
Abstract
Without the introduction of new functional groups, altering the properties of a substance, such as by changing from a non-self-healing to a rapidly self-healing material, is often difficult. In this work, we report that the properties of 2-hydroxyethyl methacrylate and acrylamide (HEMA/AAm) hydrogels can be easily altered from non-self-healing to rapidly self-healing by simply tuning the reaction temperature. Notably, the hydrogels that are prepared at room temperature do not exhibit self-healing behavior, while those treated at an elevated temperature show automatic self-healing performance within ~15 s. Interestingly, in contrast with the previous self-healing HEMA-based polymeric hydrogels, which function only above their glass transition temperatures (Tg), the hydrogels prepared herein exhibit rapid self-healing properties at room temperature, which is below their Tg. In addition, the stretching capabilities of the hydrogels can be greatly enhanced by up to 30-fold. The hydrogels also exhibit good adhesive performance and can adhere strongly onto various substrates, such as wood, glass, fabric, paper, leather, porcelain, and steel. For example, a 10 kg weight could be suspended from a wooden substrate with the aid of these hydrogels. These results may provide valuable insight regarding the design of self-healing hydrogels and their large-scale production.
Similar content being viewed by others
Introduction
Manmade materials with self-healing capabilities are highly desirable, and their development has become a goal for many scientists and engineers1,2,3,4. The term “self-healing” refers to a material’s ability to automatically recover from damage and restore its original mechanical properties5,6,7. Generally, the self-healing behavior can be classified into two categories, namely, extrinsic and intrinsic self-healing8,9,10. Extrinsic self-healing relies on pre-embedded healing agents to connect the fractured components of a damaged material via covalent bonds. However, there are several major drawbacks of extrinsic self-healing: (1) the pre-embedding process of healing agents is often complex; (2) the amounts of healing agents that can be readily incorporated are typically limited; and (3) the healing processes are often irreversible due to the formation of covalent bonds7,11,12. In contrast, intrinsic self-healing behavior depends on the inherent reversibility of dynamic chemical bonds or physical interactions between the interfaces of the damaged area, such as hydrogen bonding interactions. Thus, owing to the inherent reversibility of the latter mechanism, intrinsically self-healing materials can undergo multiple healing cycles, and the self-healing process is facile and simple without requiring additional healing agents13.
Hydrogels, a class of 3D network materials containing a large amount of water, have excellent potential for a wide range of applications, such as biological and medical applications, soft electronics, and sensors and actuators14,15. Hydrogels possessing self-healing capabilities are highly desirable and of utmost importance, especially for use as load-bearing materials16. The intrinsic ability of hydrogels to undergo self-healing from damaged states would increase their durability, service lifetimes, reliability, and safety17,18,19. Unfortunately, despite growing interest in self-healing hydrogels, these materials are still in their infancy20,21,22,23. The design and preparation of hydrogels with intrinsic self-healing capabilities still presents significant challenges because many unanswered structural questions remain regarding these 3D polymeric networks9. Currently, to fabricate self-healing hydrogels, novel monomers and crosslinkers are synthesized through processes that are often complex, tedious, and time consuming. For example, Yesilyurt et al. synthesized a four-arm PEG-NH2 star polymer incorporating various phenylboronic acid derivatives24, while Li et al. selected an Fe(III)-2,6-pyridine dicarboxamide (pdca) coordination complex as a crosslinker25. Meanwhile, Clarke et al. synthesized hybrid hydrogels consisting of a poly(γ-glutamic acid) polymer network that was physically crosslinked via the grafted self-assembly of β-sheet peptides26. As seen from the above examples, the preparation of self-healing hydrogels is rather complex. This consideration thus leaves the question: Is there any other way to develop self-healing hydrogels besides synthesizing novel monomers or crosslinkers?
Rather than attempting to investigate strategies for designing new classes of functional groups and monomers, as has been undertaken in many other studies5, we instead focus herein on addressing fundamental questions such as whether the preparation conditions would influence the self-healing properties of hydrogels. In particular, we investigate whether the properties of materials can change from one behavior to another (such as not self-healing to rapidly self-healing) either in the presence or absence of new functional groups.
In addition, the synthesis of hydrogels with adhesive properties remains a key challenge. Adhesive hydrogels can be utilized in various applications, including wound healing27, electronic devices28, and underwater adhesion in the marine sector29. However, water residing in the hydrogels can interact with the adhesive groups of hydrogels via hydrogen bonding, significantly weakening the adhesion between these hydrogels and potential substrate materials30. Therefore, the adhesive performance of hydrogels is generally rather weak.
Acrylamide (AAm) and 2-hydroxyethyl methacrylate (HEMA) have been widely used as monomers/comonomers for the preparation of hydrogels31,32,33,34. However, hydrogels based on AAm and HEMA generally cannot undergo self-healing at room temperature due to the weak interactions between these polymer chains35. Thus, researchers have attempted to introduce strong interactions, such as those found among macromolecular crystals with integrated polymer networks to achieve self-healing behavior, into hydrogels31. Herein, with the use of only simple commercially available components (HEMA and AAm), we attempted to construct self-healing, highly stretchable, and self-adhesive hydrogels without the need for chemical modification by tuning the preparation temperature. Impressively, our results revealed that we could indeed leverage temperature-regulated flexibility and polymer chain interactions to obtain hydrogels with rapidly self-healing capabilities. These hydrogels also have promising potential for use as adhesives due to their low cost, tenability, and facile synthesis.
Materials and methods
Materials
AAm, HEMA, and N,N,N,N-tetramethylethylenediamine (TEMED) were purchased from Shanghai Macklin Biochemical Co., Ltd. Ammonium persulfate (APS) was obtained from Tianjin Zhiyuan Chemical Technology Co., Ltd. Hexamethylene diisocyanate-trimer (HT) was obtained from Shenzhen Zhibang Technology Co., Ltd. N,N′-methylenebisacrylamide (MBAA) was supplied by Tianjin Damao Chemical Reagent Co., Ltd. HEMA was purified via column chromatography to remove the inhibitor. All of the hydrogels were prepared in deionized water.
Characterization
Fourier transform infrared (FTIR) spectra were recorded using an FTIR spectrometer (Tensor 27, Bruker, USA). SEM images were recorded using a Field emission scanning electron microscopy (FE-SEM) system at an acceleration voltage of 10 kV (JSM-7001F, JEOL, Japan). The thermal properties of the hydrogels were characterized via differential scanning calorimetry (DSC8000, PerkinElmer, Germany) over a temperature range of −30 to 200 °C and at a heating rate of 10 K/min. 1H NMR spectra were recorded using a Bruker AV III 500 MHz spectrometer with D2O as the solvent. The hydrodynamic diameters of the globules in the hydrogels were measured via dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern, UK) with the hydrogels formed directly in the testing models. UV-visible absorption spectroscopy was performed using a spectrophotometer (Shimadzu, model: UV-2600, Japan).
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed with a Bruker model ultrafleXtreme MALDI TOF/TOF spectrometer in a linear positive ion mode using 3-indolylacetic acid as the matrix and calibrated against Protein Calibration Standard I (Bruker). An ethanol solution of 3-indolylacetic acid (10 mg/mL), an aqueous solution of sodium chloride (10 mg/mL), and an aqueous solution of the test sample (10 mg/mL) were uniformly mixed at a respective volume ratio of 10:1:1 prior to characterization.
Preparation of the HT solution or MBAA solution
HT (0.40 g) or MBAA (0.25 g) was dissolved in 10 mL of anhydrous acetone or water and subsequently sonicated to obtain an HT/acetone solution or aqueous MBAA solution (due to the insolubility of MBAA in anhydrous acetone). The molar concentrations of the HT solution and MBAA solution were identical.
Preparation of the HEMA/AAm solutions
HEMA/AAm hydrogels were prepared as follows: APS (0.10 g) and AAm (7.10 g) were added into 23.70 mL of deionized water and dissolved. Subsequently, 2.00 g of HEMA was dispersed into the solution to obtain the desired HEMA/AAm solution.
Preparation of the HEMA/AAm hydrogels
Various amounts of the HT solution were mixed with 3.29 g of the HEMA/AAm solution described above. Subsequently, 10 μL of TEMED was added, and the solution was left at room temperature for various durations and then heated at 90 °C for 150 s. Finally, the sample was obtained after reaction at room temperature for 1 h.
As a control, different volumes of the MBAA solution were added into the HEMA/AAm solution to form hydrogels with MBAA serving as the crosslinker. All of the other hydrogels were prepared in a similar manner.
Tensile and compression measurements
The tensile properties of the hydrogels were measured using a universal mechanical testing machine (HZ-1007E, Dongguan Lixian Instrument Technology Co., Ltd, China) equipped with a 10 kg load cell at room temperature. The stretching rate was kept constant at 50 mm/min. The tensile stress and strain at break were obtained from the stress–strain measurements.
The compression measurements of the hydrogels were taken with a universal mechanical testing machine (UT4304, Shenzhen SUNS, China) at room temperature. Cylindrical hydrogel samples were used with a length of 25 mm and a diameter of 20 mm. The rate of compression was kept constant at 50 mm/min.
Adhesion tests
The hydrogels were cut into 5 × 5 mm sections prior to the adhesion tests. The hydrogels were then immobilized onto various substrates, including glass slides, cotton fabric, paper, leather, and wooden boards. Subsequently, the hydrogels were compressed by a 200 g weight for 30 min. These samples were then inverted.
Lap shear adhesion tests
The hydrogels were cut into 5 × 5 mm samples and immobilized on clear glass slides (25.4 × 76.2 mm). A single test sample was formed by covering it with another glass slide before compressing with a 100 g weight for 30 min. The area of overlap between the hydrogel and the substrate was measured using digital calipers immediately prior to testing. The samples were evaluated using a universal testing machine (HZ-1007E, Dongguan Lixian Instrument Technology Co., Ltd, China) at a rate of 5 mm/min. The hydrogels were pulled apart until their separation was achieved, and the maximum stress and strain values were recorded. The shear strength was determined via dividing the maximum stress by the initial area of the adhesive overlap. The hydrogels were also tested after they had undergone self-healing. At least three parallel samples were tested in each group.
Small-deformation oscillatory experiments
Small-deformation oscillatory experiments were conducted using an Anton Paar model MCR-92 rheometer. The polymer samples were placed under an 8-cm-diameter parallel plate with a gap of 0.1 mm. The storage moduli (G′) and loss moduli (Gʺ) were measured via temperature dispersion tests over a range from 100 to 0 °C (5 °C/min) at a constant frequency of 6.28 rad/s with an applied strain (γ) of 0.5%. Frequency sweep tests were conducted over a range from 628 to 6.28 × 10−2 rad/s at 25 °C with an applied strain (γ) of 0.5%.
Hydrogel swelling ratio tests
The swelling experiments were performed by immersing the hydrogels into a large amount of water at room temperature to reach swelling equilibrium. The swelling ratio q was calculated by the following equation:
where Ws and Wd denote the weights of the swollen hydrogel and dried hydrogel, respectively.
Results
Preparation and characterization of the HEMA/AAm hydrogels
As shown in Scheme 1, after mixing, the solution (containing 2-hydroxyethyl methacrylate (HEMA), acrylamide (AAm), hexamethylene diisocyanate-trimer (HT), APS, and TEMED) was poured into molds to form hydrogels via a one-pot method. The water content of these hydrogels was 72 wt%. The preparation process was rather simple and facile, thus providing potential for various applications as well as rapid large-scale production. The cross-linking reactions between HT, HEMA, and AAm were used to produce 3D polymer networks. In particular, HT served as a crosslinker and underwent random reactions with HEMA and AAm to form urethane groups and urea groups, respectively (see Scheme S1 in the supplementary information or SI)36, as was confirmed by FTIR spectroscopy (see Fig. S1 in the SI). 1H NMR spectroscopy also confirmed that cross-linking had indeed taken place, as indicated by the resonance at 2.14 ppm corresponding to AAm moieties that had reacted with HT (see Fig. S2 in the SI).

Schematic illustration depicting the preparation of HEMA/AAm hydrogels with self-healing properties, high stretchability, and excellent adhesion performance
Influence of the preparation temperature
The influence of the preparation parameters was first observed from the physical properties. For example, the light transparency of the hydrogels when viewed from the top differed from that when viewed from the side (Fig. 1a, b). The UV-vis spectra revealed that the hydrogels prepared at room temperature exhibited higher light transmittance than those prepared at an elevated temperature (see Fig. 1c), implying the presence of globules. The dynamic light scattering (DLS) results also confirmed the presence of globules, as depicted in Fig. S3 in the SI.

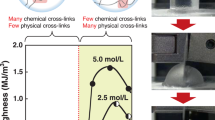
The light transparency of the hydrogels observed from the side view (a) and top view (b), and the light transmittance of the hydrogels as characterized by UV-vis spectroscopy (c). Photographs demonstrating the self-healing behavior of the hydrogels before (d) and after (e) two cleft surfaces had been in contact with one another for 15 s. One piece of hydrogel was labeled with a pink dye. The influence of the preparation temperature on the strain and self-healing behavior of the hydrogels (f). The stress (g) and strain (h) of the hydrogels that had been left at room temperature for various durations before they were placed into an oven at 90 °C. Also shown are the stress (i) and strain (j) performance of hydrogels containing different amounts of MBAA instead of HT as a crosslinker
The preparation temperature significantly influenced the self-healing behavior of the hydrogels. As shown in Movie 1, when they were prepared at room temperature, the hydrogels were soft and brittle and did not exhibit self-healing behavior even after 5 min of contact between two fractured sections at room temperature. In contrast, when the hydrogels were prepared at 90 °C, as shown in Fig. 1d, e as well as in Movie 2, the resultant HEMA/AAm hydrogels rapidly self-healed at room temperature. After ~15 s of contact between two sections of a cleaved hydrogel, the junction was sufficiently strong, and the healed hydrogel could withstand stretching and twisting. This self-healing behavior was especially impressive considering that the contacting surfaces were irregular in shape.
In view of the abovementioned influence of temperature, a series of hydrogels were further prepared at temperatures ranging from 25 to 90 °C, and their strain and self-healing performance was evaluated. With regard to these tests, the self-healing efficiency was defined as the strain ratio before and after healing37. As shown in Fig. 1f, the strain and self-healing capabilities of the hydrogels significantly increased when they were prepared at higher temperatures. In particular, the strain of the hydrogels increased from 1000 to 2800% when they were prepared at 90 °C instead of 25 °C, which corresponded to an increase of 2.8-fold. After 15 s of healing time, the strain was restored with ~80% efficiency for the hydrogels that were prepared at 90 °C.
The self-healing behavior was further studied by storing the reaction solutions at room temperature for various durations before they were placed into an oven to form hydrogels. As shown in Fig. 1g, h, as the time before thermal treatment was increased from 2 to 8 min, the stress of the hydrogels increased while the strain decreased significantly. In addition, the self-healing efficiency within 15 s decreased dramatically, from 62 to 0%. These results suggest that hydrogels should be directly prepared at high temperature to ensure their self-healing performance. Otherwise, the self-healing capabilities of those products would gradually diminish as time increased prior to the thermal curing treatment. Because the solutions formed hydrogels gradually at room temperature, the subsequent thermal treatment had little effect once the formation of the hydrogel was already complete.
We also used other hydrogels to confirm our conclusions regarding the influence of the preparation temperature on the self-healing properties, as demonstrated in Fig. 1i, j. N,N′-methylenebisacrylamide (MBAA) was used instead of HT as a crosslinker to eliminate the physical cross-linking arising from strong hydrogen interactions. As we anticipated, the hydrogels containing MBAA also exhibited self-healing behavior if they had been prepared at 90 °C, while those prepared at room temperature lacked self-healing properties (see Fig. S4 and Movies 3 and 4 in the SI). Furthermore, with increasing amounts of the chemical crosslinker MBAA, the self-healing efficiency decreased to 0%, thus demonstrating the disadvantages of excessive chemical cross-linking.
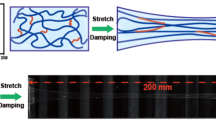
The stretchability of these HEMA/AAm hydrogels was determined via tensile experiments, and the results are depicted in Fig. 2a–c. The tensile strain at break of the hydrogels lacking HT was <2000%, while that of hydrogels incorporating 20 μL of HT exceeded 2500%, and the strain at break reached ~3000% for hydrogels containing 80 μL of HT. Conversely, the tensile stress at break increased from 9 kPa to the maximum value of 15 kPa with the incorporation of HT. Notably, these results imply that HT increased the strain and stress of the hydrogels simultaneously. However, when the amount of HT was further increased to 100 μL, both the stress and strain decreased. These surprising observations may be attributed to the reactions between HT and monomers to form urethane and urea groups, which served not only as chemical crosslinkers but also as physical crosslinkers facilitating strong hydrogen bonding interactions. Therefore, the physically crosslinked junctions could be reversibly formed and cleaved during the stretching process, thus increasing the stress and strain of the hydrogels. However, excess HT might have reacted with water to produce amino groups and then reacted with HT again, thus reducing the number of effective crosslinkers. Consequently, the performance of hydrogels incorporating 100 μL of HT was similar to those prepared with 20 μL of HT. The hydrogels also exhibited good cutting resistance. As shown in Fig. 2d, when a knife was pressed down onto the hydrogels, they were not cleaved into two sections. After the knife was removed, the impacted region slowly reverted to its original state. The loading-unloading behaviors of the hydrogels prepared at various temperatures were further evaluated via compression tests, as shown in Fig. 2e–g. During the compression tests in which the compression limit was 100%, the compression behaviors of the hydrogels prepared at various temperatures were similar, with the stress values reaching up to ~70 MPa. However, the stress values in the compression region between 80 and 96% of the hydrogels prepared at low temperature were higher than those of the samples prepared at high temperature, as shown in the inset in Fig. 2f. Meanwhile, the results of the cyclic compression tests in which the compression was limited to 95% over 10 compression cycles (Fig. 2g) indicated that the stress values of the hydrogels prepared at elevated temperatures were significantly lower than those of the hydrogels prepared at lower temperatures. In addition, the stress changed slightly during 10 repeated compression cycles, possibly because the samples was being damaged. As shown in Fig. 2e, the shapes of hydrogels prepared at 25 °C and 50 °C changed significantly, while that of the hydrogel prepared at 75 °C remained intact after 10 compression cycles, indicating that the hydrogels prepared at this temperature were robust against repeated compression. Unfortunately, the hydrogels prepared at 90 ℃ failed the compression cycle tests due to their strong adhesion to the 10 kg support plate when it was lifted during the testing process. These results demonstrated the good mechanical performance of the self-healing hydrogels.

Stress versus strain plots obtained from tensile tests performed on the hydrogels containing different amounts of HT (a). Photographs of the hydrogels before (b) and after (c) stretching. Also shown are photographs of the hydrogels that were sliced by a knife and subsequently allowed to gradually return to their original shape (d). The shapes of the hydrogels prepared at various temperatures before and after compression measurements (e) and the stress-compression curves in which the compression was limited to 100% for 1 cycle (f) and 95% for 10 cycles (g)
Adhesive properties of the HEMA/AAm hydrogels
The preparation temperature also had a significant influence on the adhesive properties of the hydrogels. Figure 3a, b demonstrates that the surfaces of the hydrogels that were prepared at room temperature lacked adhesiveness; in contrast, the surfaces of those prepared at 90 °C exhibited adhesive behavior. Therefore, as presented in Fig. 3c, the hydrogels prepared at 90 °C could readily twist around and adhere to a steel rod. Moreover, with the aid of the HEMA/AAm hydrogels, a 200 g weight could adhere to glass, wood, porcelain, leather, paper, and cotton fabric at various angles and was even suspended from these substrates (see Fig. 3d–i). After 24 h, the 200 g weight that adhered to a wood substrate was able to serve as a mount and thus support a hanging 10 kg dumbbell (Fig. 5c). This suspended dumbbell remained in place even after shaking (see Movie 5). Interestingly, when the weight was removed, the hydrogels withdrew and retreated from the surface of the weight (see Movie 6). This adhesion-and-removal process could be repeated at least 20 times. In addition, 200 g of weight could be easily removed from the wood substrate after the hydrogels absorbed water again, as shown in Movie 7. These results suggest that adhesion is a dynamic and physical process involving physical interactions, such as hydrogen interactions, in contrast to the irreversible phenomena that are achieved via covalent adhesive methods. Thus, the weight could adhere reversibly to these materials multiple times, suggesting that these hydrogels have excellent potential for applications as adhesives. For example, no traces of the hydrogels remained on the leather after they had adhered to this substrate and were subsequently removed, as shown in Fig. 4.

Photographs of the surface properties of hydrogels prepared at room temperature (a) and 90 °C (b) during evaluation with a steel rod. Photograph of a hydrogel sample that was intertwined around a steel rod (c). Photographs of a 200 g weight that adhered to glass (d), wood (e), porcelain (f), leather (g), paper (h), and cotton fabric (i) with the aid of HEMA/AAm hydrogels at angles of 180° and 90°, demonstrating the adhesive performance of the hydrogels. The leather and cotton fabric were adhered to the glass to provide these substrates with rigidity

Images of a leather sample before and after removal of a weight that adhered onto this material with the aid of HEMA/AAm hydrogels: original state with the applied weight (a); during removal of the weight (b); and after removal of the weight (c). Also shown is an image of the leather after it had been washed with water, showing a clean surface (d)
The adhesive capabilities of the hydrogels were further determined via lap shear strength tests, as shown in Fig. 5a38. For the hydrogels that had been prepared at various temperatures and subsequently adhered onto glass slides for 1 min, the shear strength increased from 0.75 kPa up to 2.33 kPa with an increase in the preparation temperature, as shown in Fig. 5d, e. With the passage of time and loss of water, the strength of each hydrogel sample increased, suggesting that the hydrogels adhered strongly to the materials, especially after the loss of water. The hydrogels prepared at 90 °C increased significantly, up to 234.68 kPa after 48 h, corresponding to a 100-fold increase, thus demonstrating the dramatic influence of the preparation temperature.

Photographs of the hydrogels that adhered to glass slides before and after the lap shear test (a). Shear strengths of the hydrogels that had adhered to the glass slides after they had adhered to the substrate for various durations and were subjected to the lap shear test (b). Photograph of a 10 kg dumbbell suspended from a 200 g weight that adhered to wood with the aid of the hydrogel (c). Shear strengths of hydrogels prepared at various temperatures after adhesion to glass for 1 min (d) and 48 h (e), and hydrogel samples prepared at 90 °C after adhesion to various substrates for 1 min (f) and 48 h (g)
In addition, we determined the shear strengths of the hydrogels that had been prepared at 90 °C and subsequently adhered onto various substrates, as shown in Fig. 5f, g. The shear strengths of the hydrogels that had adhered to various materials for 1 min were in the range of 2–11 kPa. Similarly, with the passage of time and loss of water, the shear strength of the hydrogels adhered on various substrates increased by 40–50-fold to 120–460 kPa after 48 h. This behavior was attributed to the influence of water molecules, surface roughness, and hydroxyl groups on hydrogen interactions. In particular, the interactions between the hydrogel and substrate were evidently enhanced when few competing water molecules were present and when the substrate both bore numerous hydroxyl groups and had a rough surface. Thus, higher shear strengths were observed for substrates such as paper and wood (which possess many hydroxyl groups on their porous and rough surfaces) than for leather. Based on the abovementioned results, we believe that hydrogels are promising candidates for potential use as adhesives.
Stability of the HEMA/AAm hydrogels
The stabilities of the hydrogels were determined in various extreme conditions, including high temperatures, UV radiation and corrosive pH environments based on previously published methods39, as shown in Fig. 6a–c. The transparency of the hydrogels would decrease significantly after exposure to high temperatures for 24 h, possibly due to the formation of globules, as demonstrated in Fig. 1c. In addition, the hydrogels would swell in saline solution with various pH values due to the differences in the osmotic pressures between the hydrogels and the saline solution. However, the hydrogels retained their 3D network structures after exposure to various temperatures (from 30 to 80 °C) and high UV radiation (0.96 W/cm2) for 24 h, as well as to various pH conditions (from pH = 2 to 12) for 72 h.

The stability of the hydrogels that were evaluated via treatment at various temperatures (a) and exposure to UV radiation (b) and saline solutions (1 M) at various pH values (c), including (a) pH 2; (b) pH 4; (c) pH 6; (d) pH 7; (e) pH 8; (f) pH 10; (g) pH 12, and (h) pH 7.4. Also shown are the storage (G′) and loss (G″) moduli of the hydrogels prepared at 25 and 90 °C as functions of frequency (d) and temperature (e)
The HEMA/AAm hydrogel was also compared with the other hydrogels, as shown in Table S1 in the SI. We found that the self-healing behavior of these hydrogels occurred both very rapidly and readily without the need for external input. Most importantly, conventional hydrogels do not usually exhibit both self-adhesive and self-healing properties in combination, as shown in Table S1 in the SI. Interestingly, the hydrogels presented in our work not only have great self-healing capabilities but also are remarkably adhesive on various substrate materials and at high shear strengths. Thus, these hydrogels are superior to those described in previous works with regard to their self-healing and adhesion performance, indicating the positive and valuable influence of the preparation temperature on the properties of these hydrogels. Therefore, tuning the preparation temperature can clearly provide a facile and a convenient means of preparing hydrogels with both self-healing and adhesive properties.
Rheological behavior of the hydrogels
Small-deformation oscillatory measurements were recorded and are presented in Fig. 6d, e. The storage moduli (G′) of the hydrogels exceeded the corresponding loss moduli (G″) throughout the entire frequency and temperature range, indicating that the hydrogels behaved as viscoelastic solids. In addition, the G′ values over the tested frequency and temperature ranges for the hydrogels prepared at room temperature were higher than those for their counterparts prepared at elevated temperatures, revealing that the hydrogels prepared at an elevated temperature exhibited loosely bound network structures40. The increases in G′ and G″ with higher temperature may have occurred due to the formation of clusters and globules by the thermosensitive PHEMA polymers over the temperature range from 40 to 60 °C.
Figure 7b revealed that the hydrogels prepared at different temperatures had similar Tg values (above 50 °C), in accordance with the DSC measurements (see Fig. S5 in the SI) and previous reports that the Tg values of PHEMA hydrogels were in the range of 50–145 °C41. However, the self-healing properties of conventional PHEMA hydrogels differed significantly from those reported herein. Conventional hydrogels required heating to above their Tg to enable the segments of their polymer chains to move and thus achieve the desired self-healing performance. Therefore, these hydrogels were unable to rapidly and spontaneously self-heal at room temperature. However, the hydrogels reported here can exhibit rapid self-healing behavior at temperatures below their Tg. These differences in the self-healing behavior may be attributed to the regulating effect of the preparation temperature on the flexibility of the polymer chains and the looseness of their network structures.

photographs of the original and reversal states after 20 s of pre-hydrogel polymerization at various temperatures: (a) 25 °C, (b) 50 °C, (c) 75 °C, and (d) 90 °C (a). The rheological behaviors exhibited by the products of pre-hydrogel solutions that had been polymerized at various temperatures with respective water contents of 72% (b), 86% (c), 88% (d), and 90% (e)
Discussion
Generally, an increase in temperature lowers the degree of polymerization42, and thus we predicted that the hydrogels prepared at an elevated temperature would have shorter polymer chains than their counterparts that were prepared at lower temperatures. Therefore, short and flexible polymer chains would become prerequisites for the promotion of chain interaction and the desired self-healing performance. First, as shown in Fig. 7a, prehydrogels of HEMA/AAm solutions with a water content of 86% were used to prepare hydrogels at various temperatures, and the results showed that hydrogels were formed at low temperatures while viscous solutions were obtained at high temperatures. More importantly, the viscosities of these solutions decreased with increasing temperature. These results were further confirmed via rheological analysis.
As shown in Fig. 7b–e, when the water content in the pre-hydrogel solutions was increased from 72 to 90%, the G′ and G″ values of the products decreased. When the water contents were 72%, the G′ values of all products prepared at various temperatures were higher than their corresponding G″ values, indicating the formation of hydrogels. When the water content was increased to 86%, the G′ values of the products prepared at lower temperatures were higher than their corresponding G” values; in contrast, the G′ values of those products prepared at high temperature were lower than the G″ values of their counterparts, thus indicating the typical rheological behaviors of the hydrogels and the solution. When the water contents were further increased to 88% or 90%, the G′ values of all products prepared at various temperatures were lower than their corresponding G″ values, indicating that all of these obtained samples were viscous solutions at various temperatures. Notably, the G′/G″ values of the products prepared at high temperatures were lower than those of their counterparts prepared at low temperatures for both hydrogels and viscous solutions with the same water content. These results revealed that the preparation temperature had a key role in regulating the lengths of the resultant polymer chains.
To confirm the length changes in the polymer chains, the molecular masses of the polymers in those solutions that had been polymerized at various temperatures were evaluated via matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Fig. S6 in the SI). The results demonstrated that the main molecular mass and the average molecular mass (see Table S2) of the polymer chains decreased with increasing temperature, shifting from high (~270 kDa) to low (~120 kDa) molecular weights. These results implied that the molecular weights of the polymer chains were indeed regulated by the preparation temperature.
To determine if temperature may have possibly influenced the formation of distinctly different polymers, which could also lead to molecular weight changes, the 1H NMR, FTIR, and UV-vis spectra of the polymers prepared at various temperatures were recorded. These spectra of the polymers prepared at various temperatures did not differ significantly from one another, confirming that (aside from differences in molecular weights) similar polymers were indeed obtained at various temperatures, as shown in Fig. S7 in the SI. Therefore, these results indicated that relatively short polymer chains (small molecular mass) were obtained at elevated temperatures.
Accordingly, as shown in Fig. 8a, the short polymer chains had few cross-linking junctions, so that these chains had much freedom of motion and formed a loose network. In contrast, long polymer chains prepared at low temperatures would be linked together by many cross-linking junctions and possessed less flexibility. The loose network of hydrogels comprising short polymer chains could be confirmed based on the G′/G″ values, as well as the swelling equilibria and SEM images (Fig. 7b–i)43,44. The G′/G″ value and swelling ratio q of the hydrogels prepared at high temperatures were lower than those of their counterparts prepared at low temperatures, indicating that hydrogels with fewer cross-linking junctions and looser networks could absorb more water molecules and could more readily expand their volume. Meanwhile, loose networks could enhance the swelling behavior and shorten the equilibrium time. Therefore, the hydrogels prepared at 90 °C achieved equilibria in 24 h, showing both rapid absorption behavior and high water-absorption capacities (Fig. S8 in the SI). The SEM characterization showed numerous pores and thin pore walls and provided evidence of loose network structures for the hydrogels prepared at elevated temperatures.

Schematic illustration of a long polymer chain showing more restricted motion than that in a short polymer chain (a). Also shown are the SEM images of HEMA/AAm hydrogels prepared at: (b) room temperature, 1000 ×; (c) room temperature, 2000 ×; (d) room temperature, 5000 ×; (e) room temperature, 10,000 ×; (f) 90 °C, 1000 ×; (g) 90 °C, 2000 ×, (h) 90 °C, 5000 ×, and (i) 90 °C, 10,000 ×. The yellow frames indicate the regions depicted in the higher-magnification images
The looseness of the polymer network was thus an important structural property of the hydrogels that was dependent on the lengths of the polymer chains. Our conceptual strategy presented herein demonstrated that the polymer chain length and hence the properties of the hydrogels could be conveniently controlled by simply adjusting the polymerization temperature. A loose network containing short polymer chains with a high degree of segmental motion can facilitate the exchange of hydrogen bonds, thus promoting self-healing and adhesion43,44. The high degree of segmental motion exhibited by the hydrogel prepared at an elevated temperature was also confirmed by the results of the tensile tests performed on the hydrogels at various stretching rates. As shown in Fig. S9 in the SI, when the stretching rate was increased from 20 to 500 mm/min, the stress increased dramatically for the hydrogels prepared at room temperature but negatively for those prepared at an elevated temperature, indicating that the latter hydrogels had sufficient time to relax even at a high stretching rate due to their freedom of segmental motion. Thus, we have demonstrated that manipulation of the polymerization temperature provides a facile means of achieving desirable self-healing properties and unprecedented adhesive performance for hydrogels with a high water content of 72%.
Conclusion
In summary, we have found that the preparation temperature has a significant influence on the self-healing, adhesive, and tensile properties of HEMA/AAm hydrogels. When they are formed at room temperature, the hydrogels do not exhibit self-healing behavior; in contrast, those formed at 90 °C for as short as 150s and subsequently left at room temperature for 1 h can exhibit rapid self-healing performance. The hydrogels also showed good adhesive performance on numerous substrates, such as wood, glass, fibric, paper, leather, porcelain, and steel. Moreover, HT was found to dramatically enhance both the stress and stretching performance of the hydrogels by up to 30-fold without impairing their rapid self-healing properties. We believe that these findings may provide valuable insight that will facilitate the design and large-scale production of self-healing adhesive hydrogels.
References
Cao, Y. et al. A transparent, self-healing, highly stretchable ionic conductor. Adv. Mater. 29, 1605099 (2017).
Chen, Y., Kushner, A. M., Williams, G. A. & Guan, Z. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 4, 467–472 (2012).
Wu, D. Y., Meure, S. & Solomon, D. Self-healing polymeric materials: a review of recent developments. Prog. Polym. Sci. 33, 479–522 (2008).
Ghosh, S. K. (ed.). Self-healing Materials: Fundamentals, Design Strategies, and Applications. (Wiley-VCH Verlag GmbH & Co. KGaA, 2009).
Taylor, D. L. M. in het Panhuis. Self-Heal. Hydrogels, Adv. Mater. 28, 9060–9093 (2016).
Mauldin, T. C. & Kessler, M. R. Self-healing polymers and composites. Metall. Rev. 55, 317–346 (2013).
Yuan, Y. C., Yin, T., Rong, M. Z. & Zhang, M. Q. Self healing in polymers and polymer composites. Concepts, Realiz. Outlook.: A Rev., Express Polym. Lett. 2, 238–250 (2008).
Hager, M. D., Greil, P., Leyens, C., v.d. Zwaag, S. & Schubert, U. S. Self-healing materials. Adv. Mater. 22, 5424–5430 (2010).
Yang, Y. & Urban, M. W. Self-healing polymeric materials. Chem. Soc. Rev. 42, 7446–7467 (2013).
Diesendruck, C. E., Sottos, N. R., Moore, J. S. & White, S. R. Biomimetic self-healing. Angew. Chem. Int. Ed. 54, 10428–10447 (2015).
Wang, X., Li, Y. & Sun, J. Bioinspired self-healing coatings. Handbook of biomimetics and bioinspiration:1 Bioinspired Materials 391–417 (2015).
Billiet, S., Hillewaere, X. K. D., Teixeira, R. F. A. & Prez, F. E. D. Chemistry of crosslinking processes for self-healing polymers. Macromol. Rapid Commun. 34, 290–309 (2012).
Blaiszik, B. J. et al. Self-healing polymers and composites. J. R. Soc., Interface 4, 347 (2007).
Peppas, N. A. et al. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Adv. Mater. 18, 1345–1360 (2006).
Zhang, Y. S. & Khademhosseini, A. Advances in engineering hydrogels. Science 356, eaaf3627 (2017).
Azevedo, S. et al. Bioinspired ultratough hydrogel with fast recovery, self-healing, injectability and cytocompatibility. Adv. Mater. 29, 1700759 (2017).
South, A. B. & Lyon, L. A. Autonomic self-healing of hydrogel thin films. Angew. Chem. 122, 779–783 (2010).
Mozhdehi, D., Ayala, S., Cromwell, O. R. & Guan, Z. Self-healing multiphase polymers via dynamic metal–ligand interactions. J. Am. Chem. Soc. 136, 16128–16131 (2014).
Hager, M. D., Greil, P., Leyens, C., van der Zwaag, S. & Schubert, U. S. Self‐healing materials. Adv. Mater. 22, 5424–5430 (2010).
Wei, Z. et al. Self-healing gels based on constitutional dynamic chemistry and their potential applications. Chem. Soc. Rev. 43, 8114–8131 (2014).
Amendola, V. & Meneghetti, M. Self-healing at the nanoscale. Nanoscale 1, 74–88 (2009).
Chirila, T. V. et al. Hydrogen-bonded supramolecular polymers as self-healing hydrogels: effect of a bulky adamantyl substituent in the ureido-pyrimidinone monomer. J. Appl. Polym. Sci. 131, 39932 (2014).
Strandman, S. & Zhu, X. X. Self-healing supramolecular hydrogels based on reversible physical interactions. Gels 2, 16 (2016).
Yesilyurt, V. et al. Injectable self-healing glucose-responsive hydrogels with pH-regulated mechanical properties. Adv. Mater. 28, 86–91 (2016).
Li, C.-H. et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 8, 618–624 (2016).
Clarke, D. E., Pashuck, E. T., Bertazzo, S., Weaver, J. V. M. & Stevens, M. M. Self-Healing, self-assembled β-sheet peptide–poly(γ-glutamic acid) hybrid hydrogels. J. Am. Chem. Soc. 139, 7250–7255 (2017).
Sun, L. et al. Sundew-inspired adhesive hydrogels combined with adipose-derived stem cells for wound healing. ACS Appl. Mater. Interfaces 8, 2423–2434 (2016).
Han, L. et al. A mussel-inspired conductive, self-adhesive, and self-healable tough hydrogel as cell stimulators and implantable bioelectronics. Small 13, 1601916 (2017).
Bradley, L. C. et al. Rough adhesive hydrogels (RAd gels) for underwater adhesion. ACS Appl. Mater. Interfaces 9, 27409–27413 (2017).
Liu, X., Zhang, Q., Gao, Z., Hou, R. & Gao, G. Bioinspired adhesive hydrogel driven by adenine and thymine. ACS Appl. Mater. Interfaces 9, 17645–17652 (2017).
Zhang, L., Bailey, J., Subramanian, R., Groisman, A. & Tezcan, F. Hyperexpandable, self-healing macromolecular crystals with integrated polymer networks. Nature 557, 86–91 (2018).
Ahmed, E. Hydrogel: Preparation, characterization, and applications: a review. J. Adv. Res. 6, 105–121 (2015).
Rauner, N., Meuris, M., Zoric, M. & Tiller, J. Enzymatic mineralization generates ultrastiff and tough hydrogels with tunable mechanics. Nature 543, 407–410 (2017).
Dong, L., Agarwal, A., Beebe, D. & Jiang, H. Adaptive liquid microlenses activated by stimuli-responsive hydrogels. Nature 442, 551–554 (2006).
Rapado, M. & Peniche, C. Synthesis and characterization of pH and temperature responsive poly(2-hydroxyethyl methacrylate-co-acrylamide) hydrogels. Polim.: Cienc. Tecnol. 25, 547–555 (2015).
Mathew-Krotz, J. & Mahadevan, V. Redox-initiated radical polymerization of 2-hydroxyethyl methacrylate, 3. Copolymerization with nine monomers, effect of structure and media on the reactivity of monomers. Macromol. Chem. Phys. 198, 1597–1604 (1997).
Darabi, M. A. et al. Skin-inspired multifunctional autonomic-intrinsic conductive self-healing hydrogels with pressure sensitivity, stretchability, and 3D printability. Adv. Mater. 29, 1700533 (2017).
Shan, M., Gong, C., Li, B. & Wu, G. A pH, glucose, and dopamine triple-responsive, self-healable adhesive hydrogel formed by phenylborate-catechol complexation. Polym. Chem. 8, 2997–3005 (2017).
Wang, Q. et al. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 463, 339–343 (2010).
Winter, H. H. & Chambon, F. Analysis of linear viscoelasticity of a crosslinking polymer at the gel point. J. Rheol. 30, 367–382 (1986).
Chirila, T. V., George, K. A., Ghafor, W. A. A., Pas, S. J. & Hill, A. J. Sequential homo-interpenetrating polymer networks of poly(2-hydroxyethyl methacrylate): synthesis, characterization, and calcium uptake. J. Appl. Polym. Sci. 126, E455–E466 (2012).
Chiu, W., Carratt, G. & Soong, D. A computer model for the gel effect in free-radical polymerization. Macromolecules 16, 348–357 (1983).
Yanagisawa, Y., Nan, Y., Okuro, K. & Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 359, eaam7588 (2018).
Dougherty, R. C. Temperature and pressure dependence of hydrogen bond strength: a perturbation molecular orbital approach. J. Chem. Phys. 109, 7372–7378 (1998).
Acknowledgements
We wish to thank the Talent Project of Guangzhou University (27000503169), the Natural Science Foundations of China (21878059), the Science and Technology Project of Guangdong Province (2017A050501040), and the Science and Technology Project of Guangzhou City (201610010018 and 201607010285) for sponsoring this research.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, R., Xu, X., Yu, D. et al. Temperature-regulated flexibility of polymer chains in rapidly self-healing hydrogels. NPG Asia Mater 11, 22 (2019). https://doi.org/10.1038/s41427-019-0123-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-019-0123-0
