
Abstract
Retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are RNA sensor molecules that play essential roles in innate antiviral immunity. Among the three RLRs encoded by the human genome, RIG-I and melanoma differentiation-associated gene 5, which contain N-terminal caspase recruitment domains, are activated upon the detection of viral RNAs in the cytoplasm of virus-infected cells. Activated RLRs induce downstream signaling via their interactions with mitochondrial antiviral signaling proteins and activate the production of type I and III interferons and inflammatory cytokines. Recent studies have shown that RLR-mediated signaling is regulated by interactions with endogenous RNAs and host proteins, such as those involved in stress responses and posttranslational modifications. Since RLR-mediated cytokine production is also involved in the regulation of acquired immunity, the deregulation of RLR-mediated signaling is associated with autoimmune and autoinflammatory disorders. Moreover, RLR-mediated signaling might be involved in the aberrant cytokine production observed in coronavirus disease 2019. Since the discovery of RLRs in 2004, significant progress has been made in understanding the mechanisms underlying the activation and regulation of RLR-mediated signaling pathways. Here, we review the recent advances in the understanding of regulated RNA recognition and signal activation by RLRs, focusing on the interactions between various host and viral factors.
Similar content being viewed by others


Introduction
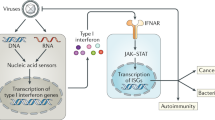
Interferons (IFNs), the family of cytokines first discovered, were identified as host factors secreted by virus-infected cells to “interfere” with viral replication.1,2 Based on the subsequent identification of their corresponding genes, they were further classified into three types in humans: type I IFNs [multiple IFN-α forms (1, 2, 4, 5, 6, 7, 8, 10, 13, 14, 16, 17, and 21), IFN-β, IFN-ε, IFN-κ, and IFN-ω], type II IFN (a single form, IFN-γ), and type III IFNs (IFNλ1-4).3 Among these types, type I and type III IFNs play essential roles in innate antiviral immunity. Infections caused by microbes, including viruses and bacteria, transiently induce the secretion of type I IFNs, which associate with the type I IFN receptor complex (IFNAR1 and IFNAR2) expressed by the infected cells themselves and surrounding noninfected cells, thereby activating cytoplasmic Janus kinases (JAK1 and TYK2). Signal transducer and activator of transcription (STAT1) and STAT2, activated by JAK-mediated tyrosine phosphorylation, together with the DNA-binding component IFN regulatory factor (IRF)-9, form a tripartite complex, referred to as IFN-stimulated gene factor 3 (ISGF3). ISGF3 then is translocated to the nucleus, where it binds to the IFN-stimulated response element (ISRE) on the promoters of hundreds of IFN-inducible genes (ISGs) and activates their transcription. Proteins encoded by ISGs induce strong antiviral innate immunity to eliminate invading viruses.4 Since the major histocompatibility complex (MHC) and costimulatory molecules are also ISGs, IFNs play critical roles in regulating acquired immunity (Fig. 1).

Interferon-mediated antiviral responses and diseases. In an antiviral innate immune system, nucleic acid sensors detect viral nucleic acids intracellularly and extracellularly, which then activate intracellular signaling pathways, leading to transcriptional activation of type I and type III interferon (IFN) genes upon the nuclear translocation of the transcription activators IFN regulatory factor (IRF)-3/7 and nuclear factor-κB (NF-κB). Secreted type I and type III IFNs activate a secondary signaling pathway by binding to their cognate IFN receptor (IFNR) complexes in an autocrine and paracrine manner. The downstream JAK-STAT pathway activates the transcription complex IFN-stimulated gene factor 3 (ISGF3), which leads to the expression of hundreds of IFN-stimulated genes (ISGs), resulting in strong antiviral responses. Since IFN-induced antiviral activity is critical for eliminating infected viruses, its weak or delayed response leads to uncontrolled infectious disease. In contrast, IFNs also play essential roles in regulating the subsequently induced adaptive immunity, and thus, their excessive or chronic activation can lead to the development of autoimmune or autoinflammatory diseases as, well as the initiation of cytokine storm, which exacerbates inflammation
Type III IFNs, similar to type I IFNs, are also activated in response to pathogenic infections; however, these cytokines are recognized by a distinct receptor complex comprising IFNLR1 and interleukin (IL)-10R2.5 The signaling pathway downstream of the type III IFN receptor complex is nearly identical to that of type I IFNs, resulting in similar antiviral activity via JAK-STAT-induced ISG production.3 The functional differences between type I and type III IFNs might be caused by the different cell types producing IFNs and their cognate receptors.5 Notably, type III IFN receptors are predominantly expressed on epithelial cells and certain myeloid cells, suggesting their critical roles in these cell types.5
IFNs have been used for the treatment of several viral infections and various immunological disorders and cancers due to their antiviral effects and cell growth inhibitory properties.6 Although type I IFNs have previously comprised the sole treatment strategy for hepatitis C virus (HCV), they have more recently been replaced by nonstructural protein (NS)3/4A and NS5A inhibitors, which have proven effective for the complete elimination of HCV. IFN therapy has been proposed as a potential strategy for the treatment of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).7 Indeed, mouse models of SARS-CoV, another pathogenic coronavirus, have exhibited delayed induction of type I IFNs during the early onset of infection, leading to potential excessive cytokine secretion, called a “cytokine storm”, which is associated with exacerbated disease.8 This outcome is also observed in COVID-19, suggesting a potential therapeutic strategy to enhance the initial production of IFNs during the early stages of SARS-CoV-2 infection.9 However, excessive IFN production in the late stages of infection is associated with the development of cytokine storm, suggesting that the therapeutic use of IFNs requires careful consideration in terms of timing and dosage.10,11 The use of type III IFNs in COVID-19 treatment has also been suggested due to their limited action on cell types such as epithelial cells;7 however, recent reports have shown that they may be detrimental to COVID-19 patients,12,13 and thus, further investigations into their suitability as therapeutic agents are warranted.
In higher vertebrates, pathogenic infections are detected by pattern recognition receptors (PRRs), which are part of the innate immune system. Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) are extracellular PRRs that detect pathogens on the plasma membrane or endosomal compartment. In addition, retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), Nod-like receptors (NLRs), and cytoplasmic DNA sensors serve as intracellular PRRs. The production of type I and type III IFNs is explicitly mediated by PRRs, such as endosomal TLRs and RLRs that detect nucleic acids. Among these PRRs, TLR-3 recognizes double-stranded RNA (dsRNA), TLR-7/8 recognizes single-stranded RNA (ssRNA), and TLR-9 recognizes unmethylated CpG DNA in the endosomal compartment. RLRs recognize cytosolic RNAs, and cyclic GMP-AMP synthase (cGAS) acts as a cytosolic DNA sensor.14 The transcription factors IRF-3/7 and nuclear factor-κB (NF-κB), downstream of IFN-inducing PRRs, are commonly activated and transiently induce IFN-encoding genes. Genes encoding proinflammatory cytokines, such as IL-6 and tumor necrosis factor (TNF)-α, are also induced by activated NF-κB. In this review, we focus on RLRs that detect cytoplasmic viral RNAs and the molecular mechanisms underlying RNA-mediated activation of RLRs and signaling regulation performed by host and viral factors.
Activation of RLR-mediated signaling
Association between RLRs and MAVS
RIG-I, which enhances IFN production in response to RNA virus infection, was isolated via expression cloning.15 The human genome encodes three RLRs, RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2), which are DExD/H box-containing RNA helicases and ubiquitously expressed in the cytoplasm.16 These RLRs commonly have a central helicase domain and a C-terminal domain (CTD) critical for RNA recognition. RIG-I and MDA5 also contain a tandem caspase recruitment domain (CARD) in their N-terminal region, which is required for their association with the adapter molecule mitochondrial antiviral-signaling protein (MAVS; also known as IPS-1, VISA, and Cardif), which also contains a single CARD at its N-terminus (Fig. 2a). The interaction between the CARDs of RIG-I/MDA5 and MAVS leads to the recruitment of downstream signaling molecules, including TNF receptor-associated factor (TRAF)3/6 and inhibitor of NF-κB kinase (IKK) family members (IKKε, TBK1, and IKKα/β), to activate IRF-3/7 and NF-κB, leading to the transcriptional activation of IFN and proinflammatory cytokine genes (Fig. 2b).17,18 More importantly, since RLR genes are ISGs, a positive feedback loop leads to further enhancement of IFN production.15

RIG-I-like receptors and the associated activation machinery. a Primary structure of retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) and mitochondrial antiviral-signaling protein (MAVS). All RLR family members contain a DExD/H box-containing RNA helicase domain composed of Hel1, Hel2i, Hel2, and pincer domains and the C-terminal domain (CTD). RIG-I and melanoma differentiation-associated gene 5 (MDA5), but not laboratory of genetics and physiology 2 (LGP2), contain repeated caspase recruitment domains (CARDs) in their N-terminal regions. MAVS has an N-terminal single CARD, three TNF receptor-associated factor (TRAF)-binding motifs (TBMs), and a transmembrane (TM) domain in the C-terminus. b RIG-I and MDA5 recognize 5′-triphosphate(ppp)-containing panhandle double-stranded (ds)RNA and long dsRNA, respectively. Recognition of substrate RNAs induces ATP-dependent conformational changes of RLRs, which induce filamentous oligomer formation on substrate RNAs. The N-terminal CARDs adopt a “lock-washer”-like tetramer with K63-linked ubiquitin chains and form signal-competent oligomers with the CARD of MAVS, which is expressed on the outer membrane of mitochondria. MAVS oligomers recruit downstream signaling molecules, such as TRAFs and inhibitors of NF-κB kinases (IKKs), which leads to the activation of IRF-3/7 and NF-κB
RIG-I detects substrate RNAs that have a panhandle double-stranded structure with a 5′-triphosphate or 5′-diphosphate moiety and activates downstream signaling in an ATP-dependent manner (Fig. 2b).19 A gene knockout (KO) study in mice revealed that RIG-I can be used for detecting multiple viral species in many cell types.20 The study showed that RIG-I KO in mice results in embryonic lethality resulting from liver degeneration; however, another study showed that KO mice with a different genomic region targeted were viable but developed spontaneous colitis.21 Although the mechanism underlying colitis development in these mice has not been fully elucidated, recent studies have demonstrated that RIG-I might participate in the maintenance of homeostasis in digestive organs by interacting with commensal microorganisms.22,23
MDA5 recognizes relatively long dsRNAs.24 Specifically, studies with MDA5-KO mice have demonstrated that MDA5 is primarily involved in detecting viruses belonging to the Picornaviridae family;25 however, it can also recognize several other viral species. In addition to their virus-sensing roles, RIG-I/MDA5 are both capable of controlling viral replication by exhibiting an “effector-like” function that competitively block the interaction between viral proteins and RNA.26,27,28
Another RLR, LGP2, does not harbor N-terminal CARD and therefore does not function as a signaling molecule. Although in vitro studies, including our own, suggested that LGP2 acts as a negative regulator of RIG-I/MDA5-mediated signaling,16,29,30 and subsequent studies with KO mice revealed a positive regulatory function for LGP2.31,32 These reports revealed that LGP2 deficiency impairs IFN production in response to picornaviruses, including encephalomyocarditis virus (EMCV), suggesting that LGP2 is involved in the MDA5-mediated signaling activation (the molecular mechanisms are described as follows). In contrast, the physiological significance of LGP2 in RIG-I-mediated signaling remains unclear because LGP2 deficiency has different effects depending on the viral infection.31,32 LGP2 has also been reported to regulate RNA silencing machinery. Specifically, it can associate with Dicer, an endoribonuclease for microRNA (miRNA) production, suggesting that LGP2 might indirectly regulate antiviral signaling by regulating gene expression.33 Moreover, LGP2 has been reported to interact with transactivation response RNA-binding protein (TRBP), a positive regulator of Dicer-mediated miRNA maturation, to inhibit the production of several miRNAs and induce the apoptotic pathway by enhancing the expression of specific genes.34,35 In the case of West Nile virus (WNV) infection, LGP2 plays an essential role in the survival and adaptation of CD8+ T cells but not in MAVS-mediated IFN production.36 Taken together, these observations suggest that LGP2 might be involved in a broad range of antiviral effects, and hence, further analysis is required.
MAVS is an adapter molecule for RLR-mediated signaling that contains an N-terminal CARD and C-terminal transmembrane (TM) domain required for its localization to intracellular membranes, including those of mitochondria, and three TRAF-binding motifs (TBMs) required for signal transduction. A MAVS-KO mouse model was used to clearly identify the essential role of MAVS in RIG-I/MDA5-mediated IFN production.37
Several studies have suggested that RLRs/MAVS are involved in inflammasome-mediated proinflammatory cytokine production, such as IL-1β and IL-18.38 RIG-I has been reported to be directly associated with apoptosis-associated speck-like protein containing CARD (ASC) and caspase-1 of the inflammasome to form virus-specific inflammasomes in response to vesicular stomatitis virus (VSV) and influenza A virus (IAV) infection.39,40 MAVS is also reportedly involved in the formation of the NLRP3 inflammasome in mitochondria.41,42 Furthermore, MAVS-mediated signaling is required to increase cell membrane permeability and the subsequent K+ ion efflux that leads to the formation of the NLRP3-containing inflammasome.43 Thus, further analysis is warranted to better understand virus-induced inflammasomes.
Activation of RLR-mediated signaling
In the steady state, RIG-I is likely to adopt an autorepressed conformation via direct interactions between CARDs and the helical insertion domain (Hel2i) located in the helicase domains.44 An initial structural analysis of RIG-I CARD showed that the CTD with substrate dsRNA forms a rigid proteolysis-resistant structure, indicating a critical role of the CTD in substrate recognition.45,46 The positively charged surface of the RIG-I CTD was also shown to be critical for recognizing the 5′-triphosphate signature of substrate dsRNA. Subsequent structural analysis of the helicase domain and CTD of RIG-I with substrate RNA revealed that both wrap around dsRNA with CTD sheathing the end.44,47,48,49 The interaction between RIG-I and its dsRNA substrate induces an ATP-dependent intramolecular conformational change and the release of N-terminal CARDs, allowing the RIG-I CARD to associate with the MAVS CARD on the mitochondria. At the same time, the translocase activity of RIG-I allows it to migrate along the RNA substrate; thus, the accumulation of multiple RIG-I molecules on RNA results in the formation of filamentous oligomers.50,51,52 Furthermore, the CARDs of the activated RIG-I proteins on the RNA form a helical tetrameric “lock washer” structure, allowing the MAVS CARD to form signal-competent aggregates (Fig. 2b).53
In contrast, the constitutive activity of MDA5 is repressed by phosphorylation.54 In contrast to RIG-I, the MDA5 CTD forms a relatively open structure, indicating that the MDA5 CTD cannot function independently to recognize RNA.55 A structural analysis of MDA5 with substrate dsRNA demonstrated that the helicase domain and CTD of MDA5 recognize internal duplexes rather than the end of the dsRNA substrate structures.49 Moreover, MDA5 molecules accumulate on long dsRNA in a head-to-tail arrangement, forming a filamentous structure and transmitting a signal by the N-terminal CARDs protruding from the filaments.49,56 Furthermore, the formation of the MDA5 filament is essential for distinguishing between self- and non-self-dsRNA via assembly/disassembly dynamics that depend on the length of the substrate dsRNA.57,58
However, the precise mechanisms by which LGP2 regulates RIG-I/MDA5-mediated signaling remain unclear. The CTD of LGP2 adopts a structure similar to that of RIG-I and recognizes the blunt end of dsRNA in a 5′-triphosphate-independent manner, reflecting the differential function of LGP2 in RIG-I- and MDA5-mediated signaling.55,59,60 A structural analysis of full-length LGP2 suggested that it forms a complex with the MDA5-like helicase domain and a RIG-I-like CTD.61 LGP2 can augment MDA5-mediated filament formation.62 Further analysis is required to elucidate the molecular machinery involved in the cooperative recognition performed by these RLRs.
Ectopic overexpression of MAVS in vitro causes constitutive IFN production, suggesting that the activity of MAVS as a signaling adapter is tightly regulated. Although several molecules have been identified as negative regulators of MAVS, their repression does not induce constitutive activation of MAVS, thus suggesting alternative inhibitory mechanisms. Qi et al. reported that the expression of multiple N-terminal truncated MAVS isoforms produced by alternative translation initiation suppresses the constitutive activation of full-length MAVS.63 The constitutive signaling capacity of full-length MAVS is repressed upon association with inactive truncated MAVS isoforms through the TM domain. However, the intramolecular interaction of MAVS suppresses constitutive activation.64 In response to the activation of RIG-I or MDA5, MAVS CARDs form protease-resistant prion-like aggregates on intracellular membranes such as mitochondria. A structural analysis revealed that tetrameric RIG-I/MDA5 CARDs act as templates for MAVS CARD filaments to which downstream signaling molecules are recruited.65,66,67
Intracellular localization of RLRs and MAVS
In the inactivated state, RLRs are uniformly expressed in the cytoplasm. Since MAVS is expressed on mitochondrial outer membranes, the activated RIG-I/MDA5 in filamentous aggregates is located close to mitochondria. MAVS is also localized in mitochondrial-associated endoplasmic reticulum membranes (MAMs) and peroxisomes and regulates RLR-mediated signaling (Fig. 3a).68,69 Peroxisomal MAVS is primarily involved in type III IFN induction.70 In addition, a recent study that performed high-resolution assays reported that microsomal fractions, such as endoplasmic reticulum membranes, but not mitochondria, play central roles in RLR-mediated signaling.71 Nuclear-localized RIG-I is required for detecting IAV, which replicates in the nucleus;72 however, the mechanism by which it activates MAVS-mediated signaling is unclear. Further analysis is required to understand the intracellular localization of RLRs.

Regulation of RLR-mediated signal transduction. A summary of the regulatory mechanisms of RLR/MAVS-mediated signaling by host and viral factors. a Activated RLRs interact with MAVS, localize to mitochondria, mitochondria-associated endoplasmic reticulum membranes (MAMs), and peroxisomes, where they activate downstream signaling. b RLRs accumulate in virus-induced stress granules (SGs) with viral RNAs, resulting in enhanced signaling. c Several host RNAs can be recognized by RLRs and are involved in the regulation of autoimmunity and cancer. Moreover, RLR/MAVS-mediated signaling is regulated by various host molecules: ubiquitin ligases/deubiquitinases (d), posttranslational modifiers (e), protein kinases/phosphatases (f), RNA-binding proteins (g), other host proteins (h), and autophagy regulators (i). These molecules are classified in square boxes (orange boxes: positive regulators; blue boxes: negative regulators). The molecules that inhibit or promote these regulatory molecules are shown on the right side of each column. Regulatory molecules of viral origin are shown in pink rounded rectangles
Stress granules (SGs) are membrane-less cytoplasmic aggregates to which RLRs are localized, and they can enhance MAVS-mediated signaling by acting as platforms for the recognition of foreign viral RNAs by RLRs (Fig. 3b).73 Under various stress conditions, including viral infection, cells produce SGs to inhibit cellular protein synthesis and store the protein synthesis machinery (translationally stalled mRNAs and RNA-binding proteins) to reinitiate translation after recovery from the stress condition.74 During viral infection, protein kinase R (PKR), a well-known dsRNA-dependent kinase that is also an antiviral ISG, phosphorylates the eukaryotic translation initiation factor eIF-2α and induces the production of SGs.75,76,77,78 The inhibition of virus-induced and PKR-dependent SG formation significantly impairs IFN production when exposed to NS1-defective IAV but not wild-type IAV, indicating that IAV-induced SGs are critical for IFN response activation and that NS1 can function as a viral inhibitor for SG-mediated signaling.73 The accumulation of MAVS-enriched mitochondria around RIG-I-containing aggregates supports the hypothesis that SGs serve as platforms for viral detection.79,80 In the case of Newcastle disease virus (NDV) infection, RIG-I is localized to the viral replication complex (vRC) to initiate primary IFN induction during the early postinfection phase, and its subsequent accumulation in SGs is required for the robust upregulation of IFN genes, suggesting the importance of intracellular dynamics of RIG-I proteins for efficient activation of the signaling pathway.80 Furthermore, growing evidence suggests that multiple RNA-binding proteins are localized in virus-induced SGs and are involved in IFN production, which strongly suggests an association between SG formation and RLR-mediated signal enhancement.75,81,82,83,84,85,86,87 Moreover, Ras-GTPase-activating protein SH3-domain-binding protein 1 (G3BP1), which is a critical component of SG formation, is implicated in RLR-mediated signaling, suggesting a functional interaction between RLRs and SG components in innate antiviral responses.88,89,90 However, several reports have revealed that RLR localization in SGs is not necessary for IFN production; thus, the requirement for SGs in RLR-mediated signaling remains unclear.91,92,93,94 Although many viruses have evolved strategies to inhibit SG formation, some exploit SG-containing proteins for their proliferation.76,95 Thus, it remains unclear whether PKR-induced SG formation is directly involved in the detection of viral RNAs by RLRs.96 To clarify the function of SGs in antiviral responses, further analysis of the relationship between the life cycle of each virus and the requirement of SG formation is needed. In addition, because SGs are membrane-less aggregates, the lack of an appropriate assay system to investigate their function has been a major problem. However, recent progress based on an in vitro reconstitution system using liquid-liquid phase separation has enabled an understanding of the molecular mechanisms of SG formation.97,98 Future analyses including those using this in vitro system are expected to clarify the physiological significance of SGs in antiviral responses.
RLRs and their involvement in autoimmune and autoinflammatory diseases
Since RLRs play essential roles in the initial production of IFNs and in regulating acquired immunity, their aberrant activation leads to various disorders, including autoimmune and autoinflammatory diseases.99,100,101 A genome-wide analysis revealed that single-nucleotide polymorphisms (SNPs) in IFIH1, which encodes MDA5, have been associated with these diseases. Smyth et al. demonstrated that an Ala to Thr mutation at 946 (A946T) of MDA5 (rs1990760) is significantly correlated with the risk of type 1 diabetes.102 Subsequent studies demonstrated that the A946T mutation is also associated with Graves’ disease,103 rheumatoid arthritis,104 multiple sclerosis,105 systemic lupus erythematosus (SLE),106,107,108 selective IgA deficiency,109 and vitiligo.110,111 In addition, a functional analysis of A946T-containing MDA5 revealed that this mutant exhibits gain-of-function properties and constitutively activates IFN production.112 Furthermore, Funabiki et al. developed a mouse strain bearing a Glu821Ser mutation that exhibited a gain-of-function phenotype similar to that in the A946T model. The mutant mice spontaneously developed autoinflammatory symptoms, including lupus-like nephritis.112 A more recent study developed a knock-in mouse model with MDA5 carrying the A946T mutation, and the results showed that it exacerbated the mouse autoimmune disease through the upregulation of basal IFN and hypersensitivity to self-RNA.113 Multiple MDA5 SNPs, in addition to A946T, have also been reported to be associated with autoimmune and autoinflammatory diseases.114,115 Several gain-of-function mutations in IFIH1 are also associated with Aicardi-Goutieres syndrome (AGS)116,117 and Singleton-Merten syndrome (SMS).118 In addition, a loss-of-function mutation in IFIH1, Lys365Glu, has been shown to cause severe recurrent respiratory viral infections.119
SNPs in DDX58, which encodes RIG-I, are also associated with SMS.120,121 Specifically, Lässig et al. reported that the Glu373Ala and Cys268Phe RIG-I mutants found in SMS, which are devoid of ATPase activity, interact with endogenous ribosomal RNA and activate unintentional ATP-independent IFN production, resulting in SMS development.122,123 However, little is known about the relationship between MAVS and autoimmune diseases, and although a loss-of-function mutation (Cys79Phe) found in MAVS has been associated with SLE, the molecular mechanism remains unclear.124
Regulation of RLR-mediated signaling by rna recognition
RNA recognition by RIG-I
An analysis of RIG-I-KO mice revealed that RIG-I is required for the detection of RNA viruses, including IAV, NDV, and Sendai virus (SeV).20 Subsequent analyses revealed that multiple types of RNA viruses, including HCV,125 WNV,126 dengue virus (DEV), Zika virus (ZIKV),127 hepatitis D virus (HDV),128 hepatitis E virus (HEV),129 and Hantaan virus (HTNV), can be recognized by RIG-I.130 Since most RNA viruses replicate their own genome using viral-encoded RNA-dependent RNA polymerase (RdRP) within the infected cells, RIG-I recognizes viral genomic RNAs based on their 5ʹ-triphosphate signature.131,132 The double-stranded structures that cannot be unwound by RIG-I helicase activity, and the short panhandle blunt-ended structures with 5′-triphosphate ends, are preferential structures for RIG-I recognition.45,133,134 A pregenomic RNA, which has been reverse-transcribed from the genomic DNA of hepatitis B virus (Hepadnaviridae) genomic DNA, is also recognized by RIG-I.27 Moreover, incoming full-length viral IAV RNAs can be recognized by RIG-I.135,136,137 Since the IAV RNA genome associates with the virus-derived nucleocapsid protein (NP) and tripartite RdRP complex at the 5′-terminal panhandle structure,138 the mechanism by which RIG-I recognizes the protected viral RNA-protein complex (RNP) remains unclear. A recent study showed that several host cosensor(s) or regulatory molecule(s) are involved in RIG-I-mediated signaling; however, the molecular machinery critical for the coordinated detection of viral RNP by RIG-I and its cofactors remains unclear. Interestingly, recent reports have demonstrated that small RNAs, such as miniviral RNA (mvRNA), produced by the aberrant replication of viral RNA via RdRP form aberrant viral RNPs, are preferentially recognized by RIG-I.139,140 In the case of SeV infection, RNA with an abnormal panhandle structure derived from defective interfering (DI) particles can be detected by RIG-I.141 Additionally, the uridine-rich region of the HCV RNA genome can be recognized by RIG-I.125 The short viral RNA suppressor of virus RNA (svRNA), generated by host ribonuclease L (RNase L)-mediated cleavage of the HCV RNA genome, is also efficiently detected by RIG-I.142
In addition to the 5′-triphosphate structure, the 5′-diphosphate moiety characteristic of the reovirus (RV) RNA genome can also be recognized by RIG-I.143 This substrate specificity for RNA recognition by RIG-I clearly shows that the 5′-cap and 5′-monophosphate structures of endogenous RNAs, including that of mRNA, tRNA, and rRNA, cannot be recognized by RIG-I as non-self-RNA.144 Schuberth-Wagner et al. further demonstrated that, in addition to the 5′-cap structure of the mRNA, 2′-O-methylation at the +1 base neighbor of the 5′-cap, the N1 position, is critical for evading recognition by RIG-I.145 They also showed that the conserved His830 residue of the RIG-I RNA-binding pocket is critical for distinguishing the 2′-O-methylated RNA at the N1 site, whereas substituting His830 with Ala enhances IFN production via the recognition of 2′-O-methylated endogenous mRNA by RIG-I. Yellow fever virus (YFV), belonging to the Flaviviridae family, encodes a 2′-O-methyltransferase that allows it to escape detection by RIG-I.145 Many viruses have developed this type of evasion strategy to avoid detection by RIG-I. For example, viruses belonging to the Picornaviridae family cannot be recognized by RIG-I due to the covalent bonding of the viral protein Vpg (viral protein genome-linked) at the 5′-end of the viral genome.146 Crimean-Congo hemorrhagic fever virus (CCHFV) and HTNV, belonging to the Bunyavirales order; and Borna disease virus (BDV), belonging to the Bornaviridae family, have a 5′-monophosphate end that is not recognized by RIG-I.147 A recent study demonstrated that the RNA of human metapneumovirus (HMPV), belonging to the Pneumoviridae family, undergoes N6-methyladenosine modification to evade recognition by RIG-I.148
Furthermore, RIG-I can also recognize RNA secreted from bacteria. Specifically, RNA of intracellular pathogenic bacteria, such as Listeria monocytogenes, which is released into the cytoplasm, can be detected by RIG-I.149,150 IFN production via RIG-I signaling has been observed in response to other intracellular bacteria, including Salmonella enterica serovar Typhimurium151 and Mycobacterium tuberculosis.152 A recent study showed that secreted L. monocytogenes RNA complexed with the bacterial RNA-binding protein lmo2686/zea can also be recognized by RIG-I.153 Furthermore, M. tuberculosis RNA is delivered into the extracellular vesicles of macrophages, leading to the activation of RIG-I-mediated signaling in uninfected cells.154 These observations strongly suggest that RIG-I-mediated signaling has a broad function in the self-defense machinery against pathogenic infection. Notably, further analysis of the maintenance of homeostasis in the intestinal and respiratory tracts via commensal microbes and RIG-I-mediated signaling is necessary.22,23,155
Interactions between RIG-I and endogenous RNAs
Since the 5′-triphosphate signature in host RNAs is apparent during transcription by RNA polymerases, RIG-I can recognize host endogenous RNAs (Fig. 3c). An analysis of IFN production in response to dsDNA showed that 5′-triphosphate RNA transcribed by RNA polymerase III from intracellular poly(dA-dT) DNA can induce IFNs via RIG-I.156,157 This pol III-mediated activation of RIG-I has been analyzed in host responses against DNA virus infections. Next-generation sequencing of RNA samples interacting with RIG-I during herpes simplex virus (HSV; Herpesviridae) infection led to the identification of endogenous RNA transcribed from 5S ribosomal RNA pseudogene 141 (RNA5SP141). RNA5SP141, transcribed by RNA pol III in the nucleus, is translocated to the cytoplasm and activates RIG-I-mediated signaling.158 In cells infected with Kaposi’s sarcoma-associated herpesvirus (KSHV; belonging to the Herpesviridae family), an endogenous RNA designated “vault RNA” transcribed by RNA pol III accumulates as a 5′-triphosphate-containing RNA due to the reduced expression of DUSP11 (dual-specificity phosphatase 11) and induces RIG-I-mediated signaling to inhibit lytic reactivation of KSHV.159 However, RIG-I can recognize KSHV RNA in a pol III-independent manner.160 These findings indicate that activation of the RIG-I pathway via pol III-induced endogenous RNA is a critical host strategy to fight against DNA virus infections. In addition, endogenous 7SL RNA transcribed by RNA pol III can activate RIG-I-mediated signaling in cancer cells. 7SL RNA, a signal recognition particle (SRP) component, is upregulated in stromal cells by Notch-Myc signaling from breast cancer cells, and an excess of 7SL RNA is secreted in exosomes, which in turn activates RIG-I in breast cancer cells, leading to increased cancer growth, metastasis, and treatment resistance.161 However, Alu-derived RNA, an endogenous retrotransposon sequence originating from 7SL RNA, exhibits anticancer effects via RIG-I activation. The inhibition of heterogeneous nuclear ribonucleoprotein C (HNRNPC), which is highly expressed in cancer cells, suppresses HNRNPC-mediated cellular splicing, resulting in the accumulation of Alu sequences within the pre-mRNA intron and Alu-mediated activation of RIG-I to inhibit cancer growth.162 Several long noncoding (lnc)RNAs repress the RIG-I pathway. Mouse-specific lnc-Lsm3b and human-specific lncATV, which are upregulated by viral infection, compete with viral RNAs for detection by RIG-I, thereby inhibiting RIG-I-mediated IFN production.163,164 However, microRNAs involved in posttranscriptional regulation are believed to be unrecognized by RIG-I, as they do not contain 5′-triphosphate and have a 3′-overhang. However, several miRNAs enriched in uridines are potential RIG-I agonists.165,166 For example, miR136, the expression of which is induced by IAV infection, can inhibit IAV growth by activating RIG-I-mediated signals. Moreover, the introduction of exogenous circular RNAs (circRNAs) into cells can activate RIG-I in a 5′-triphosphate- or double-stranded structure-independent manner.167 Chen et al. further showed that endogenous circRNAs, generated by back-splicing to covalently link the 3′ and 5′ ends of the RNA exon, avoid RIG-I recognition by exhibiting N6-methyladenosine modification.168 However, several reports have also demonstrated that contaminating short ssRNAs generated during the production of circRNAs are critical for circRNA-mediated RIG-I activation.169 Further analysis is required to elucidate the physiological significance of circRNA-mediated RIG-I activation.
Finally, based on accumulating evidence highlighting the physiological significance and molecular machinery of RIG-I-mediated signaling, research on the agonist/antagonist targeting of RIG-I has been initiated. For example, 5′-triphosphate dsRNA has been shown to exhibit antiviral activity against lethal IAV infection.170 Furthermore, a unique RIG-I agonist has been reported to exhibit significant antitumor activity.171 RIG-I antagonists have also been identified with the aim of controlling excessive IFN production.172
RNA recognition by MDA5
An analysis of MDA5-KO mice revealed that MDA5 is involved in detecting viruses belonging to the Picornaviridae family, including EMCV and Theiler’s encephalomyelitis virus (TMEV).25 A subsequent study revealed that MDA5 can recognize relatively long dsRNAs.24 In addition to picornaviruses, MDA5 also recognizes DEV (Flaviviridae), RV (Reoviridae), a segmented dsRNA virus,173 murine norovirus (MNV; Caliciviridae), with Vpg at the 5′-end of the viral RNA,174 murine hepatitis virus (MHV) belonging to the Coronaviridae family with a 5′ cap,175 as well as other viruses101. The non-self-RNA signature of these viruses is a double-stranded structure generated as a replication intermediate.176 However, the exact RNA structure recognized by MDA5 is unclear, and further analysis is required to determine the specific properties of MDA5 ligands.177,178,179 In the case of coronavirus infection, the 2′-O-methylation of viral RNA by 2′-O-methyltransferase encoded by viral nonstructural protein 16 (nsp16) suppresses MDA5-mediated IFN production;180 however, it is unclear whether 2′-O-methylation directly impacts the binding of viral RNA to MDA5.145 A recent report on MHV showed that the panhandle dsRNA structure containing 5′-polyuridines of negative-sense RNA (PUN RNA) is an agonist of MDA5.181. Since SARS-CoV-2 belongs to the Coronaviridae family as MHV, it might be recognized by MDA5 via a similar mechanism. Degradation of PUN RNA by MHV nsp15, coding for a polyU-specific endoribonuclease (EndoU), limits MAD5-mediated IFN production, suggesting a viral strategy to escape antiviral activity.181 Considering that it is conserved in the SARS-CoV-2 genome, the EndoU gene might serve as an effective potential therapeutic target for COVID-19 treatment. In addition, MAD5 can detect RNA from malaria parasites. Plasmodium infection induces type I IFN in an MDA5- and MAVS-dependent manner and limits the propagation of parasitemia.182,183 These observations suggest that MDA5-mediated IFN production modulates innate immune responses to a wide range of pathogen infections.
Interaction between MDA5 and endogenous RNA
It is becoming clear that long dsRNAs, which are substrates of MDA5, can be generated from endogenous transposable repetitive sequences, such as Alu and retroviral elements (ERVs).101,184 Alu repeats are present at more than one million sites in the human genome. Alu RNAs transcribed from inverted repeats in the genome can be recognized by MDA5 when they form a dsRNA structure.185 However, in the steady state, A-to-I modification by adenosine deaminase acting on RNA (ADAR)1 prevents the formation of immunostimulatory dsRNAs. ADAR1-deficient mice are embryonic lethal due to global IFN induction;186 however, this phenotype is partially rescued by the deletion of MAVS or MDA5 but not RIG-I,187,188 suggesting that ADAR1 is a specific suppressor of MDA5/MAVS signaling. A recent report showed that dysfunction caused by a mutation in the ADAR1 gene leads to the abnormal accumulation of Alu-derived dsRNA, resulting in the overproduction of IFNs, which leads to autoimmune diseases, including AGS.117,185,188,189,190 In addition, dsRNAs generated by the activation of intrinsic ERVs following treatment with DNA demethylating drugs or via the bidirectional transcription of specific ERV regions can also be recognized by MDA5, leading to enhanced antitumor activity through IFN production.191,192,193 In adult human cells, tripartite motif (TRIM) protein 28 (TRIM28, also known as KAP1) represses ERV transcription via epigenetic modification. However, the depletion of TRIM28 induces the RLR-mediated pathway, presumably as a result of dsRNA generation from ERV-derived RNAs.194
MDA5 can also recognize mitochondrial dsRNAs. The translocation of unstable mitochondrial dsRNAs from the mitochondria to the cytoplasm is suppressed by the mitochondrial RNA helicase SUV3 and the polynucleotide phosphorylase PNPase; however, loss of function of either protein induces cytoplasmic accumulation of dsRNAs, leading to the activation of MDA5-mediated IFN production.195 The noncoding RNA ITPRIP-1 also plays an auxiliary role in supporting MDA5 oligomer formation by binding to the C-terminus of MDA5.196
Regulation by host proteins
Regulation by ubiquitin ligases and deubiquitinases
Posttranslational modification plays an essential role in regulating RLR/MAVS-mediated signaling. In particular, regulation by ubiquitination has been extensively analyzed (Fig. 3d). In 2007, Gack et al. reported that Lys63 (K63)-linked ubiquitination at the Lys172 residue of RIG-I CARD by TRIM25, a RING-finger-containing E3 ubiquitin ligase, was required for CARD-mediated signaling activation.197 Subsequent analysis using an in vitro reconstitution assay system demonstrated that the unanchored K63-linked ubiquitin chain added by TRIM25 is also involved in RIG-I activation.198 The crystal structure of the RIG-I CARD with K63-linked ubiquitin revealed that both anchored and unanchored ubiquitin chains might participate in the stabilization of the “lock washer”-like tetramer structure formed by RIG-I CARD.53 Moreover, TRIM25 regulates RIG-I-mediated signaling in coordination with multiple host factors. Specifically, the mitochondrial-targeting chaperone 14-3-3ε forms a complex with RIG-I and TRIM25 to regulate the transition from the cytoplasm to MAM, which is implicated in the RIG-I/MAVS interaction.199 ZCCHC3, a CCHC-type zinc finger protein, recruits TRIM25 to RIG-I/MDA5 and enhances the ubiquitin-mediated activation of RIG-I/MDA5.200 Caspase-12, an inflammatory caspase, contributes to the innate response to WNV infection by enhancing the K63-linked ubiquitination of RIG-I by TRIM25.201 Furthermore, by associating with RIG-I and TRIM25, nuclear Ddf2-related 2 (NDR2), an NDR family serine/threonine kinase, also positively regulates the addition of ubiquitin chains by TRIM25.202 However, the linear ubiquitin assembly complex (LUBAC), consisting of the E3 ligase heme-oxidized IRP2 ligase-1L (HOIL-1L), and HOIL-1L-interacting protein (HOIP), negatively regulates RIG-I signaling by adding a K48-linked ubiquitin chain onto TRIM25, leading to its proteasomal degradation.203 This K48-linked ubiquitin chain on TRIM25 is eliminated by a deubiquitinating enzyme ubiquitin-specific peptidase (USP)15.204 A recent study showed that NLRP12, a member of the NLR family, binds to TRIM25 and inhibits the TRIM25-mediated ubiquitination of RIG-I while simultaneously enhancing the degradation of RIG-I via K48-linked ubiquitination of RIG-I by another E3 ligase, ring finger protein (RNF)125, demonstrating the functional interaction between the two PRR families.205 Notably, recent studies have reported that TRIM25 is capable of RNA binding, which is required for regulating signals by a mechanism that does not involve ubiquitin ligase activity.206,207,208,209
Riplet (also known as RNF135 and REUL) is another critical K63-linked E3 ligase for RIG-I.210,211,212 Riplet conjugates ubiquitin on multiple Lys residues in the CARD and CTD of RIG-I. Although a previous study suggested that Riplet functions upstream of TRIM25 through the ubiquitination of Lys788 in the RIG-I CTD,82 more recent studies have shown that Riplet, but not TRIM25, may be an essential regulator of RIG-I activation.213,214,215 Mechanistically, Cadena et al. proposed a model in which Riplet is involved in upregulating RIG-I-mediated signaling via the initial ubiquitination of oligomerized RIG-I on substrate dsRNAs and the subsequent cross-bridging of RIG-I filaments on longer dsRNAs, leading to signal amplification.214 Furthermore, Ube2D3, an E2 ligase, has been identified as a coregulator of Riplet-mediated RIG-I activation.213 However, another E3 ligase, zinc finger protein (ZNF)598, has been identified as a negative regulator of K63-linked ubiquitination by Riplet. ZNF598 conjugates the ubiquitin-like protein FAT10 (also known as ubiquitin D) on RIG-I to inhibit Riplet-mediated ubiquitination of RIG-I.216
In addition to TRIM25 and Riplet, TRIM4 and MEX3C have also been identified by expression screening methods as positive regulatory E3 ligases of RIG-I.83,217 However, recent studies suggest that these ligases are not involved in RIG-I-mediated MAVS aggregation in vitro.213 The physiological function of the four E3 ligases, specifically TRIM25, Riplet, TRIM4 and MEX3C, in the activation of RIG-I remains unclear. This issue has been discussed in detail in recent excellent review articles.218,219 Briefly, although all of these molecules may be involved in the positive regulation of RIG-I activation, differing analytical conditions of recent reports has led to conflicting interpretations. Future analysis that takes into account differences in e viral, animal, and cell types is expected to reveal the exact regulatory mechanisms based on specific usage and/or redundancy of the molecules in RIG-I-mediated signal activation.
Several E3 ligases that negatively regulate RIG-I have been identified. For example, TRIM40 promotes proteasomal degradation by conjugating K27- and K48-linked ubiquitin chains on both RIG-I and MDA5,220 whereas RNF125 and RNF122 negatively regulate RIG-I activation via K48-linked ubiquitination of RIG-I CARD.221,222 The RNF125-mediated degradation of RIG-I is antagonized by the SG component G3BP1 via enhanced self-ubiquitination of RNF125.89 After viral infection, the STUB1 E3 ligase associates with the CARD of RIG-I via the trithorax group protein mixed linage leukemia (MLL5), promoting K48-linked ubiquitin-mediated degradation of RIG-I.223 Another E3 ligase, c-Cbl, is recruited, together with RIG-I, to Siglec-G, which is expressed on the plasma membrane and promotes the proteasomal degradation of RIG-I via K48-linked ubiquitin conjugation on the Lys813 residue of RIG-I.224 Moreover, Siglec-G belongs to the CLR family, suggesting an interaction between different PRRs.
Regulation by several deubiquitinating enzymes has been demonstrated. USP4 and ovarian tumor-domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) stabilize RIG-I proteins by eliminating the K48-linked ubiquitin chains conjugated to RIG-I.225,226 Conversely, CYLD, a tumor suppressor gene expressed in cylindromatosis, negatively regulates RIG-I activation by deubiquitinating the K63-linked ubiquitin chains on RIG-I.227 Moreover, syndecan-4 (SDC4), which is a TM protein, complexes with both RIG-I and CYLD to promote the CYLD-mediated deubiquitination of RIG-I, leading to subsequent signaling inhibition.228 Furthermore, USP3,229 USP21,230 USP14,231 and USP27X232 also deubiquitinate the K63-linked ubiquitin of RIG-I to inhibit RIG-I function.
An in vitro reconstitution analysis revealed that MDA5 CARD also requires K63-linked ubiquitin chains to activate the signaling pathway.233 However, the precise mechanism associated with ubiquitin-mediated regulation of MDA5 has remained unclear. TRIM65 promotes K63-linked ubiquitination at Lys743 of MDA5 to enhance the oligomerization of MDA5 on dsRNAs.234 Moreover, arrestin domain-containing 4 (ARRDC4) interacts with MDA5 to recruit TRIM65 in response to enterovirus 71 (EV71) infection, leading to the activation of MDA5-mediated signaling via Lys63-linked ubiquitination of MDA5.235
The regulation of MAVS by ubiquitin has also been extensively analyzed. K63-linked ubiquitination at Lys19, Lys311, and Lys461 of MAVS by TRIM31 is critical for the formation of prion-like aggregates of MAVS.236 The scaffold protein FAS-associated factor 1 (FAF1) negatively regulates TRIM31-mediated ubiquitination of MAVS by disrupting the interactions between MAVS and TRIM31.237 The phosphorylation of FAF1 by IKKε, which is activated by RLR-mediated signaling, induces FAF1 degradation and promotes the activation of MAVS-mediated signaling. OTUD4, a deubiquitinase, enhances MAVS-mediated signaling by stabilizing MAVS and by eliminating K48-linked ubiquitins.238
Several studies have demonstrated that the ubiquitination of MAVS negatively regulates MAVS-mediated signaling. In contrast to RIG-I, TRIM25 induces the degradation of MAVS via K48-linked ubiquitination at Lys7 and Lys10 of MAVS.239 The HECT domain-containing E3 ligase ITCH (also known as AIP4) binds to MAVS via poly(C)-binding protein (PCBP) 1/2 and TAX1 binding protein (TAX1BP1), which triggers K48-linked ubiquitination of MAVS and its subsequent degradation.240,241,242 Membrane-associated ring-CH-type finger 5 (MARCH5), a mitochondrial ubiquitin ligase, and RNF5 promote MAVS degradation via K48-linked ubiquitination at Lys7 and 500, as well as at Lys362 and 461, respectively. Interestingly, ER-associated inactive rhomboid protein (iRhom) 2 inhibits the MARCH5- and RNF5-mediated degradation of MAVS and positively regulates MAVS-mediated signaling.243 The TRIM29 protein conjugates with the K11-linked ubiquitin chain on MAVS for its degradation.244 However, MARCH8 is recruited by Tetherin (also known as BST2) to bind to MAVS and promote K27-linked ubiquitination at Lys7 of MAVS.245 Moreover, RNF34 conjugates with K27- and K29-linked ubiquitin chains at multiple Lys/Arg positions of MAVS. This K27/K29-linked ubiquitination induces NDP52/CALCOCO2-dependent autophagic degradation of MAVS.246 The activity of MAVS is also regulated by deubiquitinases, including YOD1,247 and OTUD3,248 which repress MAVS-mediated signaling by removing the K63-linked ubiquitin chains. Although a considerable number of studies have reported the regulation of RLR/MAVS-mediated signaling via ubiquitination/deubiquitination, the mechanisms underlying these regulatory processes remain unclear, and further studies are warranted.
Roles of other ubiquitin-like modifiers
Regulation via ubiquitin-like modifications has also been demonstrated (Fig. 3e). An early study showed that RIG-I protein levels are negatively regulated by conjugation with the IFN-inducible ubiquitin-like protein ISG15 (ISGylation).249 Very recently, it has been reported that ISGylation of MDA5 is essential for triggering IFN production in response to several RNA viruses, including coronaviruses.250 Furthermore, TRIM38 positively regulates both RIG-I and MDA5 via SUMOylation at K43/K865 and K96/K888, respectively.251 The SUMOylation of RLRs inhibits degradation by K48-linked ubiquitination. However, in the late stage of infection, de-SUMOylation by SUMO-specific protease 2 (SENP2) enhances degradation, which leads to disrupted signaling. As mentioned earlier, Riplet-mediated RIG-I activation is attenuated by the conjugation of FAT10 with RIG-I.216 Moreover, the noncovalent interaction between FAT10 and RIG-I suppresses RIG-I activation.252
Regulation by kinases and phosphatases
RIG-I is negatively regulated by phosphorylation at Thr170 and Ser8 (Fig. 3f).253,254 Protein kinase C (PKC)-α and PKC-β are involved in phosphorylation, which suppresses the TRIM25 interaction and K63-linked ubiquitination, resulting in the inhibition of RIG-I-mediated signaling.255 However, as mentioned earlier, the requirement of TRIM25 in RIG-I activation remains unclear and requires further analysis.213,214,215 Casein kinase 2 (CK2) phosphorylates RIG-I at Thr770, Ser854, and Ser855,256 which might be necessary for maintaining RIG-I in the autorepressed state. Phosphorylation of RIG-I by DAPK1 at Thr667, located within the helicase domain, also suppresses RIG-I-mediated signaling. Since DAPK1 activity is induced by RIG-I-mediated signaling, negative feedback regulation by DAPK1 is suggested.257 However, MDA5 is phosphorylated by RIO kinase 3 (RIOK3) at Ser828, which inhibits MDA5 oligomerization.258 Moreover, phosphorylation at Ser88 in the MDA5 CARD also inhibits MDA5 activation.54 Importantly, dephosphorylation of RIG-I and MDA5 by PP1α and PP1γ induces the transition of RLRs to an activated state, indicating that dephosphorylation is one of the fundamental regulatory mechanisms for RLR activation.54 The regulation of MAVS by phosphorylation has also been reported. Protein kinase A (PKA) promotes the degradation of MAVS by MARCH5 by phosphorylating Thr54 of MAVS and suppressing IFN induction.259 In contrast, phosphorylation of MAVS Tyr by c-Abl positively regulates MAVS-mediated IFN production.260,261
Regulation by other modifications
RIG-I is regulated by acetylation (Fig. 3e). For example, the deacetylation of the Lys858 and Lys909 residues of RIG-I via the deacetylating enzyme HDAC6 is implicated in viral RNA detection by RIG-I;262,263 however, the molecular mechanisms underlying RIG-I acetylation remain unclear. Moreover, MAVS is positively regulated by O-GlcNAcylation.264,265 In fact, Li et al. demonstrated that O-GlcNAcylation at Ser366 of MAVS in response to VSV infection promotes K63-linked ubiquitination of MAVS by TRIM31, leading to increased IFN production.
Regulation by RNA-binding proteins
Since RLRs are RNA-binding proteins, activation of RLR-mediated signaling is regulated by the coordination of various host RNA-binding proteins (Fig. 3g). Members of the 2′-5′ oligoadenylate (2-5 A) synthetase (OAS) family, which are classical antiviral ISGs, are activated by viral dsRNA and eliminate viral RNA via the activation of the host ribonuclease RNase L.266 Interestingly, human OAS-like protein (OASL) is a member of the OAS family that contains a unique ubiquitin-like domain but lacks oligoadenylation activity. The ubiquitin-like domain of OASL mimics the K63-linked polyubiquitin moiety and induces the oligomerization of RIG-I on RNA substrates, thereby enhancing RIG-I-mediated signaling.267
PKR, another classic antiviral ISG, has been extensively analyzed.268 PKR inhibits viral growth by potently repressing host translation through the phosphorylation of eIF-2α. In terms of the regulatory function of RLRs, as mentioned earlier, SGs formed via PKR mediation might play critical roles as platforms for viral RNA detection by RLRs, suggesting an indirect regulatory function of PKR in RLR-mediated signaling.269,270 Moreover, the RNA helicase DHX36 facilitates PKR-dependent SG formation, resulting in increased RLR-mediated IFN production.75
Protein activators of PKR (PACT) and TRBP are dsRNA-binding proteins that exert positive and negative regulatory functions of PKR, respectively.271 Initially, PACT was reported to participate in IFN production272 but was subsequently shown to also physically associate with both RIG-I and MDA5, effectively augmenting their signaling by enhancing ATPase function and oligomerization, respectively.273,274 Moreover, recent studies have indicated that the PACT-LGP2 interaction is required for upregulated MDA5-mediated signaling.275,276 However, the regulation of RLR-mediated signaling by PACT is antagonized by viral factors, including VP35 of Ebola virus (EBOV), NS1 of IAV, and NP of arenavirus.277,278,279 Importantly, the 4a protein of Middle East respiratory syndrome coronavirus (MERS-CoV) and the nucleocapsid (N) protein of MHV and SARS-CoV directly interact with PACT to inhibit RLR-mediated signaling,280,281 suggesting that it is a possible therapeutic target for COVID-19. In addition to the inhibitory function of PKR, TRBP enhances MDA5-mediated innate responses by physically interacting with LGP2 in response to infection by cardiovirus, which belongs to the Picornaviridae family.282 However, TRBP plays an essential role in miRNA processing from precursor RNAs in conjunction with the ribonuclease Dicer.283 Recent studies have shown that the enhanced expression of LGP2 in response to viral infection suppresses Dicer-mediated miRNA maturation via the LGP2-induced sequestration of TRBP from the Dicer-TRBP complex.34 Genome-wide analysis revealed that the suppression of RNA silencing by TRBP-dependent miRNA leads to enhanced specific gene expression, including cell death-related gene expression, suggesting a potential novel antiviral defense mechanism induced by the LGP2-TRBP interaction.35
The involvement of multiple RNA helicases in RLR-mediated signaling has been demonstrated. For example, DDX60 can enhance RIG-I-mediated signaling by associating with RIG-I, indicating its role as a sentinel RNA sensor.284,285 However, another report revealed that DDX60 deficiency does not affect IFN production in response to several RNA viruses or dsRNAs, suggesting that it might function in antiviral responses against very specific viral species.286 In addition, DDX6, DHX29, and DHX15 can also function as cosensors of RIG-I and MDA5 by directly interacting with RLRs.287,288,289,290 However, DDX3, which was initially identified as a MAVS-interacting helicase via two-hybrid screening,291 participates in the detection of aberrant human immunodeficiency virus type 1 (HIV-1) RNA in dendritic cells (DCs), which induces MAVS-mediated signaling independent of RLRs.292 Furthermore, DHX9 is also involved in the RLR-independent activation of MAVS-mediated signaling in response to dsRNA in DCs.293 Since the molecular mechanisms of these observations remain unclear, further analysis is necessary.
Regulation by other host proteins
14-3-3 proteins, including 14-3-3ε and 14-3-3η, can function as mitochondrial transporting chaperones for RIG-I and MDA5, respectively (Fig. 3h).199,294 NS3 proteins of DENV, ZIKV, and presumably WNV antagonize RLR-mediated signaling by sequestrating 14-3-3 proteins.295 Zyxin, which is involved in actin polymerization, localizes with MAVS on the mitochondria and functions as a scaffold protein for the interaction between RLRs and MAVS.296 SIDT2, the mammalian ortholog of the Caenorhabditis elegans dsRNA transporter systemic RNA interference defective protein 1 (SID-1), has been shown to enhance RLR signaling by enhancing the transport of internalized dsRNA from endosomal compartments to the cytoplasm for viral detection by RLRs.297
RLR/MAVS-mediated signaling is inhibited by multiple host factors. Soonthornvacharin et al. identified K-homology splicing regulatory protein (KHSRP) as a negative regulatory molecule for RIG-I-mediated signaling by genome-wide RNAi screening.298 KHSRP suppresses the early activation of RIG-I by directly associating with RIG-I CTD. The predicted methyltransferase TTLL12, which was identified as a host regulator that inhibits SeV-induced RIG-I-mediated signaling, binds to MAVS to suppress signaling.299 DNAJ homolog subfamily B member 1 (DNAJB1), which is a heat shock protein (HSP) 40 family member protein, was identified as an MDA5-interacting molecule.87 In response to viral infection, DNAJB1 is translocated with HSP70 to SGs, where it inhibits the formation of signal-competent MDA5/MAVS filaments. The zinc finger FYVE domain-containing protein ZFYVE1 specifically associates with MDA5 and inhibits MDA5-mediated signaling.300 FK506-binding protein 8 (FKBP8) inhibits the RLR-MAVS pathway by directly binding to MAVS.301 Eckard et al. demonstrated that SKIV2L, which is a cytosolic 3′-5′ RNA exosome, prevents the constitutive activation of RLRs via the degradation of endogenous RNAs, which are produced by inositol requiring enzyme-1 (IRE-1)-mediated cytoplasmic splicing in response to the unfolded protein response.302 They also observed that patients with human trichohepatoenteric syndrome (THES), caused by a SKIV2L loss-of-function mutation, exhibit an excessive IFN signature, indicating that the metabolic machinery of endogenous RLR substrates consists of exosomal RNAs.303 NLRX1, a mitochondrial-localized NLR family member, suppresses RIG-I-mediated signaling by interrupting RIG-I and MAVS.304,305 A subsequent study demonstrated that NLRX1 not only suppresses RIG-I-mediated signaling but also promotes autophagosome formation in association with mitochondrial Tu translation elongation factor (TUFM), leading to enhanced VSV replication.306 The involvement of autophagy and related proteins, such as Atg5-Atg12, in RLR-mediated signaling has been demonstrated (Fig. 3i).307,308 Recently, leucine-rich repeat-containing protein 25 (LRRC25) has been reported to interact with ISGylated RIG-I and to promote the degradation of RIG-I in autophagosomes to suppress IFN production.309 However, LRRC59 inhibits the degradation of ISGylated RIG-I by competing with LRRC25.310 Coiled-coil domain-containing 50 (CCDC50), a novel autophagy receptor, also induces the degradation of RIG-I/MDA5 by recruiting it as an autophagic cargo in a K63-linked ubiquitin-dependent manner to inhibit the IFN pathway.311 Immunity-related GTPase M (IRGM), in which loss-of-function mutations are known to be genetically associated with various immune diseases,312 binds to RIG-I/MAVS to promote autophagic degradation.313 Finally, a recent study revealed that caspase-3 negatively regulates this signaling pathway via the degradation of MAVS and IRF-3.314
Regulation by viral proteins
Most viruses have evolved strategies that favor their proliferation by suppressing the host innate immune system.315 In particular, the target molecules range from RLR/MAVS to downstream signaling molecules such as TRAFs, IKKs, IRFs, NF-κBs, IFNRs, JAKs, and STATs. In this section, we summarize recent findings regarding direct interactions between RLR/MAVS and pathogenic viruses (Fig. 3).
Negative-sense ssRNA viruses
RNA viruses with single-stranded, negative-sense genomes inhibit RLR/MAVS in various ways. For example, the V protein of viruses belonging to the Paramyxoviridae family suppresses IFN production by interacting with MDA5 and LGP2 but not with RIG-I.316,317,318 Moreover, the V proteins of measles virus (MV) and Nipah virus (NiV) inhibit dephosphorylation-mediated activation of MDA5 by directly associating with PP1α/γ.319 However, since paramyxoviruses have a 5′-triphosphate moiety, which is a non-self-RNA signature specific for RIG-I, the biological significance of the V-mediated repression of MDA5 is unclear. Notably, MDA5 has been suggested to be involved in SeV-induced IFN production in the late stages of infection; thus, the mechanism of action of V proteins might differ depending on the time points of the antiviral response.320 The V proteins of NiV, SeV, and parainfluenzavirus (PIV) can suppress RIG-I-mediated signaling by preventing TRIM25-mediated ubiquitin conjugation on RIG-I via complex formation with both RIG-I and TRIM25.321 However, the V proteins of several paramyxoviruses, including NDV, suppress IFN signaling by enhancing MAVS degradation by the RNF5 E3 ligase.322
Moreover, the NS1 protein of human respiratory syncytial virus (RSV), a member of the Pneumoviridae family, also interacts with TRIM25 and inhibits ubiquitin-conjugation of RIG-I.323 In addition, the NS2 protein of RSV directly binds to RIG-I CARD, which inhibits the association between activated RIG-I and MAVS.324 The VP35 protein of EBOV and Marburgvirus (MARV), which belongs to the Filoviridae family, can bind dsRNA and antagonize RLR-mediated signaling.325,326 In addition, VP35 can suppress RIG-I-mediated signaling by suppressing RIG-I-PACT interactions.277 BDV, a member of the Bornaviridae family, has a strategy to trim several nucleotides at the 5′ end of the viral genome to evade recognition by RIG-I, resulting in latent infection.147,327
Viruses belonging to the Orthomyxoviridae family, including IAV, also have negative-sense ssRNAs. The IAV NS1 protein, derived from the smallest viral genome of the eight segments that make up the IAV genome, is a well-known multifunctional virulence factor. NS1 contains an N-terminal dsRNA-binding domain and C-terminal effector domain that suppress antiviral responses through distinct actions.328 While the N-terminal dsRNA-binding domain antagonistically represses RIG-I-mediated IFN production,132,329 NS1 also inhibits RIG-I via direct interaction in an RNA-independent manner.330 NS1 also associates with TRIM25 and Riplet and represses the K63-linked ubiquitination of RIG-I.331,332 The C-terminal effector domain of NS1 interacts with several host factors, including PKR.333 IAVs that lack NS1 can efficiently activate PKR and enhance RIG-I-mediated IFN production via SG formation.73 The PB1-F2 protein is a mitochondrial-localized peptide (87 amino acids) translated from the PB1-encoding RNA segment and contributes to virulence.334 Although PB1-F2 inhibits MAVS-mediated signaling via direct interaction,335 PB1-F2 of H7N9 highly pathogenic IAVs inhibits K63-linked ubiquitination of MAVS by suppressing the TRIM31-MAVS interaction.336 PB1-F2 can also repress MAVS-mediated signaling by degrading MAVS via mitophagy.337 However, M2, a membrane protein generated from the M-encoding RNA segment, enhances MAVS-mediated signaling by antagonizing reactive oxygen species (ROS)-induced autophagy activation and increasing the number of MAVS aggregates on the mitochondria.338
Positive-sense ssRNA viruses
HCV, a member of the Flaviviridae family, encodes protease NS3/4A, which is essential for the cleavage of the viral polyprotein. NS3/4A inhibits IFN production via the degradation of MAVS.339,340 NS3/4A also disrupts RIG-I-mediated signaling by degrading Riplet.82 Since many flaviviruses are transmitted by arthropods (known as arboviruses), they share several standard features. The phosphomimetic amino acid sequences in the NS3 proteins of ZIKV, DENV, and WNV repress RLR-mediated signaling by associating with the scaffold proteins 14-3-3ε and η.295,341 The capsid proteins of DENV and WNV suppress IFN-λ gene expression via the degradation of peroxisome-localized MAVS in virus-infected cells.342 NS4A of DENV and ZIKV inhibits IFN signaling by directly interacting with MAVS.343,344 Flaviviruses produce unique subgenomic flavivirus RNA (sfRNA), a noncoding RNA derived from the 3′ UTR, which is associated with viral pathogenicity.345 A comparative analysis of the DENV viral clade revealed that an increase in sfRNA inhibits the deubiquitination of the K48-linked ubiquitin chains of TRIM25, which suppresses the RIG-I-mediated signaling induced by TRIM25.206
Viruses belonging to the Picornaviridae family terminate innate immune signaling using their protease activity, which is essential for processing viral polyproteins. Enteroviruses have two proteases, 2A and 3C. EV71, coxsackievirus 3 (CBV3), and poliovirus (PV) degrade MDA5 via their respective 2A protease.346 Moreover, MAVS can also be degraded by 2A and 3C proteases.346,347,348,349 In the case of PV infection, degradation of MDA5 by the host proteasome and caspase has been suggested.350 Furthermore, RIG-I can also be processed by enteroviral proteases; however, its biological significance is unclear.351 In addition, 3C of PV and EMCV inhibits SG formation by degrading G3BP1 (an essential regulator of SGs)352 and represses SG-mediated enhancement of RLR-mediated signaling.353 Foot-and-mouth disease virus (FMDV), an Aphthovirus genus member, encodes leader protease (Lpro), a unique protein that degrades LGP2 and indirectly suppresses MDA5-mediated signaling.354 A recent study indicated that Lpro inhibits signaling by degrading MAVS and TBK1.355 However, Lpro has been shown to deubiquitinate RIG-I, thereby terminating RIG-I-mediated signaling.356 Furthermore, the 2B protein of FMDV specifically inhibits RIG-I;357 however, it remains unclear why these picornavirus proteins target RIG-I.
Viruses belonging to the Bunyavirales order replicate their genomic RNA using a “prime-and-realign” machinery.358 The genomic RNA of CCHFV (Nairoviridae) and HTNV (Hantaviridae) avoids RIG-I recognition upon the removal of the triphosphate moiety at the 5′ end of the single G overhang and generating a 5′-monophosphate by “prime-and-realign” replication.147 However, viruses belonging to the Arenaviridae family have been reported to escape RIG-I detection precisely because of its 5′ overhang structure, even though the 5′-triphosphate G is not removed.359 Moreover, the Z protein of Arenaviridae viruses such as Junin virus (JUNV) suppresses the IFN response via direct interaction with RIG-I.360 This suppression mechanism is common among pathogenic arenaviruses, suggesting that it is an essential factor for their pathogenicity. However, the NS protein of severe fever with thrombocytopenia syndrome virus (SFTSV), a family member of the Phenuiviridae family, associates with RIG-I and TRIM25 and suppresses RIG-I-mediated signaling by repressing the K63-linked ubiquitination of RIG-I.361,362 Although inhibition of RIG-I by its direction interaction with the NS protein of Toscana virus (TOSV) has been suggested,363 a recent report revealed that NS can function as an E3 ubiquitin ligase and degrade the RIG-I protein to terminate signaling.364
Finally, we describe recent findings on the interaction between Coronaviridae family members and RLR/MAVS. Pathogenic human coronaviruses, including SARS-CoV, SARS-CoV-2, and MERS-CoV, are classified as betacoronaviruses. Since the production of IFNs is significantly attenuated in cells infected with these viruses,365,366,367 suppression of the RLR/MAVS pathway by viral proteins might be a critical therapeutic target for coronaviral infectious diseases, including COVID-19. ORF9 of SARS-CoV, which encodes the N protein, interacts with TRIM25 and inhibits RIG-I via the ubiquitination of RIG-I.368 SARS-CoV ORF-9b, a short peptide with 98 amino acids translated based on a discordant ORF in the N gene, localizes to the mitochondria, altering their morphology by inhibiting mitochondrial fission and thereby promoting the PCBP2-mediated degradation of MAVS by the E3 ubiquitin ligase ITCH/AIP4.369 In addition, MERS-CoV is closely related to SARS-CoVs; however, the genomic structures differ slightly. Through its dsRNA-binding activity, the unique 4a protein of MERS-CoV represses MDA5 signaling.370 However, 4a also associates with PACT to suppress IFN production independent of RIG-I/MDA5.280 Since 4a can act antagonistically on PKR,371 it might be involved in PKR/PACT-mediated SG formation to regulate IFN production. Currently, reports demonstrating the suppression of RLR-mediated signaling by SARS-CoV-2-encoded proteins are accumulating. Recent studies revealed that ORF-9b of SARS-CoV-2 represses IFN induction via an interaction with the mitochondrial protein TOM70, which is a critical regulator of MAVS-mediated antiviral signaling.372 ORF-3b, a 22-amino acid short peptide encoded by SARS-CoV-2, was identified as a potent antagonist of IFN production; however, the molecular machinery associated with its antagonistic function remains unclear.373 Very recently, it was demonstrated that membrane protein (M), encoded by ORF5 of SARS-CoV-2, functions as an IFN antagonist via direct interactions with RLRs and MAVS to prevent RLR/MAVS/TRAF3/TBK1-containing signal complex formation.374,375 Furthermore, multiple viral factors have been reported to inhibit RLR/MAVS and its downstream signaling pathway.376,377,378 Further analysis of these interactions might reveal effective therapeutic targets for COVID-19 treatment.
Concluding remarks
The world is currently battling a significant health crisis in the form of the COVID-19 pandemic caused by SARS-CoV-2. In addition, infectious diseases caused by pathogenic RNA viruses such as EBOV, avian IAV, and other unidentified viruses are expected to occur in the future. The RLR/MAVS-mediated signaling pathway plays an essential role in the antiviral innate immune response against these viruses. However, RLR/MAVS pathway activation can also cause autoimmune diseases and trigger cytokine storms, as observed in COVID-19 patients, indicating that this pathway functions as a double-edged sword in the human immune system. Moreover, as the regulation of this pathway is significantly affected by the interaction between individual viruses and host factors, elucidating the detailed regulatory mechanisms underlying each viral infection from a spatiotemporal perspective and understanding them in an integrated manner are crucial. An improved understanding of the regulatory mechanisms associated with this pathway can inform the development of novel therapeutic strategies against infectious diseases, cancer, and immunological disorders in the future.
References
Nagano, Y. & Kojima, Y. [Immunizing property of vaccinia virus inactivated by ultraviolets rays]. Comptes Rendus Des. Séances De. La Soci.été De. Biologie Et. De. Ses. Fil. 148, 1700–1702 (1954).
Isaacs, A. & Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B - Biol. Sci. 147, 258–267 (1957).
Mesev, E. V., LeDesma, R. A. & Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 4, 914–924 (2019).
Schneider, W. M., Chevillotte, M. D. & Rice, C. M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev. Immunol. 32, 513–545 (2014).
Ye, L., Schnepf, D. & Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 19, 614–625 (2019).
Parker, B. S., Rautela, J. & Hertzog, P. J. Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144 (2016).
Park, A., Iwasaki, A. Type I and Type III Interferons – Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 27, 870–878 (2020).
Channappanavar, R. et al. Dysregulated Type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19, 181–193 (2016).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Zhou, Z. et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 1–16 (2020).
Lucas, C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020).
Broggi, A. et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 369, 706–712 (2020).
Major, J. et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369, 712–717 (2020).
Ablasser, A. & Hur, S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 21, 1–13 (2019).
Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004).
Yoneyama, M. et al. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J. Immunol. 175, 2851–2858 (2005).
Yoneyama, M., Onomoto, K., Jogi, M., Akaboshi, T. & Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 32, 48–53 (2015).
Rehwinkel, J. & Gack, M. U. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 147, 1–15 (2020).
Kell, A. M. & Gale, M. RIG-I in RNA virus recognition. Virology 479–480, 110–121 (2015).
Kato, H. et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28 (2005).
Wang, Y. et al. Rig-I−/− mice develop colitis associated with downregulation of G alpha i2. Cell Res. 17, 858–868 (2007).
Liu, L. et al. Commensal viruses maintain intestinal intraepithelial lymphocytes via noncanonical RIG-I signaling. Nat. Immunol. 20, 1–17 (2019).
Zhu, H. et al. RNA virus receptor Rig-I monitors gut microbiota and inhibits colitis-associated colorectal cancer. J. Exp. Clin. Cancer Res. 36, 2 (2017).
Kato, H. et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205, 1601–1610 (2008).
Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006).
Yao, H. et al. ATP-Dependent Effector-like Functions of RIG-I-like Receptors. Mol. Cell 58, 541–548 (2015).
Sato, S. et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 (2015).
Weber, M. et al. Influenza Virus Adaptation PB2-627K Modulates Nucleocapsid Inhibition by the Pathogen Sensor RIG-I. Cell Host Microbe 17, 1–27 (2015).
Rothenfusser, S. et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175, 5260–5268 (2005).
Komuro, A. & Horvath, C. M. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 80, 12332–12342 (2006).
Venkataraman, T. et al. Loss of DExD/H Box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 178, 6444–6455 (2007).
Satoh, T. et al. From the Cover: LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl Acad. Sci. USA 107, 1512–1517 (2010).
Veen, A. G. Vder et al. The RIG‐I‐like receptor LGP2 inhibits Dicer‐dependent processing of long double‐stranded RNA and blocks RNA interference in mammalian cells. EMBO J. 37, e97479–14 (2018).
Takahashi, T. et al. LGP2 virus sensor regulates gene expression network mediated by TRBP-bound microRNAs. Nucleic Acids Res. 42, D68–14 (2018).
Takahashi, T., Nakano, Y., Onomoto, K., Yoneyama, M. & Ui-Tei, K. LGP2 virus sensor enhances apoptosis by upregulating apoptosis regulatory genes through TRBP-bound miRNAs during viral infection. Nucleic Acids Res. 48, 1494–1507 (2019).
Suthar, M. S. et al. The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity 37, 235–248 (2012).
Kumar, H. et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 203, 1795–1803 (2006).
Lupfer, C., Malik, A. & Kanneganti, T.-D. Inflammasome control of viral infection. Curr. Opin. Virol. 12, 38–46 (2015).
Poeck, H. et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 11, 63–69 (2009).
Pothlichet, J. et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 9, e1003256 (2013).
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z. & Germain, R. N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013).
Park, S. et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366 (2013).
Franchi, L. et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ Efflux. J. Immunol. 193, 4214–4222 (2014).
Kowalinski, E. et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by Viral RNA. Cell 147, 423–435 (2011).
Takahasi, K. et al. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29, 428–440 (2008).
Cui, S. et al. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol. Cell 29, 169–179 (2008).
Luo, D. et al. Structural insights into RNA recognition by RIG-I. Cell 147, 409–422 (2011).
Jiang, F. et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 (2011).
Wu, B. et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152, 276–289 (2013).
Myong, S. et al. Cytosolic viral sensor RIG-I is a 5’-triphosphate-dependent translocase on double-stranded RNA. Science 323, 1070–1074 (2009).
Peisley, A., Wu, B., Yao, H., Walz, T. & Hur, S. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 51, 573–583 (2013).
Patel, J. R. et al. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep. 14, 780–787 (2013).
Peisley, A., Wu, B., Xu, H., Chen, Z. J. & Hur, S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509, 110–114 (2014).
Wies, E. et al. Dephosphorylation of the RNA Sensors RIG-I and MDA5 by the Phosphatase PP1 Is Essential for Innate Immune Signaling. Immunity 38, 437–449 (2013).
Takahasi, K. et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 284, 17465–17474 (2009).
Berke, I. C. & Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 31, 1714–1726 (2012).
Peisley, A. et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl Acad. Sci. USA 108, 21010–21015 (2011).
Peisley, A. et al. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc. Natl Acad. Sci. USA 109, E3340–E3349 (2012).
Li, X. et al. The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J. Biol. Chem. 284, 13881–13891 (2009).
Pippig, D. A. et al. The regulatory domain of the RIG-I family ATPase LGP2 senses double-stranded RNA. Nucleic Acids Res. 37, 2014–2025 (2009).
Uchikawa, E. et al. Structural analysis of dsRNA binding to anti-viral pattern recognition receptors LGP2 and MDA5. Mol. Cell 62, 586–602 (2016).
Bruns, A. M. et al. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 55, 771–781 (2014).
Qi, N. et al. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat. Commun. 8, 1–16 (2017).
Takahasi, K. et al. Identification of a new autoinhibitory domain of interferon-beta promoter stimulator-1 (IPS-1) for the tight regulation of oligomerization-driven signal activation. Biochem. Biophys. Res. Commun. 517, 662–669 (2019).
Hou, F. et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461 (2011).
Xu, H. et al. Structural basis for the prion-like MAVS filaments in antiviral innate immunity. Elife 3, e01489 (2014).
Wu, B. et al. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 55, 511–523 (2014).
Horner, S. M. et al. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl Acad. Sci. USA 108, 14590–14595 (2011).
Dixit, E. et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681 (2010).
Odendall, C. et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 15, 717–726 (2014).
Esser-Nobis, K., Hatfield, L. D. & Gale, M. Spatiotemporal dynamics of innate immune signaling via RIG-I-like receptors. Proc. Natl Acad. Sci. USA 117, 15778–15788 (2020).
Liu, G. et al. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 9, 343–344 (2018).
Onomoto, K. et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 7, 1–21 (2012).
Panas, M. D., Ivanov, P. & Anderson, P. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 215, 313–323 (2016).
Yoo, J.-S. et al. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog. 10, e1004012 (2014).
Gaete-Argel, A., Márquez, C. L., Barriga, G. P., Soto-Rifo, R. & Valiente-Echeverría, F. Strategies for success. Viral infections and membraneless organelles. Front Cell Infect. Microbiol 9, 336 (2019).
Zhang, Q., Sharma, N. R., Zheng, Z.-M. & Chen, M. Viral regulation of RNA granules in infected cells. Virol. Sin. 34, 175–191 (2019).
Liu, Y. et al. The role of host eIF2α in viral infection. Virol. J. 17, 112 (2020).
Onoguchi, K. et al. Virus-Infection or 5′ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 6, e1001012 (2010).
Oh, S.-W. et al. Leader-containing uncapped viral transcript activates RIG-I in antiviral stress granules. PLoS Pathog. 12, e1005444–22 (2016).
Okonski, K. M. & Samuel, C. E. Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J. Virol. 87, 756–766 (2012).
Oshiumi, H., Miyashita, M., Matsumoto, M. & Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 9, e1003533 (2013).
Kuniyoshi, K. et al. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl Acad. Sci. USA 111, 5646–5651 (2014).
Narita, R. et al. A novel function of human pumilio proteins in cytoplasmic sensing of viral infection. PLoS Pathog. 10, e1004417 (2014).
Zhang, P. et al. IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J. Cell Sci. 127, 2471–2482 (2014).
Nikolic, J., Civas, A., Lama, Z., Lagaudrière-Gesbert, C. & Blondel, D. Rabies virus infection induces the formation of stress granules closely connected to the viral factories. PLoS Pathog. 12, e1005942 (2016).
Takashima, K., Oshiumi, H., Matsumoto, M. & Seya, T. DNAJB1/HSP40 suppresses melanoma differentiation-associated gene 5-mitochondrial antiviral signaling protein function in conjunction with HSP70. J. Innate Immun. 10, 44–55 (2017).
Kim, S. S.-Y., Sze, L. & Lam, K.-P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-β response. J. Biol. Chem. 294, 6430–6438 (2019).
Yang, W. et al. G3BP1 inhibits RNA virus replication by positively regulating RIG-I-mediated cellular antiviral response. Cell Death Dis. 10, 946 (2019).
Yang, W. et al. Foot-and-mouth disease virus 3A protein causes upregulating of autophagy-related protein LRRC25 to inhibit the G3BP1-mediated RLH signaling pathway. J. Virol. 94, e02086-19 (2020).
Schulz, O. et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7, 354–361 (2010).
Sen, A., Pruijssers, A. J., Dermody, T. S., Garcia-Sastre, A. & Greenberg, H. B. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 85, 3717–3732 (2011).
Langereis, M. A., Feng, Q. & van Kuppeveld, F. J. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J. Virol. 87, 6314–6325 (2013).
Beauclair, G. et al. Retinoic Acid Inducible Gene I and Protein Kinase R, but Not Stress Granules, Mediate the Proinflammatory Response to Yellow Fever Virus. J. Virol. 94, e00403–20 (2020).
Pène, V., Li, Q., Sodroski, C., Hsu, C.-S. & Liang, T. J. Dynamic interaction of stress granules, DDX3X, and IKK-α mediates multiple functions in hepatitis C virus infection. J. Virol. 89, 5462–5477 (2015).
McCormick, C. & Khaperskyy, D. A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 17, 1–14 (2017).
Yoo, H., Triandafillou, C. & Drummond, D. A. Cellular sensing by phase separation: Using the process, not just the products. J. Biol. Chem. 294, 7151–7159 (2019).
Alberti, S., Gladfelter, A. & Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434 (2019).
Crowl, J. T., Gray, E. E., Pestal, K., Volkman, H. E. & Stetson, D. B. Intracellular nucleic acid detection in autoimmunity. Annu Rev. Immunol. 35, 313–336 (2017).
Kato, H., Oh, S.-W. & Fujita, T. RIG-I-like receptors and type I interferonopathies. J. Interferon Cytokine Res. 37, 207–213 (2017).
Junior, A. G. D., Sampaio, N. G. & Rehwinkel, J. A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 27, 75–85 (2019).
Smyth, D. J. et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 38, 617–619 (2006).
Sutherland, A. et al. Genomic Polymorphism at the Interferon-Induced Helicase (IFIH1) Locus Contributes to Graves’ Disease Susceptibility. J. Clin. Endocrinol. Metab. 92, 3338–3341 (2007).
Martinez, A. et al. Association of the IFIH1-GCA-KCNH7 chromosomal region with rheumatoid arthritis. Ann. Rheum. Dis. 67, 137–138 (2008).
Enevold, C. et al. Multiple sclerosis and polymorphisms of innate pattern recognition receptors TLR1-10, NOD1-2, DDX58, and IFIH1. J. Neuroimmunol. 212, 125–131 (2009).
Gateva, V. et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233 (2009).
Graham, D. S. C. et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with Systemic Lupus Erythematosus. PLoS Genet. 7, e1002341 (2011).
Robinson, T. et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J. Immunol. 187, 1298–1303 (2011).
Ferreira, R. C. et al. Association of IFIH1 and other autoimmunity risk alleles with selective IgA deficiency. Nat. Genet. 42, 777–780 (2010).
Jin, Y. et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 44, 676–680 (2012).
Jin, Y., Andersen, G. H. L., Santorico, S. A. & Spritz, R. A. Multiple Functional Variants of IFIH1, a Gene Involved in Triggering Innate Immune Responses, Protect against Vitiligo. J. Invest. Dermatol 137, 522–524 (2017).
Funabiki, M. et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40, 199–212 (2014).
Gorman, J. A. et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat. Immunol. 18, 744–752 (2017).
Liu, S. et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum. Mol. Genet. 18, 358–365 (2009).
Nejentsev, S., Walker, N., Riches, D., Egholm, M. & Todd, J. A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324, 387–389 (2009).
Oda, H. et al. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet. 95, 121–125 (2014).
Rice, G. I. et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet 44, 1243–1248 (2012).
Rutsch, F. et al. A Specific IFIH1 gain-of-function mutation causes singleton-merten syndrome. Am. J. Hum. Genet. 96, 275–282 (2015).
Lamborn, I. T. et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 214, 1949–1972 (2017).
Jang, M.-A. et al. REPOR TMutations in DDX58, which Encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 96, 266–274 (2015).
Ferreira, C. R. et al. DDX58 and classic singleton-merten syndrome. J. Clin. Immunol. 39, 75–80 (2018).
Lässig, C. et al. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. Elife 4, e10859 (2015).
Lässig, C. et al. Unified mechanisms for self-RNA recognition by RIG-I Singleton-Merten syndrome variants. Elife 7, e38958 (2018).
Pothlichet, J. et al. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol. Med. 3, 142–152 (2011).
Saito, T., Owen, D. M., Jiang, F., Marcotrigiano, J. & Gale, M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527 (2008).
Fredericksen, B. L. & Gale, M. West nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 80, 2913–2923 (2006).
Chazal, M. et al. RIG-I recognizes the 5′ region of dengue and zika virus genomes. Cell Rep. 24, 320–328 (2018).
Zhang, Z. et al. Hepatitis D virus replication is sensed by MDA5 and induces IFN-β/λ responses in hepatocytes. J. Hepatol. 69, 25–35 (2018).
Sooryanarain, H., Heffron, C. L. & Meng, X.-J. The U-Rich untranslated region of the Hepatitis E virus induces differential type I and type III interferon responses in a host cell-dependent manner. mBio 11, e03103–19 (2020).
Kell, A. M., Hemann, E. A., Turnbull, J. B. & Gale, M. RIG-I-like receptor activation drives type I IFN and antiviral signaling to limit Hantaan orthohantavirus replication. PLoS Pathog. 16, e1008483 (2020).
Hornung, V. et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314, 997–1001 (2006).
Schlee, M. et al. Recognition of 5’ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34 (2009).
Schmidt, A. et al. 5’-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl Acad. Sci. USA 106, 12067–12072 (2009).
Rehwinkel, J. et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140, 397–408 (2010).
Weber, M. et al. Incoming RNA virus nucleocapsids containing a 5’-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 13, 336–346 (2013).
Liu, G., Park, H.-S., Pyo, H.-M., Liu, Q. & Zhou, Y. Influenza A virus panhandle structure is directly involved in RIG-I activation and interferon induction. J. Virol. 89, 6067–6079 (2015).
Arranz, R. et al. The Structure of Native Influenza Virion Ribonucleoproteins. Science 338, 1634–1637 (2012).
Velthuis, A. J. Wte et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 3, 1234–1242 (2018).
Liu, G., Lu, Y., Liu, Q. & Zhou, Y. Inhibition of ongoing influenza A virus replication reveals different mechanisms of RIG-I activation. J Virol. 93, e02066–18 (2019).
Martinez-Gil, L. et al. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J. Virol. 87, 1290–1300 (2013).
Malathi, K. et al. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 16, 2108–2119 (2010).
Goubau, D. et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature 514, 372–375 (2014).
Ren, X., Linehan, M. M., Iwasaki, A. & Pyle, A. M. RIG-I selectively discriminates against 5′-monophosphate RNA. Cell Rep. 26, 2019–2027 (2019).
Schuberth-Wagner, C. et al. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1-2’O-methylated Self RNA. Immunity 43, 41–51 (2015).
Nomoto, A., Detjen, B., Pozzatti, R. & Wimmer, E. The location of the polio genome protein in viral RNAs and its implication for RNA synthesis. Nature 268, 208–213 (1977).
Habjan, M. et al. Processing of genome 5’ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 3, e2032 (2008).
Lu, M. et al. N6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 5, 584–598 (2020).
Abdullah, Z. et al. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J. 31, 4153–4164 (2012).
Hagmann, C. A. et al. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS ONE 8, e62872 (2013).
Schmolke, M. et al. RIG-I detects mRNA of intracellular Salmonella enterica serovar Typhimurium during bacterial infection. mBio 5, e01006–e01014 (2014).
Cheng, Y. & Schorey, J. S. Mycobacterium tuberculosis–induced IFN-β production requires cytosolic DNA and RNA sensing pathways. J. Exp. Med. 215, 2919–2935 (2018).
Pagliuso, A. et al. An RNA-binding protein secreted by a bacterial pathogen modulates RIG-I signaling. Cell Host Microbe 26, 823–834 (2019).
Cheng, Y. & Schorey, J. S. Extracellular vesicles deliver Mycobacterium RNA to promote host immunity and bacterial killing. EMBO Rep. 20, e46613 (2019).
Bradley, K. C. et al. Microbiota-Driven Tonic Interferon Signals in Lung Stromal Cells Protect from Influenza Virus Infection. Cell Rep. 28, 245–256 (2019).
Chiu, Y.-H., Macmillan, J. B. & Chen, Z. J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009).
Ablasser, A. et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072 (2009).
Chiang, J. J. et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 19, 1–13 (2017).
Zhao, Y., Ye, X., Dunker, W., Song, Y. & Karijolich, J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun. 9, 4841 (2018).
Zhang, Y. et al. RIG-I detects Kaposi’s sarcoma-associated herpesvirus transcripts in a RNA polymerase III-independent manner. mBio 9, e00823–18 (2018).
Nabet, B. Y. et al. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in. Cancer Cell 170, 352–366 (2017).
Wu, Y. et al. Function of HNRNPC in breast cancer cells by controlling the dsRNA‐induced interferon response. EMBO J. 37, e99017 (2018)
Jiang, M. et al. Self-Recognition of an Inducible Host lncRNA by RIG-I Feedback Restricts Innate Immune Response. Cell 173, 906–912 (2018).
Fan, J., Cheng, M., Chi, X., Liu, X. & Yang, W. A human long non-coding RNA LncATV Promotes Virus Replication Through Restricting RIG-I–Mediated Innate Immunity. Front Immunol. 10, 1711 (2019).
Karlsen, T. A. & Brinchmann, J. E. Liposome delivery of microRNA-145 to mesenchymal stem cells leads to immunological off-target effects mediated by RIG-I. Mol. Ther. 21, 1169–1181 (2013).
Zhao, L. et al. Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Sci. Rep. 5, 14991 (2015).
Chen, Y. G. et al. Sensing self and foreign circular RNAs by Intron Identity. Mol. Cell 67, 228–238 (2017).
Chen, Y. G. et al. N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96–109 (2019).
Wesselhoeft, R. A. et al. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell 74, 508–520 (2019).
Chiang, C. et al. Sequence-specific modifications enhance the broad-spectrum antiviral response activated by RIG-I agonists. J. Virol. 89, 8011–8025 (2015).
Jiang, X. et al. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J. Exp. Med. 216, 2854–2868 (2019).
Rawling, D. C., Jagdmann, G. E., Potapova, O. & Pyle, A. M. Small-molecule antagonists of the RIG-I innate immune receptor. Acs Chem. Biol. 15, 311–317 (2020).
Loo, Y.-M. et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82, 335–345 (2008).
Mccartney, S. A. et al. MDA-5 recognition of a murine norovirus. PLoS Pathog. 4, e1000108 (2008).
Roth-Cross, J. K., Bender, S. J. & Weiss, S. R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 82, 9829–9838 (2008).
Triantafilou, K. et al. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci. 125, 4761–4769 (2012).
Runge, S. et al. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 10, e1004081 (2014).
Deddouche, S. et al. Identification of an LGP2-associated MDA5 agonist in picornavirus-infected cells. Elife 3, e01535–20 (2014).
David, R. Y. S. et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. Elife 5, e11275 (2016).
Züst, R. et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12, 137–143 (2011).
Hackbart, M., Deng, X. & Baker, S. C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl Acad. Sci. USA 117, 8094–8103 (2020).
Liehl, P. et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat. Med. 20, 47–53 (2014).
Wu, J. et al. Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc. Natl Acad. Sci. USA 111, E511–E520 (2014).
Lamers, M. M., Hoogen, B. G. van den & Haagmans, B. L. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol. 10, 1763 (2019).
Ahmad, S. et al. Breaching self-tolerance to alu duplex RNA underlies MDA5-Mediated Inflammation. Cell 172, 797–802 (2018).
Hartner, J. C., Walkley, C. R., Lu, J. & Orkin, S. H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 10, 109–115 (2008).
Mannion, N. M. et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494 (2014).
Pestal, K. et al. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944 (2015).
Liddicoat, B. J. et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015).
Chung, H. et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824 (2018).
Roulois, D. et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015).
Chiappinelli, K. B. et al. Inhibiting DNA methylation causes an interferon response in cancer via dsrna including endogenous retroviruses. Cell 162, 974–986 (2015).
Cañadas, I. et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med. 24, 1143–1150 (2018).
Tie, C. H. et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 19, e45000 (2018).
Dhir, A. et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 1–24 (2018).
Xie, Q. et al. Long Noncoding RNA ITPRIP-1 Positively Regulates the Innate Immune Response through Promotion of Oligomerization and Activation of MDA5. J. Virol. 92, e00507–e00518 (2018).
Gack, M. U. et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 (2007).
Zeng, W. et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141, 315–330 (2010).
Liu, H. M. et al. The mitochondrial targeting chaperone 14-3-3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 11, 528–537 (2012).
Lian, H. et al. The zinc-finger protein ZCCHC3 binds RNA and facilitates viral RNA sensing and activation of the RIG-I-like receptors. Immunity 49, 438–448 (2018).
Wang, P. et al. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat. Immunol. 11, 912–919 (2010).
Liu, Z. et al. NDR2 promotes the antiviral immune response via facilitating TRIM25-mediated RIG-I activation in macrophages. Sci. Adv. 5, eaav0163 (2019).
Inn, K.-S. et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 41, 354–365 (2011).
Pauli, E.-K. et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal 7, ra3–ra3 (2014).
Chen, S.-T. et al. NLRP12 regulates anti-viral RIG-I activation via interaction with TRIM25. Cell Host Microbe 25, 602–616 (2019).
Manokaran, G. et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221 (2015).
Choudhury, N. R. et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 15, 105 (2017).
Sanchez, J. G. et al. TRIM25 binds RNA to modulate cellular anti-viral defense. J. Mol. Biol. 430, 5280–5293 (2018).
Lin, H. et al. The long noncoding RNA Lnczc3h7a promotes a TRIM25-mediated RIG-I antiviral innate immune response. Nat. Immunol. 20, 812–823 (2019)
Oshiumi, H., Matsumoto, M., Hatakeyama, S. & Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-β induction during the early phase of viral infection. J. Biol. Chem. 284, 807–817 (2008).
Gao, D. et al. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE 4, e5760 (2009).
Oshiumi, H. et al. The ubiquitin ligase riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8, 496–509 (2010).
Shi, Y. et al. Ube2D3 and Ube2N are essential for RIG-I-mediated MAVS aggregation in antiviral innate immunity. Nat. Commun. 8, 1–14 (2017).
Cadena, C. et al. Ubiquitin-dependent and -independent roles of E3 Ligase RIPLET in innate immunity. Cell 177, 1–31 (2019).
Hayman, T. J. et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 97, 840–852 (2019).
Wang, G., Kouwaki, T., Okamoto, M. & Oshiumi, H. Attenuation of the innate immune response against viral infection due to ZNF598-promoted binding of FAT10 to RIG-I. Cell Rep. 28, 1961–1970 (2019).
Yan, J., Li, Q., Mao, A.-P., Hu, M.-M. & Shu, H.-B. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol. 6, 154–163 (2014).
Hage, A. & Rajsbaum, R. To TRIM or not to TRIM: the balance of host–virus interactions mediated by the ubiquitin system. J. Gen. Virol. 100, 1641–1662 (2019).
Oshiumi, H. Recent advances and contradictions in the study of the individual roles of ubiquitin ligases that regulate RIG-I-like receptor-mediated antiviral innate immune responses. Front Immunol. 11, 1296 (2020).
Zhao, C. et al. The E3 ubiquitin ligase TRIM40 attenuates antiviral immune responses by targeting MDA5 and RIG-I. Cell Rep. 21, 1613–1623 (2017).
Arimoto, K. et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl Acad. Sci. USA 104, 7500–7505 (2007).
Wang, W. et al. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl Acad. Sci. USA 113, 9581–9586 (2016).
Zhou, P. et al. MLL5 suppresses antiviral innate immune response by facilitating STUB1-mediated RIG-I degradation. Nat. Commun. 9, 1243 (2018).
Chen, W. et al. Induction of Siglec-G by RNA Viruses Inhibits the Innate Immune Response by Promoting RIG-I Degradation. Cell 152, 467–478 (2013).
Wang, L. et al. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J. Virol. 87, 4507–4515 (2013).
Jahan, A. S. et al. OTUB1 is a key regulator of RIG-I-dependent immune signaling and is targeted for proteasomal degradation by Influenza A NS1. Cell Rep. 30, 1570–1584 (2020).
Friedman, C. S. et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9, 930–936 (2008).
Lin, W. et al. Syndecan-4 negatively regulates antiviral signalling by mediating RIG-I deubiquitination via CYLD. Nat. Commun. 7, 1–15 (2016).
Cui, J. et al. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 24, 400–416 (2014).
Fan, Y. et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 211, 313–328 (2014).
Li, H. et al. USP14 promotes K63-linked RIG-I deubiquitination and suppresses antiviral immune responses. Eur. J. Immunol. 49, 42–53 (2018).
Tao, X., Chu, B., Xin, D., Li, L. & Sun, Q. USP27X negatively regulates antiviral signaling by deubiquitinating RIG-I. Plos Pathog. 16, e1008293 (2020).
Jiang, X. et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36, 959–973 (2012).
Lang, X. et al. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 214, 459–473 (2016).
Meng, J. et al. ARRDC4 regulates enterovirus 71-induced innate immune response by promoting K63 polyubiquitination of MDA5 through TRIM65. Cell Death Dis. 8, e2866–e2866 (2017).
Liu, B. et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 18, 214–224 (2016).
Dai, T. et al. FAF1 regulates antiviral immunity by inhibiting MAVS but is antagonized by phosphorylation upon viral infection. Cell Host Microbe 24, 776–790 (2018).
Liuyu, T. et al. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res. 29, 67–79 (2018).
Castanier, C. et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 10, 44 (2012).
You, F. et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 10, 1300–1308 (2009).
Zhou, X., You, F., Chen, H. & Jiang, Z. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS). Cell Res. 22, 717–727 (2012).
Choi, Y. B., Shembade, N., Parvatiyar, K., Balachandran, S. & Harhaj, E. W. TAX1BP1 restrains virus-induced apoptosis by facilitating Itch-mediated degradation of the mitochondrial adaptor MAVS. Mol. Cell Biol. 37, e00422–16 (2016).
Luo, W.-W. et al. iRhom2 is essential for innate immunity to RNA virus by antagonizing ER- and mitochondria-associated degradation of VISA. PLoS Pathog. 13, e1006693 (2017).
Xing, J., Zhang, A., Minze, L., Li, X. & Zhang, Z. TRIM29 Negatively Regulates the Type I IFN Production in Response to RNA Virus. J. Immunol. 201, 183–192 (2018).
Jin, S. et al. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol. Cell 68, 308–322 (2017).
He, X. et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 38, e100978 (2019).
Liu, C. et al. The otubain YOD1 suppresses aggregation and activation of the signaling adaptor MAVS through Lys63-linked deubiquitination. J. Immunol. 202, 2957–2970 (2019).
Zhang, Z. et al. Acetylation-dependent deubiquitinase OTUD3 controls MAVS activation in innate antiviral immunity. Mol. Cell 79, 304–319 (2020).
Kim, M.-J., Hwang, S.-Y., Imaizumi, T. & Yoo, J.-Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 82, 1474–1483 (2008).
Liu, G. et al. ISG15-dependent activation of the RNA sensor MDA5 and its antagonism by the SARS-CoV-2 papain-like protease. Preprint at https://www.biorxiv.org/content/10.1101/2020.10.26.356048v1 (2020).
Hu, M.-M., Liao, C.-Y., Yang, Q., Xie, X.-Q. & Shu, H.-B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J. Exp. Med. 214, 973–989 (2017).
Nguyen, N. T. H., Now, H., Kim, W.-J., Kim, N. & Yoo, J.-Y. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci. Rep. 6, 23377 (2016).
Gack, M. U., Nistal-Villán, E., Inn, K.-S., García-Sastre, A. & Jung, J. U. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J. Virol. 84, 3220–3229 (2010).
Nistal-Villán, E. et al. Negative role of RIG-I Serine 8 phosphorylation in the regulation of interferon-β production. J. Biol. Chem. 285, 20252–20261 (2010).
Maharaj, N. P., Wies, E., Stoll, A. & Gack, M. U. Conventional protein kinase C-α (PKC-α) and PKC-β negatively regulate RIG-I antiviral signal transduction. J. Virol. 86, 1358–1371 (2012).
Sun, Z., Ren, H., Liu, Y., Teeling, J. L. & Gu, J. Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J. Virol. 85, 1036–1047 (2011).
Willemsen, J. et al. Phosphorylation-dependent feedback inhibition of RIG-I by DAPK1 Identified by kinome-wide siRNA screening. Mol. Cell 65, 403–415.e8 (2017).
Takashima, K., Oshiumi, H., Takaki, H., Matsumoto, M. & Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 11, 192–200 (2015).
Yan, B.-R. et al. PKACs attenuate innate antiviral response by phosphorylating VISA and priming it for MARCH5-mediated degradation. PLoS Pathog. 13, e1006648 (2017).
Song, T. et al. c-Abl tyrosine kinase interacts with MAVS and regulates innate immune response. FEBS Lett. 584, 33–38 (2009).
Cheng, J. et al. Autophagy regulates MAVS signaling activation in a phosphorylation-dependent manner in microglia. Cell Death Differ. 24, 276–287 (2016).
Choi, S. J. et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 35, 429–442 (2016).
Liu, H. M. et al. Regulation of retinoic acid inducible gene-I (RIG-I) activation by the histone deacetylase 6. Ebiomedicine 9, 195–206 (2016).
Li, T. et al. O-GlcNAc transferase links glucose metabolism to MAVS-mediated antiviral innate immunity. Cell Host Microbe 24, 791–803 (2018).
Song, N. et al. MAVS O-GlcNAcylation is essential for host antiviral immunity against lethal RNA viruses. Cell Rep. 28, 2386–2396 (2019).
Kristiansen, H., Gad, H. H., Eskildsen-Larsen, S., Despres, P. & Hartmann, R. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. 31, 41–47 (2011).
Zhu, J. et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40, 936–948 (2014).
Williams, B. R. PKR; a sentinel kinase for cellular stress. Oncogene 18, 6112–6120 (1999).
Onomoto, K., Yoneyama, M., Fung, G., Kato, H. & Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 35, 420–428 (2014).
Yoneyama, M., Jogi, M. & Onomoto, K. Regulation of antiviral innate immune signaling by stress-induced RNA granules. J. Biochem. 159, 279–286 (2016).
Heyam, A., Lagos, D. & Plevin, M. Dissecting the roles of TRBP and PACT in double-stranded RNA recognition and processing of noncoding RNAs. Wiley Interdiscip. Rev. RNA 6, 271–289 (2015).
Iwamura, T. et al. PACT, a double-stranded RNA binding protein acts as a positive regulator for Type I Interferon gene induced by newcastle disease virus. Biochem. Biophys. Res. Commun. 282, 515–523 (2001).
Kok, K. H. et al. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9, 299–309 (2011).
Lui, P.-Y. et al. PACT facilitates RNA-induced activation of MDA5 by promoting MDA5 oligomerization. J. Immunol. 199, 1846–1855 (2017).
Miyamoto, M. & Komuro, A. PACT is required for MDA5-mediated immunoresponses triggered by Cardiovirus infection via interaction with LGP2. Biochem Biophys. Res Commun. 494, 227–233 (2017).
David, R. Y. S. et al. LGP2 binds to PACT to regulate RIG-I- and MDA5-mediated antiviral responses. Sci. Signal 12, eaar3993 (2019).
Luthra, P. et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14, 74–84 (2013).
Tawaratsumida, K. et al. Quantitative proteomic analysis of the influenza A virus nonstructural proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J. Virol. 88, 9038–9048 (2014).
Shao, J. et al. Arenaviral Nucleoproteins Suppress PACT-Induced Augmentation of RIG-I Function To Inhibit Type I Interferon Production. J. Virol. 92, e00482–18 (2018).
Siu, K.-L. et al. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol. 88, 4866–4876 (2014).
Ding, Z. et al. The nucleocapsid proteins of mouse hepatitis virus and severe acute respiratory syndrome coronavirus share the same IFN-β antagonizing mechanism: attenuation of PACT-mediated RIG-I/MDA5 activation. Oncotarget 8, 49655–49670 (2017).
Komuro, A., Homma, Y., Negoro, T., Barber, G. N. & Horvath, C. M. The TAR-RNA binding protein is required for immunoresponses triggered by Cardiovirus infection. Biochem Biophys. Res Commun. 480, 187–193 (2016).
Daniels, S. M., Daniels, S. M., Gatignol, A. & Gatignol, A. The multiple functions of TRBP, at the hub of cell responses to viruses, stress, and cancer. Microbiol Mol. Biol. Rev. 76, 652–666 (2012).
Miyashita, M., Oshiumi, H., Matsumoto, M. & Seya, T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell Biol. 31, 3802–3819 (2011).
Oshiumi, H. et al. DDX60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep. 11, 1193–1207 (2015).
Goubau, D. et al. Mouse superkiller-2-like helicase DDX60 is dispensable for type I IFN induction and immunity to multiple viruses. Eur. J. Immunol. 45, 3386–3403 (2015).
Núñez, R. et al. The RNA Helicase DDX6 Associates with RIG-I to Augment Induction of Antiviral Signaling. Int. J. Mol. Sci. 19, 1877–14 (2018).
Sugimoto, N., Mitoma, H., Kim, T., Hanabuchi, S. & Liu, Y.-J. Helicase proteins DHX29 and RIG-I cosense cytosolic nucleic acids in the human airway system. Proc. Natl Acad. Sci. USA 111, 7747–7752 (2014).
Zhu, Q. et al. DHX29 functions as a RNA co-sensor for MDA5-mediated EMCV-specific antiviral immunity. PLoS Pathog. 14, e1006886 (2018).
Pattabhi, S., Knoll, M. L., Gale, M. & Loo, Y.-M. DHX15 Is a Coreceptor for RLR Signaling That Promotes Antiviral Defense Against RNA Virus Infection. J. Interferon Cytokine Res. 39, 331–346 (2019).
Oshiumi, H., Sakai, K., Matsumoto, M. & Seya, T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-β-inducing potential: HIGHLIGHTS. Eur. J. Immunol. 40, 940–948 (2010).
Gringhuis, S. I. et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 18, 225–235 (2016).
Zhang, Z., Yuan, B., Lu, N., Facchinetti, V. & Liu, Y. J. DHX9 Pairs with IPS-1 To Sense Double-Stranded RNA in Myeloid Dendritic Cells. J. Immunol. 187, 4501–4508 (2011).
Lin, J.-P., Fan, Y.-K. & Liu, H. M. The 14-3-3η chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathog. 15, e1007582 (2019).
Riedl, W. et al. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 26, 493–503 (2019).
Kouwaki, T. et al. Zyxin stabilizes RIG-I and MAVS interactions and promotes type I interferon response. Sci. Rep. 7, 11905 (2017).
Nguyen, T. A. et al. SIDT2 transports extracellular dsRNA into the cytoplasm for innate immune recognition. Immunity 47, 498–509 (2017).
Soonthornvacharin, S. et al. Systems-based analysis of RIG-I-dependent signalling identifies KHSRP as an inhibitor of RIG-I receptor activation. Nat. Microbiol. 2, 17022 (2017).
Ju, L.-G. et al. TTLL12 inhibits the activation of cellular antiviral signaling through interaction with VISA/MAVS. J. Immunol. 198, 1274–1284 (2016).
Zhong, X. et al. ZFYVE1 negatively regulates MDA5- but not RIG-I-mediated innate antiviral response. PLoS Pathog. 16, e1008457 (2020).
Xu, S.-S. et al. FKBP8 inhibits virus-induced RLR-VISA signaling. J. Med. Virol. 91, 482–492 (2018).
Eckard, S. C. et al. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol. 15, 839–845 (2014).
Fabre, A. et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am. J. Hm. Genetics 90, 689–692 (2012).
Moore, C. B. et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451, 573–577 (2008).
Allen, I. C. et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity 34, 854–865 (2011).
Lei, Y. et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36, 933–946 (2012).
Jounai, N. et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl Acad. Sci. USA 104, 14050–14055 (2007).
Richetta, C. & Faure, M. Autophagy in antiviral innate immunity. Cell Microbiol. 15, 368–376 (2013).
Du, Y. et al. LRRC25 inhibits type I IFN signaling by targeting ISG15‐associated RIG‐I for autophagic degradation. EMBO J. 37, 351–366 (2017).
Xian, H., Yang, S., Jin, S., Zhang, Y. & Cui, J. LRRC59 modulates type I interferon signaling by restraining the SQSTM1/p62-mediated autophagic degradation of pattern recognition receptor DDX58/RIG-I. Autophagy 16, 408–418 (2019).
Hou, P. et al. A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell Res. https://doi.org/10.1038/s41422-020-0362-1. (2020).
Kim, S., Eun, H. S. & Jo, E.-K. Roles of autophagy-related genes in the pathogenesis of inflammatory bowel disease. Cells 8, 77 (2019).
Jena, K. K. et al. Autoimmunity gene IRGM suppresses cGAS-STING and RIG-I-MAVS signaling to control interferon response. EMBO Rep. 21, e50051 (2020).
Ning, X. et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol. Cell 74, 19–31 (2019).
Chan, Y. K. & Gack, M. U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373 (2016).
Childs, K. S., Andrejeva, J., Randall, R. E. & Goodbourn, S. Mechanism of mda-5 Inhibition by paramyxovirus V proteins. J. Virol. 83, 1465–1473 (2008).
Parisien, J.-P. et al. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J. Virol. 83, 7252–7260 (2009).
Motz, C. et al. Paramyxovirus V proteins disrupt the fold of the RNA sensor MDA5 to inhibit antiviral signaling. Science 339, 690–693 (2013).
Davis, M. E. et al. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 16, 19–30 (2014).
Gitlin, L. et al. Melanoma differentiation-associated gene 5 (MDA5) is involved in the innate immune response to paramyxoviridae infection in vivo. PLoS Pathog. 6, e1000734 (2010).
Sanchez-Aparicio, M. T., Feinman, L. J., García-Sastre, A. & Shaw, M. L. Paramyxovirus V Proteins Interact with the RIG-I/TRIM25 Regulatory Complex and Inhibit RIG-I Signaling. J. Virol. 92, 396–21 (2018).
Sun, Y. et al. Newcastle disease virus V protein degrades mitochondrial antiviral signaling protein to inhibit host type I interferon production via E3 ubiquitin ligase RNF5. J. Virol. 93, e00322–19 (2019).
Ban, J. et al. Human respiratory syncytial virus NS 1 targets TRIM25 to suppress RIG-I ubiquitination and subsequent RIG-I-mediated antiviral signaling. Viruses 10, 716 (2018).
Ling, Z., Tran, K. C. & Teng, M. N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 83, 3734–3742 (2009).
Cárdenas, W. B. et al. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 80, 5168–5178 (2006).
Ramanan, P. et al. Structural basis for Marburg virus VP35–mediated immune evasion mechanisms. Proc. Natl Acad. Sci. USA 109, 20661–20666 (2012).
Schneider, U., Schwemmle, M. & Staeheli, P. Genome trimming: a unique strategy for replication control employed by Borna disease virus. Proc. Natl Acad. Sci. USA 102, 3441–3446 (2005).
Marc, D. Influenza virus non-structural protein NS1: interferon antagonism and beyond. J. Gen. Virol. 95, 2594–2611 (2014).
Donelan, N. R., Basler, C. F. & García-Sastre, A. A recombinant influenza A virus expressing anRNA-binding-defective NS1 protein induces high levels of betainterferon and is attenuated in Mice. J. Virol. 77, 13257–13266 (2003).
Mibayashi, M. et al. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81, 514–524 (2007).
Gack, M. U. et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449 (2009).
Rajsbaum, R. et al. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 8, e1003059 (2012).
Li, S., Min, J.-Y., Krug, R. M. & Sen, G. C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349, 13–21 (2006).
Chen, W. et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7, 1306–1312 (2001).
Varga, Z. T. et al. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 7, e1002067 (2011).
Cheung, P.-H. H. et al. Virus subtype-specific suppression of MAVS aggregation and activation by PB1-F2 protein of influenza A (H7N9) virus. PLoS Pathog. 16, e1008611 (2020).
Wang, R. et al. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy https://doi.org/10.1080/15548627.2020.1725375 (2020).
Wang, R. et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 15, 1163–1181 (2019).
Meylan, E. et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 (2005).
Li, K. et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl Acad. Sci. USA 102, 2992–2997 (2005).
Chan, Y. K. & Gack, M. U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 17, 523–530 (2016).
You, J. et al. Flavivirus infection impairs peroxisome biogenesis and early antiviral signaling. J. Virol. 89, 12349–12361 (2015).
He, Z. et al. Dengue virus subverts host innate immunity by targeting adaptor protein MAVS. J. Virol. 90, 7219–7230 (2016).
Ma, J. et al. Zika virus non-structural protein 4A blocks the RLR-MAVS signaling. Front Microbiol 9, 1350 (2018).
Pijlman, G. P. et al. A highly structured, nuclease-resistant, noncoding rna produced by flaviviruses is required for pathogenicity. Cell Host Microbe 4, 579–591 (2008).
Feng, Q. et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 88, 3369–3378 (2014).
Drahos, J. & Racaniello, V. R. Cleavage of IPS-1 in cells infected with human rhinovirus. J. Virol. 83, 11581–11587 (2009).
Mukherjee, A. et al. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7, e1001311 (2011).
Wang, B. et al. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 9, e1003231 (2013).
Barral, P. M. et al. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 81, 3677–3684 (2007).
Barral, P. M., Sarkar, D., Fisher, P. B. & Racaniello, V. R. RIG-I is cleaved during picornavirus infection. Virology 391, 171–176 (2009).
White, J. P., Cardenas, A. M., Marissen, W. E. & Lloyd, R. E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2, 295–305 (2007).
Ng, C. S. et al. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 87, 9511–9522 (2013).
Pulido, M. R. et al. Innate immune sensor LGP2 is cleaved by the Leader protease of foot-and-mouth disease virus. PLoS Pathog. 14, e1007135 (2018).
Visser, L. J. et al. Dissecting distinct proteolytic activities of FMDV Lpro implicates cleavage and degradation of RLR signaling proteins, not its deISGylase/DUB activity, in type I interferon suppression. PLoS Pathog. 16, e1008702 (2020).
Wang, D. et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85, 3758–3766 (2011).
Zhu, Z. et al. Foot-and-mouth disease virus viroporin 2B antagonizes RIG-I-mediated antiviral effects by inhibition of its protein expression. J. Virol. 90, 11106–11121 (2016).
Guu, T. S. Y., Zheng, W. & Tao, Y. J. Viral molecular machines. Adv. Exp. Med. Biol. 726, 245–266 (2011).
Marq, J.-B., Kolakofsky, D. & Garcin, D. Unpaired 5’ ppp-nucleotides, as found in arenavirus double-stranded RNA panhandles, are not recognized by RIG-I. J. Biol. Chem. 285, 18208–18216 (2010).
Fan, L., Briese, T. & Lipkin, W. I. Z proteins of new world arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 84, 1785–1791 (2009).
Min, Y.-Q., Ning, Y.-J., Wang, H. & Deng, F. A RIG-I–like receptor directs antiviral responses to a bunyavirus and is antagonized by virus-induced blockade of TRIM25-mediated ubiquitination. J. Biol. Chem. 295, 9691–9711 (2020).
Santiago, F. W. et al. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J. Virol. 88, 4572–4585 (2014).
Gori-Savellini, G., Valentini, M. & Cusi, M. G. Toscana virus NSs protein inhibits the induction of type I interferon by interacting with RIG-I. J. Virol. 87, 6660–6667 (2013).
Savellini, G. G., Anichini, G., Gandolfo, C., Prathyumnan, S. & Cusi, M. G. Toscana virus non-structural protein NSs acts as E3 ubiquitin ligase promoting RIG-I degradation. PLoS Pathog. 15, e1008186 (2019).
Spiegel, M. et al. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 79, 2079–2086 (2005).
Kindler, E. et al. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. mBio. 4, e00611–e00612 (2013).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1–20 (2020).
Hu, Y. et al. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 91, 101–115 (2017).
Shi, C.-S. et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193, 3080–3089 (2014).
Niemeyer, D. et al. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J. Virol. 87, 12489–12495 (2013).
Rabouw, H. H. et al. Middle east respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PLoS Pathog. 12, e1005982 (2016).
Jiang, H. et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol. Immunol. 17, 998–1000 (2020).
Konno, Y. et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Reports 32, 108185 (2020).
Fu, Y.-Z. et al. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol. Immunol. https://www.nature.com/articles/s41423-020-00571-x (2020).
Zheng, Y. et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Preprint at https://www.biorxiv.org/content/10.1101/2020.07.26.222026v1 (2020).
Banerjee, A. K. et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339 (2020)
Lei, X. et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 11, 3810 (2020).
Yang, Z. et al. Suppression of MDA5-mediated antiviral immune responses by NSP8 of SARS-CoV-2. Preprint at https://www.biorxiv.org/content/10.1101/2020.08.12.247767v1 (2020).
Acknowledgements
We would like to thank Editage (www.editage.com) for English language editing. This work is supported by Grants-in-Aids for Scientific Research (B) (18H02660) and (C) (19K07589 and 20K07534) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Onomoto, K., Onoguchi, K. & Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell Mol Immunol 18, 539–555 (2021). https://doi.org/10.1038/s41423-020-00602-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-020-00602-7
Keywords
This article is cited by
-
Psoriasis may increase the risk of idiopathic pulmonary fibrosis: a two-sample Mendelian randomization study
Respiratory Research (2024)
-
A tumor suppressor protein encoded by circKEAP1 inhibits osteosarcoma cell stemness and metastasis by promoting vimentin proteasome degradation and activating anti-tumor immunity
Journal of Experimental & Clinical Cancer Research (2024)
-
Innate immune responses in pneumonia
Pneumonia (2023)
-
Flavivirus prM interacts with MDA5 and MAVS to inhibit RLR antiviral signaling
Cell & Bioscience (2023)
-
Innate immune evasion strategies of SARS-CoV-2
Nature Reviews Microbiology (2023)
