported method14. The Fe3O4 NPs were coated with an amphiphilic polymer (PMA) using Parak’s procedure15. Briefly, the Fe3O4 NPs and PMA were mixed in a round-bottom flask with a ratio of RP/Area = 300 monomers/nm2, where RP/Area was the ratio of the polymer per nanoparticle surface area. After the solvent was evaporated slowly from the round-bottom flask, the polymer-coated samples were dissolved in sodium borate buffer (SBB) 12 (50 mM, pH = 12). The PMA-coated Fe3O4 NPs were then purified by ultrafiltration (3000 rpm, 10 min) and dispersed in Milli-Q water.
Bioconjugation of Fe3O4 NPs with VEGF
PMA-coated Fe3O4 NPs were gradually functionalized via EDC chemistry. First, 6 mg of N-hydroxysuccinimide (NHS) dissolved in 1 mL of SBB 9.0 (50 mM, pH 9.0) was mixed with 6 mg of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) in 1 mL of SBB 9.0. Then, PMA-coated Fe3O4 NPs (1 μM, 2 mL) were added. The mixture was incubated at 37 °C for 20 min on a shaking bed to activate the carboxyl groups. Afterward, 0.5 mL of VEGF (His-tagged, 1 mg/mL) was added to the activated PMA-coated Fe3O4 NP solution, which was then maintained at room temperature for 12 h on a shaking bed. Finally, the VEGF-conjugated Fe3O4 NPs (Fe3O4 NP–VEGF) were concentrated by a centrifuge filter (Millipore, Mw = 100 kDa) with centrifugation (2000 rpm, 10 min).
Characterization of Fe3O4 NPs, ASC–ECM, and Fe3O4 NP–VEGF
The morphology of the synthesized Fe3O4 NPs and Fe3O4 NP–VEGF was imaged with transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) (JEOL, Tokyo, Japan). The ultraviolet-visible (UV–vis) spectra of Fe3O4 NPs, VEGF, and Fe3O4 NP–VEGF were measured using a UV–vis spectrophotometer (Varian Inc., Palo Alto, CA, USA). The Fourier transform infrared (FTIR) spectra of Fe3O4 NPs, ASC–ECM, and Fe3O4 NP–VEGF were captured by a Nicolet 6700 spectrometer using KBr-supported pellets (Thermo Electron Corporation, Madison, WI, USA).
Fabrication and characterization of functionalized bladder grafts
The loading of Fe3O4 NP–VEGF onto the ASC–ECM grafts was realized by incubation in a Fe3O4 NP–VEGF solution (100 μg/mL). The Fe3O4 NP–VEGF solution was first mixed with EDC (6 mg/mL) and NHS (6 mg/mL) solutions by gently shaking for 20 min. The ASC–ECM grafts were then transferred to the mixture for 12 h to fabricate the ASC–ECM–Fe3O4 NP–VEGF scaffold. The FTIR spectra of ASC–ECM–Fe3O4 NP–VEGF were obtained using a Nicolet 6700 FTIR spectrometer. The intrinsic VEGF in the ASC–ECM and ASC–ECM–Fe3O4 NP–VEGF scaffolds was quantified using enzyme-linked immunosorbent assay (ELISA) kits (Lianke Biotech, Hangzhou, China) according to the manufacturer’s instructions. The electrospun SF was prepared as previously described by our research group16. Furthermore, the ASC–ECM–Fe3O4 NP–VEGF was layered on both the top and bottom surfaces of the electrospun SF to obtain the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold. The SF/ASC–ECM–Fe3O4 NP–VEGF scaffold was viewed with SEM and TEM to evaluate the distribution of Fe3O4 NP–VEGF. The distribution of iron in the samples was assessed by an energy dispersive spectroscopy (EDS) unit attached to the SEM and TEM setups.
Cell viability and attachment to the bladder scaffolds
Endothelial cells were cultured on SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds. After cultivation for 1, 3, and 7 days, the cell viability was assayed using a live/dead cell kit according to the manufacturer’s instructions, and the cells were observed using a confocal immunofluorescence microscope. The experiment was repeated four times. The number and total percentage of viable cells were calculated at four independent sites of each group to evaluate the biocompatibility of the scaffolds.
Morphological characterization of the scaffolds
For the electron microscopic study, SF, ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds were washed twice with PBS. After fixing with 2% glutaraldehyde and washing with PBS, the specimens were dehydrated in a gradient series of ethanol and then dried by lyophilization. The samples were sputter-coated with gold and examined with SEM. The SF/ASC–ECM–Fe3O4 NP–VEGF scaffold was also viewed with TEM to evaluate the distribution of Fe3O4 NP–VEGF. The iron element distribution of the samples was assessed by EDS. The surface topography features of the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold were detected using atomic force microscopy (AFM; Agilent, Chandler, AZ, USA).
In vivo bladder reconstruction study
All animal experimental procedures were approved by the Animal Care and Use Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. Twelve-week-old SD female rats were anesthetized using isoflurane inhalation, and then a ventral lower midline incision was made on the lower abdomen to expose the bladder. The dome of the bladder was incised longitudinally (~1 cm). Forty SD rats were randomly classified into four groups (10 rats per group) receiving different treatments: (1) cystotomy control group: the bladder was treated with suture repair alone; (2) SF group: the bladder was augmented by the electrospun SF scaffold; (3) SF/ASC–ECM group: the bladder was augmented by the SF/ASC–ECM scaffold; and (4) SF/ASC–ECM–Fe3O4 NP–VEGF group: the bladder was augmented by the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold. After the treatment, the incisions on the abdomen were closed using interrupted sutures. Twelve weeks post-implantation, animals were harvested for the endpoint evaluations described below.
Magnetic resonance imaging and urodynamics
The gross morphology of the bladders was determined by MRI. Rats were anesthetized with 2.5% isoflurane and placed inside the MRI receiver coil. Respiration was monitored throughout the experiment. Longitudinal T2-weighted MRI scans from the abdomen to the back were obtained using the Rapid Acquisition with Relaxation Enhancement sequence with the following parameters: effective echo time = 76 ms, repetition time = 2600 ms, matrix size = 320 × 320, field of view = 120 × 120 mm, and slice thickness = 1.0 mm.
During urodynamic testing, the bladder was exposed through the lower abdomen under pentobarbital anesthesia. A flared-end polyethylene-90 catheter was delicately placed into the bladder. The other end of the bladder catheter was attached to a physiological pressure transducer to measure intravesical pressure, while sterile PBS was continuously infused into the bladder. The urodynamic parameters were recorded and analyzed using LabChart software (ADInstruments, Castle Hill, New South Wales, Australia). Bladder compliance was calculated by the ratio of the introduced bladder volume and the change in intravesical pressure.
Histological and immunofluorescence analysis
The bladders with scaffolds were entirely harvested from the rats. After fixing the bladder specimens in 4% paraformaldehyde, they were processed for dehydration and paraffin embedding. Tissue sections were then separately stained with hematoxylin and eosin (H&E) and Masson’s trichrome. Moreover, immunofluorescent staining was also performed. Briefly, the sections were incubated with primary antibodies against cytokeratin (CK), α-SMA, and CD31 (Abcam, Cambridge, MA, USA) and then with Alexa Fluor 488 and Alexa Fluor 594 secondary antibodies (Invitrogen, Carlsbad, CA, USA). The cell nucleus was counterstained with DAPI. Afterward, the blood vessel density and hierarchical structure of the bladder specimens were observed using a Nikon 400 fluorescence microscope (Nikon Inc., NY, USA). Primary organs (heart, lung, liver, spleen, kidney, and bladder) were collected from the cystotomy control and SF/ASC–ECM–Fe3O4 NP–VEGF groups for further HE staining.
Statistical analysis
All data are expressed as the mean ± standard deviation. Student’s t-test and one-way analysis of variance were used to analyze the data. Statistical evaluation of the data were performed using SPSS and GraphPad Prism software. A p value less than 0.05 was considered statistically significant.
Results
Characterization of ASC sheet-derived ECM (ASC–ECM)
ASCs that were isolated from fresh rat adipose tissue exhibited a spindle- or stellate-shaped morphology after five days of culture (Fig. 2A). When cultured in an osteogenic induction medium, the cells showed Alizarin red-positive calcium deposits (Fig. 2B). Cells cultured in adipoinductive medium accumulated lipid droplets, as evidenced by oil red O staining (Fig. 2C). Cells treated with chondrocyte differentiation culture medium differentiated into chondrocytes, as demonstrated by Alcian Blue staining of sulfated glycosaminoglycans (Fig. 2D).

A ASCs at passage 1. B Osteogenic differentiation of ASCs. C Adipogenic differentiation of ASCs. D Chondrogenic differentiation of ASCs. E The fabrication of the ASC sheet. F Light microscopy image of the ASC sheet. G H&E staining of the ASC sheet. H H&E staining of ASC sheet-derived ECM after decellularization. I, J Representative SEM images of the architectures in the surface layer of the ECM bioscaffolds. K, L Cross-sectional SEM images of the ECM bioscaffolds. Scale bars = 200 μm (A, B, D, F), 100 μm (C), 50 μm (G, H), 10 μm (I, K), and 4 μm (J, L).
ASCs were cultured for 14 days to form a continuous cell sheet (Fig. 2E). Cell sheets had dense ASCs and ECM (Fig. 2F). To evaluate the decellularization effects, H&E staining was performed before and after decellularization. A high density of cells was observed in the ECM before decellularization (Fig. 2G). After decellularization, the cellular components were completely removed, and the ECM structure was preserved in the decellularized matrices (Fig. 2H and Fig. S1). The SEM images showed that the lyophilized decellularized matrices exhibited a relatively flat surface and abundant ECM proteins (Fig. 2I–L).
Synthesis of Fe3O4 NP–VEGF
TEM analysis shows that the synthesized Fe3O4 nanoparticles before and after bioconjugation of VEGF had a spherical morphology (Fig. 3A, B). The high-resolution TEM shown in Fig. 3C indicates that the Fe3O4 nanoparticles had a well-defined crystal structure. The saturated magnetization values were ~60.2 and 40.6 emu g−1 before and after bioconjugation, respectively (Fig. 3D). UV–vis and FTIR spectra were used to evaluate the conjugation of the Fe3O4 NPs and VEGF (Fig. 3E, F). Compared with the Fe3O4 NPs, the characteristic absorption peak in the UV–vis absorption curve of Fe3O4 NP–VEGF appeared at ~260 nm. The peak at 1641 cm−1 in the FTIR spectra of the Fe3O4 NP–VEGF indicates the presence of an amide bond, which confirms successful covalent coupling between the Fe3O4 NPs and VEGF. In addition, compared with the FTIR spectra of ASC–ECM, new absorption peaks at 586 cm−1 appeared for ASC–ECM–Fe3O4 NP–VEGF, which indicates bonding between the Fe3O4 NP–VEGF and ASC Sheet-Derived ECM.

A TEM micrograph of Fe3O4 NPs. B TEM micrograph of VEGF-bioconjugated Fe3O4 NPs. C HRTEM micrograph of VEGF-bioconjugated Fe3O4 NPs. D Magnetization loops of the Fe3O4 NPs and Fe3O4 NP–VEGF analyzed at 300 K. E UV–vis spectra of Fe3O4 NPs, VEGF, and Fe3O4 NP–VEGF. F FTIR spectra of Fe3O4 NPs, Fe3O4 NP–VEGF, ASC–ECM, and ASC–ECM–Fe3O4 NP–VEGF. Scale bars = 30 nm (A, B) and 5 nm (C).
Characterization of the Fe3O4 NP–VEGF-modified ASC–ECM
The surface of Fe3O4 NP–VEGF-modified ASC–ECM was densely and homogeneously covered by nanoparticles (Fig. 4A–C). The iron distribution and content on the surfaces of the scaffold were detected by SEM-EDC (Fig. 4D, E). The cross-sectional TEM image of the scaffold indicated that Fe3O4 NP–VEGF closely bonded with the collagen fibers in ASC–ECM, which was consistent with the SEM image (Fig. 4F). The Fe element of the nanoparticles was further confirmed by element mapping (Fig. 4G, H). VEGF is a strong angiogenic factor that stimulates endothelial cell proliferation and migration. The level of VEGF, which plays an important role in angiogenesis during the tissue regeneration process, in protein extracts from the ASC–ECM–Fe3O4 NP–VEGF group was significantly greater than that in protein extracts from the ASC–ECM group (Fig. S2).

A–C SEM images of the scaffolds. D, E SEM images and EDS spectra of the distribution of Fe in the scaffolds. F TEM images of the scaffolds. G, H TEM images and EDS spectra showing Fe element distribution in the scaffolds. Scale bars = 10 μm (A), 2 μm (B), 1 μm (C, D), 200 nm (F), and 30 nm (G, H).
Morphology and mechanical property characterization of the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold
The gross appearances of SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF are shown in Fig. 5A–C. The surface morphology and topographic features of the scaffolds were examined via SEM (Fig. 5D–F) and AFM (Fig. 5G–L). The SF/ASC–ECM scaffold exhibited a flat and smooth surface. The surface morphology of the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold was similar to that of the SF/ASC–ECM, which indicates that the surface morphology of ASC–ECM was not affected by the pretreatments of Fe3O4 NP–VEGF. The cross-sectional SEM image of the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold showed that the ASC–ECM–Fe3O4 NP–VEGF scaffold adhered tightly to the SF scaffold (Fig. S3). The mechanical properties of the scaffolds are vital for bladder reconstruction. Representative stress/strain curves of the scaffolds are shown in Fig. 5M. The maximum load of different scaffolds was calculated (Fig. 5N), and the results suggest that the mechanical strength of the SF/ASC–ECM scaffold decreased slightly after Fe3O4 NP bioconjugation (p > 0.05).

A–C Gross appearance of SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds. D–F Low-magnification SEM images of SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds. Scale bars = 50 μm. G–I Two-dimensional AFM images of SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds. Scale bars = 0.5 μm. J–L Three-dimensional AFM images of SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds. M Representative stress/strain curves for SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds. N Comparison of the maximum load of the three scaffolds. Statistical analysis is shown as ★p < 0.05, ns no significance.
Attachment, spreading, and proliferation of endothelial cells on scaffolds
The influence of the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold on the attachment and proliferation of endothelial cells was assessed by live/dead cell staining. As shown in Fig. 6A, the number of endothelial cells on the three scaffolds increased over the 7-day observation period, while endothelial cells (ECs) on SF/ASC–ECM and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds were more proliferative than those on SF scaffolds, indicating the positive role of ASC–ECM in cell proliferation. Furthermore, endothelial cells grown on the ASC–ECM–Fe3O4 NP–VEGF scaffold had a higher viability (93%) on Day 7 of the incubation compared with cells cultured on ASC–ECM (87%) and SF (76%) scaffolds (Fig. 6B). In addition, the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold improved endothelial cell proliferation the most (Fig. 6C). These results suggest that Fe3O4 NP–VEGF promoted endothelial cell proliferation and increased cell viability on the scaffolds.

A Live/dead fluorescence labeling of endothelial cells. Green: live cells. Red: dead cells. Scale bars = 50 µm. B Relative cell viability based on live/dead staining. C The number of cells in SF, SF/ASC–ECM, and SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds at different time points. Statistical analysis is shown as ★p < 0.05, ns no significance.
Inflammatory cell distribution in bladder tissue regeneration
At 12 weeks post augmentation, CD68-positive macrophages indicative of chronic inflammatory reactions were evaluated in a de novo bladder wall. No chronic inflammatory cells were observed in the cystotomy control group (Fig. S4A), while the treatment groups had mild infiltration of macrophages in the regenerated bladder tissue (Fig. S4B–D). The macrophage numbers of the SF/ASC–ECM and SF/ASC–ECM–Fe3O4 NP–VEGF groups were not significantly different (Fig. S4E) (p > 0.05). These data suggest that the addition of Fe3O4 NP–VEGF did not aggravate the inflammatory reaction at the implantation site.
Urodynamic study and histological evaluation of angiogenesis
All of the rats survived the duration of the study without significant postoperative complications. At 12 weeks postoperation, MR scans were conducted for the cystotomy control group and treatment groups (Fig. 7A). MR imaging showed that bladders reconstructed in the treatment groups exhibited increased bladder capacity with regular morphology. No evidence of urethral leakage in the reconstructed bladders was noted. Bladders underwent urodynamic testing at 12 weeks post augmentation. Bladder augmentation with the SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds led to a significant increase in bladder capacity in comparison to the nonaugmented control, SF, and SF/ASC–ECM groups (Fig. 7B). In addition, bladder compliance in the SF/ASC–ECM and SF/ASC–ECM–Fe3O4 NP–VEGF groups reached nearly the same level, which was significantly greater than that in the control group (Fig. 7C). Evidence of de novo vascularization with CD31 was observed in regenerated bladder tissues. In comparison to the SF and SF/ASC–ECM groups, significant vascularization was observed in the SF/ASC–ECM–Fe3O4 NP–VEGF group (Fig. 7D). The mean vessel diameter of the regenerated bladders in the SF/ASC–ECM–Fe3O4 NP–VEGF group was significantly greater than that in the control group, and the density of the vessels was not significantly different between the two groups (Fig. 7E, F). Therefore, these results indicate that the SF/ASC–ECM–Fe3O4 NP–VEGF scaffold promotes the vascularization of the scaffold in the repair area and supports the regeneration of the original bladder wall structure.

A Representative MRI images of the regenerated bladder. B Capacity of the regenerated bladder at 12 weeks. C Bladder compliance of the engineered neobladder in different groups. D Central regions of regenerated bladders were immunofluorescence-stained with CD31 to observe the neovascularization conditions. The SF/ASC–ECM–Fe3O4 NP–VEGF group showed evident regeneration of vessels. Scale bar = 100 µm. E, F Histomorphometric assessment of the extent of regenerated neovascularization in the central regions of the regenerated bladders. Statistical analysis is shown as ★p < 0.05, ns no significance.
Histological evaluation of regenerated bladder tissue
In all augmented bladders, the edges of the implantation area were marked by nonabsorbable 7-0 polypropylene sutures, and host tissue ingrowth spanning the entire original implanted areas was observed between the marking sutures. Gross histological examinations (HE and MTS analysis) were performed 12 weeks after the operation in both the control and treatment groups (Fig. 8A). The de novo bladder wall in the treatment groups had a trilayered tissue architecture consisting of the urothelium, lamina propria, and smooth muscle bundles. SF fibers degraded completely in the SF/ASC–ECM and SF/ASC–ECM–Fe3O4 NP–VEGF groups. MTS analysis revealed pronounced fibrosis and increased collagen deposition in the SF groups, while thicker muscle layer regeneration was observed in the SF/ASC–ECM–Fe3O4 NP–VEGF group than in the SF/ASC–ECM group.

A (First row) Gross appearances of bladder longitudinal sections of each scaffold group and the nonaugmented control group 12 weeks postoperation. Reconstructed areas of the scaffold implantation are bracketed by dashed lines. Scale bars = 5 mm. (Second and third rows) Magnification of the representative urothelium (UE) and smooth muscle (SM) tissue architecture bracketed in the first row. Scale bars = 200 μm. (fourth row) Masson’s trichrome staining of the magnified regenerated bladder areas is bracketed in the first row. Black arrows indicate SF remnants. Scale bars = 200 μm. B Cytokeratin and α-SMA markers that reflect urothelial and smooth muscle, respectively, in the regenerated bladder tissue of the four groups at 12 weeks postoperation. Scale bar = 100 µm. C, D Histomorphometric assessment of the extent of regenerated urothelium and smooth muscle in central regions of the regenerated bladders. Statistical analysis is shown as ★p < 0.05, ns no significance.
The histological observations were further substantiated by immunofluorescence staining (Fig. 8B). The multilayered urothelium in the treatment groups stained positive for cytokeratin. Histomorphometric analyses showed that the degree of urothelial proliferation in the de novo bladder tissues was significantly greater than that in the control bladder tissues (Fig. 8C). With regard to smooth muscle, α-SMA-positive organized smooth muscle bundle formation was observed in the reconstructed area of the SF/ASC–ECM and SF/ASC–ECM–Fe3O4 NP–VEGF groups, but not in the SF group (Fig. 8B). Furthermore, the histomorphometric analysis showed that α-SMA expression in regenerated tissues in the SF/ASC–ECM–Fe3O4 NP–VEGF group was similar to that in the control group, indicating smooth muscle maturation (Fig. 8D). Compared with the cystotomy control group, no lesions or abnormalities were identified in the rat organs treated with SF/ASC–ECM–Fe3O4 NP–VEGF scaffolds (Fig. S5).
Discussion
Extracellular matrix (ECM)-derived biomaterials have been used as raw materials for targeted tissue-specific regeneration in a variety of applications17. ECMs derived from native tissues/organs have been widely studied because of their favorable bioactivity and biocompatibility18. The ECM, as a bioactive scaffold, provides growth signals to cells, promotes their survival, and stimulates new vessel formation via endogenous growth factors19,20. The progress in cell sheet technology holds great promise in the field of regenerative medicine21. Because the procedure for harvesting the cell sheet avoids enzymatic treatment, important growth factors and the associated ECM remain intact within the cell sheet22. As a result, ECM scaffolds can be fabricated by decellularizing cell sheets23,24. ECM derived from autologous ASC sheets can overcome the risk of immune rejection by the host tissue. Additionally, the flat and smooth ASC–ECM surface can act as a waterproof barrier that prevents penetration of urine, reducing urine crystal precipitation that decreases the inflammatory response. Although decellularized cell sheets retain the ECM structure and some angiogenic factors, they are difficult to apply directly to urinary bladder tissue engineering without further modification.
Rapid angiogenesis of engineered scaffolds is crucial for constructive bladder remodeling. To promote angiogenesis, decellularized ECM can be further modified with bioactive factors25. In this case, exogenous angiogenic factors within the decellularized ECM can be released according to cellular demand, thus mimicking the natural protease-mediated release from the ECM. Iron-based nanomaterials have received clinical approval from the Food and Drug Administration. Fe3O4 NPs have been used broadly in biological and medical studies because of their excellent biocompatibility and versatility of surface modification26,27. In our previous study, Fe3O4 nanoparticles in a certain concentration range exhibited no cytotoxicity toward cells12. Thus, we chose Fe3O4 nanoparticles to enrich ECM with VEGF. The results of this study demonstrate that VEGF could be conjugated using Fe3O4 NPs for attachment to collagen fibers in the ASC–ECM. The Fe3O4 NP–VEGF-modified scaffold showed a surface morphology similar to that of the unmodified ECM, except the surface was covered by a layer of small nanoparticles. The large quantities of Fe3O4 NP–VEGF, which were distributed inside the ECM, suggest that Fe3O4 NP–VEGF could also penetrate deep into the matrix through voids and bind to the collagen fibers in ASC–ECM scaffolds.
VEGF appears to play an essential role in regulating angiogenesis28,29. In vitro studies showed that SF/ASC–ECM–Fe3O4 NP–VEGF bioscaffolds and SF/ASC–ECM bioscaffolds were capable of promoting endothelial cell attachment and proliferation. Moreover, cells on the SF/ASC–ECM–Fe3O4 NP–VEGF bioscaffolds proliferated faster than those on SF/ASC–ECM bioscaffolds. Thus, the Fe3O4 NP–VEGF scaffolds might activate VEGF receptors on the cell membrane, thereby exerting the significant biological role of VEGF, which is to induce the proliferation of endothelial cells. In addition, surface topographical cues of scaffolds may play a role in cell attachment30,31,32. Compared with SF scaffolds, ECM scaffolds before and after Fe3O4 NP–VEGF modification had a smooth and compact surface morphology, which may promote endothelial cell attachment.
The mechanical properties of the scaffolds are key for bladder regeneration33,34,35. However, the mechanical properties of the ECM–Fe3O4 NP–VEGF bioscaffolds in this study did not completely meet the requirements for the repair of bladder defects. Therefore, we further reinforced the mechanical strength of the ECM–Fe3O4 NP–VEGF bioscaffolds by incorporating SF nanomaterials. The breaking strength showed that the addition of electrospun SF nanomaterials enhanced the mechanical strength of the ECM–Fe3O4 NP–VEGF bioscaffolds, which provided strong support for their use in an in vivo experiment. Moreover, SF also has the advantage of high porosity, which facilitates the growth of blood vessels and cells.
Various studies have demonstrated that angiogenesis is crucial for successful tissue construction in vivo36,37. To achieve the desired vascularization, previous studies have focused on the application of high doses of angiogenic factors, which increases the cost and may increase the risk of systemic cancer7,38. The biomimetic ECM-like proangiogenic scaffolds designed in this study promote physiological angiogenesis, which could allow a reduction in the therapeutic dose of VEGF. In our animal study, we used a bladder augmentation model to demonstrate the function of the proangiogenic scaffold in vivo. The blood vessel density in the SF/ASC–ECM–Fe3O4 NP–VEGF bioscaffold group was significantly greater than that in the other groups. In addition, the SF/ASC–ECM–Fe3O4 NP–VEGF bioscaffold promoted the regeneration of urothelium and smooth muscle within the implantation sites, thus improving certain urodynamic parameters in the bladder augmentation model. Our findings collectively demonstrate that SF/ASC–ECM–Fe3O4 NP–VEGF may be a platform for the intricate coordination of topographical and bioactive cues in regulating angiogenesis in vitro and in vivo, which are important for improving constructive tissue remodeling. Previous studies have revealed that cells on a proper scaffold can accelerate the regeneration process. Thus, in future studies, a bladder augmentation model is needed to evaluate the effects of seeding SF/ASC–ECM–Fe3O4 NP–VEGF bioscaffolds with bladder cells and the contribution of transplanted cells in bladder reconstruction.
Our study has several limitations. First, we demonstrated the feasibility of creating a proangiogenic ECM based on covalently linking VEGF-conjugated Fe3O4 and a decellularized cell sheet. A study of dose ranges is required to further determine a safe minimal dose of ECM-bound VEGF that is sufficient to induce functional angiogenesis. Second, this is a short-term observational study evaluating tissue-engineered bladder grafts in rat models. The safety and efficacy of the proangiogenic scaffolds should be tested in larger animals before clinical consideration. Third, iron oxide nanoparticles could induce macrophage polarization. Further trials are necessary to assess the effect of macrophage polarization in all phases of scaffold-induced tissue regeneration.
Conclusion
We developed a biomimetic platform for conjugating VEGF to ECM scaffolds using Fe3O4 NPs. The modified ECM demonstrated improved mechanical properties after combination with SF. The in vitro study demonstrated that the ASC–ECM–Fe3O4 NP–VEGF bioscaffold promotes the proliferation of endothelial cells. The in vivo study further confirmed the biological effect of the ASC–ECM–Fe3O4 NP–VEGF bioscaffold in enhancing vascularization and supporting bladder tissue regeneration. Thus, the ASC–ECM–Fe3O4 NP–VEGF bioscaffold has potential application in bladder defect repair.
References
Chung, Y. G. et al. The use of bi-layer silk fibroin scaffolds and small intestinal submucosa matrices to support bladder tissue regeneration in a rat model of spinal cord injury. Biomaterials 35, 7452–7459 (2014).
Xiao, S. et al. Bi-layer silk fibroin skeleton and bladder acellular matrix hydrogel encapsulating adipose-derived stem cells for bladder reconstruction. Biomater. Sci. 9, 6169–6182 (2021).
Hussey, G. S., Dziki, J. L. & Badylak, S. F. Extracellular matrix-based materials for regenerative medicine. Nat. Rev. Mater. 3, 159–173 (2018).
Zhang, D. et al. Cryopreserved skin epithelial cell sheet combined with acellular amniotic membrane as an off-the-shelf scaffold for urethral regeneration. Mater. Sci. Eng. C. Mater. Biol. Appl. 122, 111926 (2021).
Xing, Q. et al. Decellularization of fibroblast cell sheets for natural extracellular matrix scaffold preparation. Tissue Eng. Part C. Methods 21, 77–87 (2015).
Jakus, A. E. et al. “Tissue papers” from organ-specific decellularized extracellular matrices. Adv. Funct. Mater. 27, 1700992 (2017).
Tan, Q. et al. Controlled release of chitosan/heparin nanoparticle-delivered VEGF enhances regeneration of decellularized tissue-engineered scaffolds. Int. J. Nanomed. 6, 929–942 (2011).
Liu, X. L. et al. Magnetic nanomaterials for advanced regenerative medicine: the promise and challenges. Adv. Mater. 31, e1804922 (2018).
Chen, H. et al. Magnetic cell-scaffold interface constructed by superparamagnetic IONP enhanced osteogenesis of adipose-derived stem cells. ACS Appl. Mater. Interfaces 10, 44279–44289 (2018).
Brassart-Pasco, S. et al. Tumor microenvironment: extracellular matrix alterations influence tumor progression. Front. Oncol. 10, 397 (2020).
Umuhoza, D. et al. Strategies for tuning the biodegradation of silk fibroin-based materials for tissue engineering applications. ACS Biomater. Sci. Eng. 6, 1290–1310 (2020).
Wang, Y. et al. Bioengineered bladder patches constructed from multilayered adipose-derived stem cell sheets for bladder regeneration. Acta Biomater. 85, 131–141 (2019).
Xiong, Y. et al. Retention of the stemness of mouse adipose-derived stem cells by their expansion on human bone marrow stromal cell-derived extracellular matrix. Tissue Eng. Part A. 21, 1886–1894 (2015).
Zhou, S. et al. Fabrication of tissue-engineered bionic urethra using cell sheet technology and labeling by ultrasmall superparamagnetic iron oxide for full-thickness urethral reconstruction. Theranostics 7, 2509–2523 (2017).
Lin, C. A. et al. Design of an amphiphilic polymer for nanoparticle coating and functionalization. Small 4, 334–341 (2008).
Huang, J. W. et al. Tissue performance of bladder following stretched electrospun silk fibroin matrix and bladder acellular matrix implantation in a rabbit model. J. Biomed. Mater. Res. Part A. 104, 9–16 (2016).
Garreta, E. et al. Tissue engineering by decellularization and 3d bioprinting. Mater. Today 20, 166–178 (2017).
Marinkovic, M. et al. Native extracellular matrix, synthesized ex vivo by bone marrow or adipose stromal cells, faithfully directs mesenchymal stem cell differentiation. Matrix Biol. 8, 100044 (2020).
Fercana, G. R. et al. Perivascular extracellular matrix hydrogels mimic native matrix microarchitecture and promote angiogenesis via basic fibroblast growth factor. Biomaterials 123, 142–154 (2017).
Chun, S. Y. et al. Identification and characterization of bioactive factors in bladder submucosa matrix. Biomaterials 28, 4251–4256 (2007).
Yoon, J. et al. Thermosensitive, stretchable, and piezoelectric substrate for generation of myogenic cell sheet fragments from human mesenchymal stem cells for skeletal muscle regeneration. Adv. Funct. Mater. 27, 1703853 (2017).
Hsu, M. et al. Crispr-based activation of endogenous neurotrophic genes in adipose stem cell sheets to stimulate peripheral nerve regeneration. Theranostics 9, 6099–6111 (2019).
Xing, Q., Vogt, C., Leong, K. W. & Zhao, F. Highly aligned nanofibrous scaffold derived from decellularized human fibroblasts. Adv. Funct. Mater. 24, 3027–3035 (2014).
Wang, X. et al. Cell-sheet-derived ECM coatings and their effects on BMSCs responses. Acs Appl. Mater. Inter. 10, 11508–11518 (2018).
Jiang, X. et al. Co-delivery of VEGF and bFGF via a PLGA nanoparticle-modified bam for effective contracture inhibition of regenerated bladder tissue in rabbits. Sci. Rep. 6, 20784 (2016).
Wolf, F. et al. Mr and pet-ct monitoring of tissue-engineered vascular grafts in the ovine carotid artery. Biomaterials 216, 119228 (2019).
Zhou, Z., Yang, L., Gao, J. & Chen, X. Structure-relaxivity relationships of magnetic nanoparticles for magnetic resonance imaging. Adv. Mater. 31, e1804567 (2019).
Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V. & Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309 (1989).
Wu, L. et al. Hierarchical micro/nanofibrous membranes of sustained releasing VEGF for periosteal regeneration. Biomaterials 227, 119555 (2019).
Naganuma, T. The relationship between cell adhesion force activation on nano/micro-topographical surfaces and temporal dependence of cell morphology. Nanoscale 9, 13171–13186 (2017).
Gui, N. et al. The effect of ordered and partially ordered surface topography on bone cell responses: a review. Biomater. Sci. 6, 250–264 (2018).
Kyle, D. J., Oikonomou, A., Hill, E. & Bayat, A. Development and functional evaluation of biomimetic silicone surfaces with hierarchical micro/nano-topographical features demonstrates favourable in vitro foreign body response of breast-derived fibroblasts. Biomaterials 52, 88–102 (2015).
Ajalloueian, F., Lemon, G., Hilborn, J., Chronakis, I. S. & Fossum, M. Bladder biomechanics and the use of scaffolds for regenerative medicine in the urinary bladder. Nat. Rev. Urol. 15, 155–174 (2018).
Song, L. et al. Bladder acellular matrix and its application in bladder augmentation. Tissue Eng. Part B Rev. 20, 163–172 (2014).
Baker, S. C., Rohman, G., Southgate, J. & Cameron, N. R. The relationship between the mechanical properties and cell behaviour on PLGA and PCL scaffolds for bladder tissue engineering. Biomaterials 30, 1321–1328 (2009).
Myu, M. J. K. et al. Construction of a vascularized hydrogel for cardiac tissue formation in a porcine model. J. Tissue Eng. Regen. Med. 12, e2029–e2038 (2018).
Jia, W. et al. Urethral tissue regeneration using collagen scaffold modified with collagen binding VEGF in a beagle model. Biomaterials 69, 45–55 (2015).
Chung, Y. I. et al. Efficient revascularization by VEGF administration via heparin-functionalized nanoparticle-fibrin complex. J. Control. Release 143, 282–289 (2010).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 82100714, 82170694, and 81671737), the Shanghai Outstanding Academic Leaders Plan (No. 17XD1403100), the Natural Science Foundation of Shanghai (No. 20ZR1442100), the Interdisciplinary Program of Shanghai Jiao Tong University (No. YG2021QN102, YG2019QNB31, and YG2022ZD020), and the Program of Shanghai Sixth People’s Hospital (No. ynts202004).
Author information
Authors and Affiliations
Contributions
Q.F., G.G., and Y.W. conceived the project and designed the experiments. Y.W., J.C., M.D., H.C., M.Y., M.L., and J.H. performed the experiments. Q.F. and W.Z. analyzed the data. Y.W. and J.C. wrote the original manuscript. G.G. and Q.F. meticulously scrutinized and revised the manuscript. Y.W. and Q.F. supported the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Chen, J., Duan, M. et al. Extracellular matrix sheet modified with VEGF-loaded nanoparticles for bladder regeneration. NPG Asia Mater 14, 93 (2022). https://doi.org/10.1038/s41427-022-00442-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-022-00442-z
