
Abstract
The three isoforms of apolipoprotein E (APOE2, APOE3, and APOE4) only differ in two amino acid positions but exert quite different immunomodulatory effects. The underlying mechanism of such APOE isoform dependence remains enigmatic. Here we demonstrate that APOE4, but not APOE2, specifically interacts with the leukocyte immunoglobulin-like receptor B3 (LilrB3). Two discrete immunoglobin-like domains of the LilrB3 extracellular domain (ECD) recognize a positively charged surface patch on the N-terminal domain (NTD) of APOE4. The atomic structure reveals how two APOE4 molecules specifically engage two LilrB3 molecules, bringing their intracellular signaling motifs into close proximity through formation of a hetero-tetrameric complex. Consistent with our biochemical and structural analyses, APOE4, but not APOE2, activates human microglia cells (HMC3) into a pro-inflammatory state in a LilrB3-dependent manner. Together, our study identifies LilrB3 as a putative immune cell surface receptor for APOE4, but not APOE2, and may have implications for understanding the biological functions as well as disease relevance of the APOE isoforms.
Similar content being viewed by others


Introduction
The APOE gene product APOE plays a key role in lipid metabolism, immune regulation and neurology.1,2,3 The three most common APOE alleles ε2, ε3 and ε4 correspond to three protein isoforms: APOE2, APOE3, and APOE4, which differ only in two amino acid positions 112 and 158 (Fig. 1a).4,5 These APOE isoforms exhibit distinct roles in physiology and pathophysiology.3,6,7 As one of the major lipid carriers, APOE transports lipids to cells and tissues, and regulates plasma lipid levels.8,9,10 Consistent with its function in lipid metabolism, APOE binds with high affinity to a number of cell surface receptors, including LDL receptor and HSPG receptor.11,12,13,14,15,16,17,18 APOE2 (Cys112/Cys158), which shows weaker binding to the LDL receptor compared to APOE3 (Cys112/Arg158) or APOE4 (Arg112/Arg158), is linked to the recessive form of type III hyperlipoproteinemia.19,20,21

a Schematic diagram of APOE. APOE consists of an NTD for receptor binding and a CTD for lipid binding. The three APOE isoforms only differ in two amino acid positions (112 and 158) that may confer isoform-specific biological ramifications. b Schematic diagram of the AlphaLISA assay. The two interacting proteins are tagged with Flag and Strep, which are captured by monoclonal antibodies on the donor and acceptor beads, respectively. The interaction strength is reflected by the intensity of the luminescence that occurs between the donor and acceptor. c APOE4 specifically interacts with the ECD of LilrB3. Shown here are results of the AlphaLISA assays, in which pairwise interactions between Strep-tagged APOE and the ECD of Flag-tagged LilrB/PirB proteins were individually examined. These experiments were repeated six times (n = 6). Data are presented as means ± SD. d APOE4 binds to LilrB3-ECD in a concentration-dependent manner. Each of the three APOE isoforms was exposed to increasing concentrations of LilrB3-ECD in the AlphaLISA assay. Data are presented as means ± SD (n = 3). e APOE4 binds to LilrB3-ECD with a dissociation constant of 22.0 ± 2.7 μM. Compared to APOE4, APOE3 exhibits a weaker affinity of 57.8 ± 14.1 μM. In contrast, APOE2 shows no detectable binding to LilrB3-ECD. Shown here are results of ITC.
The immunoregulatory role of APOE was originally discovered as part of the inhibitory effect of plasma lipoproteins on T cell proliferation.22,23,24 APOE is known to suppress T cell proliferation and neutrophil activation,25 regulate macrophage function,26,27,28,29 facilitate lipid antigen presentation,30,31 and modulate inflammation and oxidation.32 Multiple sclerosis (MS) patients with APOE ε4, but not APOE ε2, display accelerated loss of brain tissue, resulting in early cognitive deficits.33,34,35
APOE isoforms also have distinct roles in neurology and neurodegeneration such as Alzheimer’s disease (AD).3,36,37 Compared to the most prevalent isoform APOE3, APOE4 drastically increases AD risk with a relatively earlier age of onset.38,39 In contrast, APOE2 is protective because it confers a remarkably lower AD risk compared to APOE3.40 Early studies suggest a role of APOE in β-amyloid deposition. Besides the extensive studies of APOE isoform-dependent amyloid-β clearance,41,42,43,44 increasing evidences suggest that the dysfunction of immune system play a pivotal role in AD pathology.45,46,47,48,49,50 In particular, aberrant activation of microglia, which leads to neurotoxic cytokine release and synapse pruning, has been linked to memory loss and the development of AD.51,52,53,54,55,56,57 In the tauopathy mouse model, knock-in of APOE4 results in markedly stronger activation of microglia compared to that of APOE2.58
In summary, the immunomodulatory function of APOE appears to be strongly isoform dependent. APOE is a secreted protein and may exert its function through engagement with an immune cell surface receptor. Macrophage/microglia are a key player of the immune system. The leukocyte immunoglobulin-like receptor B (LilrB) family of checkpoint proteins are abundantly expressed in macrophage/microglia and have been reported to regulate their activation.59 Notably, APOE3 was reported to interact with LilrB4 to suppress T cell proliferation;60 however, we have been unable to detect this interaction. APOE also interacts with the triggering receptor expressed on myeloid cells 2 (TREM2); however, TREM2 binds indiscriminately to all three APOE isoforms.61,62
In this study, we identify LilrB3 as a candidate immune cell surface receptor for APOE4, but not APOE2. We provide a structural basis for the specific recognition of APOE4 by LilrB3 through cryo-EM reconstruction of the binary complex at an average resolution of 3.0 Å. This is the first time that an APOE-bound receptor complex has been structurally elucidated, revealing tantalizing clues about APOE isoform specificity and receptor-mediated signaling.
Results
APOE4 specifically interacts with LilrB3 in vitro
A series of checkpoint proteins expressed on microglial surface are known to regulate activation of the macrophage and microglia (the resident macrophage in central nervous system).63,64 Notably, the LilrB family members have been reported to regulate the phagocytosis of macrophage.65 Ablation of PirB, the murine homolog of LilrB, prevents synapse pruning during normal mouse development and rescues memory deficit in AD model mice,66,67 identifying LilrB/PirB as candidate APOE receptors. Although we failed to confirm the reported interaction between APOE3 and LilrB4,60 we decided to broaden our search. We assessed the potential interactions between all three APOE isoforms and all six members of the LilrB/PirB family in a combinatorial fashion (Supplementary information, Tables S1, S2).
The extracellular domains (ECDs) of all six LilrB/PirB family members, each with a C-terminal Flag tag, were individually expressed in HEK293F cells and purified to homogeneity (Supplementary information, Fig. S1a, b). All three human APOE isoforms, each with a C-terminal Strep tag, were similarly expressed and purified to homogeneity (Supplementary information, Fig. S1b). These recombinant APOE proteins appear as particles of varying sizes (Supplementary information, Fig. S1c). The overall sizes of these APOE particles are similar to those purified from immortalized astrocytes.68 Potential interaction between each Strep-tagged APOE and each Flag-tagged LilrB-ECD was examined using the AlphaLISA assay,69 which yields luminescence in case of close physical proximity between two target proteins (Fig. 1b). Higher luminescence values are correlated with stronger interactions.
Based on the AlphaLISA assay, APOE2 fails to interact with any ECD of the LilrB/PirB family members, as reflected by the basal-level values of luminescence (Fig. 1c). In contrast, APOE4 exhibits strong interaction with LilrB3-ECD, but not with any other LilrB/PirB-ECD. APOE3 also interacts with LilrB3-ECD, but with weaker binding strength compared to APOE4 (Fig. 1c). These results contradict with a previous study60 and identify LilrB3 as the only candidate receptor for APOE binding. To further characterize the interaction, we titrated increasing concentrations of LilrB3-ECD into a fixed amount of APOE2/3/4. LilrB3-ECD displays tighter binding to APOE4 compared to APOE3 and undetectable binding to APOE2 (Fig. 1d).
Results of the AlphaLISA assay suggest that the ECD of the orphan receptor LilrB3 may specifically recognize APOE4, but not APOE2. LilrB3-ECD may also recognize APOE3, but to a lesser extent compared to APOE4. To confirm these preliminary findings, we quantified the interactions between LilrB3-ECD and each of the three APOE isoforms using isothermal titration calorimetry (ITC) (Fig. 1e). Consistent with results of the AlphaLISA assays, APOE4 binds LilrB3-ECD more tightly compared to APOE3, with dissociation constants of 22.0 ± 2.7 μM and 57.8 ± 14.1 μM, respectively. APOE2 fails to bind LilrB3-ECD.
APOE4 is specifically recognized by LilrB3 on cell surface
Next, we investigated whether APOE4 can be specifically recognized by LilrB3 on the cell surface. Following expression of RFP-tagged APOE4 in HEK293F cells, the APOE-containing supernatant was collected and incubated individually with six different HEK293F cultures, each expressing a specific GFP-tagged LilrB/PirB receptor (Fig. 2a). These cells were sorted through fluorescein isothiocyanate (FITC)/phycoerythrin (PE) dual channels using FACS analysis. The presence of HEK293F cells in the upper right quadrant and concurrent absence in the upper left quadrant are indicative of concentration-dependent interaction between APOE4 in the medium and LilrB3 on the cell surface (Fig. 2b). Strikingly, 22.0% of the LilrB3-expressing cells are sorted into the upper right quadrant. In contrast, no more than 3% of the other LilrB/PirB-expressing cells appear in this quadrant. Thus, APOE4 specifically associates with LilrB3 on the cell surface, but not with any other LilrB/PirB receptors.

a Schematic diagram of the cell-based binding assay. HEK293F cells that express each of the LilrB/PirB proteins were incubated with APOE4-containing supernatant. The resulting cells were analyzed by FACS (b) and confocal microscopy (c). b APOE4 specifically associates with LilrB3-expressing cells in the FACS analysis. APOE with a C-terminal RFP expressed in HEK293F cells is secreted into the medium. The full-length LilrB receptor with a C-terminal GFP tag is expressed in HEK293F cells. The LilrB-expressing cells were incubated with the supernatant of the APOE-expressing medium and sorted by FACS. c APOE4 specifically co-localizes with LilrB3 on the cell surface. APOE4 also co-localizes with PirB, but to a much lesser extent compared to LilrB3. Scale bars, 10 μm.
To confirm the FACS results, we examined a subset of the APOE4-incubated HEK293F cells using confocal microscopy (Fig. 2c). As anticipated, each of the six LilrB/PirB receptors is predominantly localized to the cell surface (Fig. 2c, left column). In contrast, APOE4 is abundantly present only on the surface of LilrB3-expressing cells (Fig. 2c, middle column). The cell surface localization of APOE4 coincides with that of LilrB3 (Fig. 2c, right column). In addition, APOE4 is also detectable on the surface of PirB-expressing cells, but at a much lower level compared to that on LilrB3-expressing cells (Fig. 2c, middle column).
LilrB3 recognizes a positively charged surface patch on APOE-NTD
Human APOE consists of an N-terminal domain (NTD, residues 1–167), which constitutes the putative receptor binding region, and a C-terminal domain (CTD, residues 207–299), which harbors the lipid-binding region (Fig. 1a). We expressed and purified to homogeneity the NTDs of all three APOE isoforms (Supplementary information, Fig. S1b) and examined their interactions with LilrB3-ECD using ITC. Remarkably, each of the three NTDs associates stably with LilrB3-ECD with similar binding affinities: 20.3 ± 3.9 μM, 14.3 ± 2.5 μM, and 17.9 ± 2.9 μM for APOE2, APOE3, and APOE4, respectively (Fig. 3a). The interaction between LilrB3-ECD and APOE4-NTD was further confirmed using gel filtration (Supplementary information, Fig. S2a). In contrast to LilrB3, none of the other LilrB/PirB family proteins interacts with APOE4-NTD using the same assay (Supplementary information, Fig. S2b–f).

a The NTD of each APOE isoform interacts with LilrB3-ECD with a similar binding affinity as determined by ITC. The binding affinities for the NTDs of APOE2, APOE3, and APOE4 are 20.3 ± 3.9 μM, 14.3 ± 2.5 μM, and 17.9 ± 2.9 μM, respectively. The ITC results are shown. b Differential impacts of 52 missense mutations in APOE4 on its interaction with LilrB3. Shown here is a summary of the binding results using FACS analysis. The binding strength between WT APOE4 and LilrB3-ECD is normalized as 100. Data are presented as means ± SD (n = 3). c The vast majority of the deleterious mutations map to a specific surface area in the NTD of APOE4. Except four mutations in helix α1, all other 10 mutations are clustered to a specific surface area on helices α3 and α4 of APOE4. d Structure of the full-length APOE3 reveals how the CTD interacts with the NTD. Shown here is the NMR structure of APOE3, with its CTD colored blue. Two close-up views are shown to illustrate the specific residues at the intra-molecular interface. Importantly, the surface elements of the APOE3-NTD that are recognized by its CTD are the same as those required for interaction with LilrB3. This finding suggests a negative regulation on LilrB3 binding by the CTD. e Mutations in APOE2-CTD result in enhanced interaction with LilrB3-ECD. Guided by the NMR structure of APOE3, we designed mutations to destabilize the interactions between the NTD and CTD of APOE2. The binding strength between WT APOE2 and LilrB3-ECD is normalized as 100. Data are presented as means ± SD (n = 3). f Mutations in APOE4-CTD have little impact on its interaction with LilrB3-ECD. Because the NTD and CTD of APOE4 may no longer associate with each other, these mutations in APOE4 have little impact on its binding to LilrB3. The binding strength between WT APOE4 and LilrB3-ECD is normalized as 100. Data are presented as means ± SD (n = 3).
The fact that each of the three APOE-NTDs, but not the full-length APOE isoform, binds equally well to LilrB3-ECD identifies the CTD as a negative regulator of APOE recognition by LilrB3. Obviously, the amino acid identity at positions 112 and 158 of the NTD governs such regulation.
To identify specific residues that interact with LilrB3-ECD, we generated 52 missense mutations in APOE4, each targeting a surface-exposed residue in its NTD (Fig. 3b). All 52 APOE4 variants were individually examined for their recognition by LilrB3. For 38 APOE4 variants, each retains at least 50% binding to LilrB3 compared to wild-type (WT) APOE4 (Fig. 3b). Neither R112A nor R158A has any appreciable impact on APOE4 recognition by LilrB3, confirming a dispensable role for residues 112/158 in receptor binding. Remarkably, however, for 14 APOE4 variants, each displays no more than 30% binding level compared to WT APOE4 (Fig. 3b). Twelve of these 14 mutations map to helices 1 and 4 of the NTD, mainly affecting a cluster of basic residues (Fig. 3c). These residues define a positively charged surface patch that is responsible for binding to LilrB3-ECD.
To explain the negative regulation on LilrB3 binding by the CTD of APOE2, we analyzed the NMR structure of the full-length APOE3,70 the only experimentally determined structure of any full-length APOE. An extended α-helical element from the CTD of APOE3 closely stacks against the NTD through an interface that includes the bulk of the positively charged surface patch (Fig. 3d). Therefore, this surface patch of APOE-NTD is likely required for binding to both its CTD and LilrB3-ECD. In APOE2, the intramolecular interaction may lock the CTD onto NTD, thus precluding APOE2 binding to LilrB3 (Supplementary information, Fig. S3). The replacement of Cys158 by Arg in APOE3 likely weakens the interaction, giving the NTD some leeway to be recognized by LilrB3. Replacement of Cys112 by Arg in APOE4 may further disable the intramolecular interaction, allowing the NTD to bind LilrB3 constitutively. In fact, the locations of residues 112/158 in APOE-NTD are consistent with this explanation; for example, the side chain of Arg158 in APOE3/4 would sterically clash with Ala256 of the CTD in the closed state (Supplementary information, Fig. S3).
Based on this explanation, mutations in the CTD that weaken its interaction with the NTD may allow the full-length APOE2 to re-gain binding to LilrB3-ECD. We generated seven missense mutations in APOE2, each affecting a surface residue of the CTD at the interface of the intramolecular interaction and near the LilrB3-binding patch. Supporting our hypothesis, all seven APOE2 variants show improved binding to LilrB3-ECD compared to WT APOE2 (Fig. 3e). In particular, five mutations, R226A, D227A, E234A, K242A, and Q246A, each result in > 50% improvement compared to WT APOE2. In contrast, none of these mutations has an impact on APOE4 binding to LilrB3-ECD (Fig. 3f).
The D2/D4 domains of LilrB3 recognize APOE4
LilrB3-ECD, but not LilrB2-ECD, specifically recognizes APOE4 in the cell-based binding assay (Fig. 2b). The ECD of LilrB2 or LilrB3 comprises four tandem domains, D1–D4 (Supplementary information, Fig. S4a). To identify the region of LilrB3-ECD that recognizes APOE4-NTD, we generated four LilrB3 chimeric variants by individually replacing each of the four tandem domains by that from LilrB2. These LilrB3 variants were individually examined for their abilities to bind APOE4. The replacement of D1 or D3 domain of LilrB3 has little impact on its interaction with APOE4 (Fig. 4a). In contrast, the swapping of D2 or D4 domain of LilrB3 by that from LilrB2 results in abrogation of the interaction.

a The D2 and D4 domains of LilrB3 are required for APOE4 interaction. The D1/2/3/4 domains of LilrB3 were individually replaced by those of LilrB2. The resulting LilrB3 chimeric variants were examined for their abilities to bind APOE4. Shown here are results of the FACS analysis. Data are presented as means ± SD (n = 3). b The D2 and D4 domains of LilrB3 in the correct orientation are sufficient for interaction with APOE4. The D1/2/3/4 domains of LilrB2 were replaced by those of LilrB3 as indicated. The resulting LilrB2 chimeric variants were examined for their abilities to bind APOE4. Data are presented as means ± SD (n = 3). c Replacement of selected sequence elements of LilrB3 by those from LilrB2 weakens its interaction with APOE4. Shown here is the summary of the cell-based binding assays. Data are presented as means ± SD (n = 3). d The APOE4-binding elements of LilrB3-D4 are clustered on the same side of D4. Seven sequence elements are mapped onto the modeled structure of LilrB3-ECD. e The APOE4-binding elements of LilrB3-ECD define a novel protein–protein interface. Eight such elements, five from the D4 domain and three from the D2 domain, are clustered on the same side of D2 and D4 in the modeled structure of LilrB3-ECD.
Next, we performed the reverse domain-swapping experiments. Individual domains of LilrB3 were transferred onto the framework of LilrB2. Replacement of all four domains D1/D2/D3/D4 (abbreviated as D1234) or three consecutive domains D2/D3/D4 (D234) of LilrB2 by those from LilrB3 led to restoration of full interaction with APOE4 (Fig. 4b). Swapping of D2/D4 (D24) of LilrB2 by those from LilrB3 also largely restored APOE4 interaction. Replacement of D2 only or D2/D3 (D23) of LilrB2 by those from LilrB3 still allowed some interaction with APOE4. All other LilrB2 chimeras exhibit sharply reduced interaction with APOE4.
The ECDs of LilrB2 and LilrB3 share 81% sequence identity (Supplementary information, Fig. S4b). To pinpoint the LilrB3 residues that bind APOE4, we generated 13 LilrB3 chimeras by individually replacing sequence elements of LilrB3 with those of LilrB2. Eleven of these 13 LilrB3 chimeras involve sequence elements in D2 (D2-F1 through D2-F4) and D4 (D4-F1 through D4-F7). The LilrB3 variant D4-F2, which has two residues Arg/His replaced by Trp/Asp from LilrB2, nearly abolished its interaction with APOE4 (Fig. 4c). Altogether, three elements in D2 and five elements in D4 are found to be important, as the replacement of each of these elements led to at least 70% reduction of APOE4 binding. Next, we mapped these eight elements onto the structure of LilrB3-ECD predicted by AlphaFold.71,72 The five elements of D4 are located on the same side of the structure (Fig. 4d). All eight elements define a surface patch on the same side of LilrB3-ECD (Fig. 4e).
Structural basis of APOE4 recognition by LilrB3
To understand the structural basis of APOE4 recognition by LilrB3, we purified a binary complex between APOE4-NTD and LilrB3-ECD (Supplementary information, Fig. S2a). The sample was subjected to cryo-electron microscopy (cryo-EM) analysis (Supplementary information, Fig. S5). 5,029 micrographs were collected, resulting in 1,996,689 particles. After three-dimensional classifications, a final EM reconstruction of the APOE4–LilrB3 complex was obtained at an average resolution of 3.0 Å (Supplementary information, Fig. S6). The EM density map allowed unambiguous identification of amino acids and atomic modeling of the complex (Supplementary information, Fig. S7 and Table S3).
In the structure, two molecules of APOE4-NTD associate with two molecules of LilrB3-ECD (Fig. 5a). Each APOE4-NTD engages both copies of LilrB3-ECD, binding to D2 of one LilrB3 molecule and D4 of the other. Remarkably, the two APOE4-NTD molecules make no direct contacts with each other and the two LilrB3-ECDs are separated by a distance of nearly 100 Å. Nonetheless, simultaneous recognition of two APOE4 molecules tethers these two LilrB3 molecules, allowing their intracellular immunoreceptor tyrosine-based inhibitory motifs (ITIMs) to be placed on the same side (Fig. 5a). Such a previously unanticipated 2:2 binding mode is likely conducive to intracellular signaling.

a Cryo-EM structure of the binary complex between APOE4-NTD and LilrB3-ECD. In the complex, two molecules of APOE4-NTD associate with two molecules of LilrB3-ECD. Two perpendicular views are shown. b Close-up view of one interface between APOE4-NTD and LilrB3-ECD. This interface, involving the D2 domain of LilrB3, features a network of inter-molecular hydrogen bonds. c Close-up view of the other interface between APOE4-NTD and LilrB3-ECD. This interface, involving the D4 domain of LilrB3, contains van der Waals contacts at its center. d The CTD of APOE2 in the closed conformation likely blocks the ability of the NTD to interact with LilrB3-ECD. The NTD of the full-length APOE3 (PDB: 2L7B) is aligned to APOE4-NTD (shown in orange cartoon) of the APOE4-NTD/LilrB3-ECD complex (left panel). In the closed conformation of APOE3, the positively charged residue Arg158 in the NTD is located close to the non-polar Ala256 in the CTD (right panel), creating an unfavorable environment.
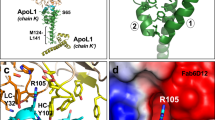
At the APOE4 interface with the D2 domain (Fig. 5b), three polar or charged amino acids (Arg114, Gln117, and Glu121) from helix α4 of APOE4 mediate four hydrogen bonds (H-bonds) with three residues from LilrB3 (Arg143, Ser174, and Gln178). His140 of APOE4 donates an H-bond to Gln178 of LilrB3. In the center of the APOE4 interface with the D4 domain (Fig. 5c), Trp39 of APOE4 makes van der Waals contacts with Trp348 and Trp349 of LilrB3. These contacts are flanked by H-bonds on both sides. In particular, Asp35 and Arg38 from helix α1 of APOE4 make three H-bonds, one each to Asp323, Thr324, and Trp349 of LilrB3. These structural observations nicely corroborate our biochemical analyses (Figs. 3, 4). For example, each of the three mutations D35A/R38A/W39A in APOE4, which affects the D4 interface (Fig. 5c), results in severe reduction of LilrB3 binding (Fig. 3b). Similarly, the mutation R114A in APOE4, which affects the D2 interface (Fig. 5b), nearly abrogates LilrB3 binding (Fig. 3b).
Structural comparison between the APOE4-NTD/LilrB3-ECD complex and the full-length APOE3 of the closed conformation (PDB: 2L7B) provides a plausible explanation for the observation that APOE4, but not APOE2, is recognized by LilrB3 (Fig. 5d). With the NTD of the full-length APOE3 aligned to APOE4-NTD of the APOE4-NTD/LilrB3-ECD complex, the CTD of the full-length APOE3 sterically clashes with the D2 domain of LilrB3-ECD (Fig. 5d, left panel). Therefore, the full-length APOE2 or APOE3 in its closed conformation is unable to engage the LilrB3 receptor. In the closed conformation of APOE3, the positively charged residue Arg158 of the NTD is located close to the non-polar Ala256 of the CTD (Fig. 5d, right panel), creating an unfavorable environment. In contrast, Cys158 in the NTD of APOE2 would be favorably accommodated by the local environment. This analysis provides a plausible explanation for the observation that the replacement of Cys158 by Arg in APOE3 or APOE4 allows LilrB3 recognition.
LilrB3-dependent microglia activation by APOE4
LilrB3-ECD specifically recognizes APOE4, but not APOE2, in the in vitro binding assays (Fig. 1). APOE4 binds preferentially to LilrB3, but not other LilrB receptors, on the cell surface (Fig. 2). To examine the potential consequence of such interaction, we eliminated both alleles of the LilrB3 gene in human microglia cell line 3 (HMC3) cells (Supplementary information, Fig. S8) and assessed the impact of APOE isoform treatment on both WT and LilrB3-null HMC3 cells. Total RNAs were extracted from these cells and processed for RNA-seq analysis. For each APOE isoform, three independent replicates were performed.
Compared to the PBS-treated control, 167 differentially expressed genes (DEGs) were identified in the APOE4-treated WT HMC3 cells using the criteria of fold change (FC) > 1.5 and false discovery rate (FDR) < 0.05 (Fig. 6a; Supplementary information, Tables S4, S5). Of these genes, 95 are up-regulated and 72 are down-regulated (Fig. 6c). In contrast, the numbers of up/down-regulated genes are 76/9 and 34/1, for APOE3- and APOE2-treated groups, respectively (Fig. 6a, c). These numbers suggest more pronounced changes induced by APOE4 compared to APOE3 or APOE2. Consistent with these results, principal component analysis (PCA) suggests distinct responses of the HMC3 cells upon treatment by the three APOE isoforms (Fig. 6b). The heatmaps reveal a clear pattern of APOE isoform dependence (Fig. 6c), which is corroborated by the Venn diagram (Fig. 6d). Of the 95 APOE4-stimulated genes, 80 display isoform dependence that is defined by a stimulation order of APOE4 > APOE3 > APOE2. Of the 72 APOE4-suppressed genes, 62 display such isoform dependence.

a Treatment by APOE2, APOE3, and APOE4 results in progressively more pronounced alteration of the gene expression pattern in HMC3 cells. Shown here are the volcano plots of the RNA-seq results. b PCA reveals distinct response patterns to treatment by APOE2, APOE3, and APOE4. Tabulated here are the heat maps of 95 up-regulated genes and 72 down-regulated genes in APOE4-treated HMC3 cells. c Treatment by APOE2, APOE3, and APOE4 leads to distinct patterns of gene expression in HMC3 cells. d A large majority of the up-/down-regulated genes display APOE isoform dependence. 83% (80 of 95) and 86% (62 of 72) of the up- and down-regulated genes, respectively, in APOE4-treated HMC3 cells show APOE isoform dependence. Shown here are results of the Venn diagram on the up-/down-regulated genes to identify APOE isoform dependence. e The up- and down-regulated genes mainly cluster in pro-inflammatory pathways. f Results of qPCR of APOE4-responsive genes in WT HMC3 cells. Shown here are the results of qPCR from three independent experiments (means ± SD).
Consistent with previous studies, a large majority of the APOE4-stimulated genes participate in pro-inflammatory responses, including interferon, cytokine, and antiviral signalings, on the basis of Reactome pathway analysis (REAC)73,74 (Fig. 6e; Supplementary information, Table S4). In particular, the 30 type I interferon-stimulated genes (ISGs) include IFITM3, which inhibits viral entry;75 BST2, which fights viral infection and plays a role in microglial engulfment;76 MX1, which blocks influenza virus infection; ISG15, which shapes the host antiviral response;77 and STAT1, which drives microglia activation toward the M1 phenotype78 (Supplementary information, Table S4). These representative APOE-responsive genes were validated by quantitative PCR (qPCR) analysis (Fig. 6f).
Then we assessed the impact of APOE isoforms on the LilrB3-null HMC3 cells. In contrast to the WT HMC3 cells, the numbers of up- and down-regulated genes in response to APOE4 treatment have been sharply reduced to 18 and 1, respectively, in the LilrB3-null cells (Fig. 7a). Among the 18 APOE4-stimulated genes, only 4 are found in the previously defined set of 80 genes that display APOE isoform dependence (Fig. 7b). The transcription patterns of the 167 DEGs in the WT HMC3 cells have largely disappeared in the LilrB3-null cells (Fig. 7c). The APOE4-stimulated pro-inflammatory genes in the WT HMC3 cells are no longer responsive to APOE4 treatment in the LilrB3-null cells (Fig. 7a; Supplementary information, Table S5).

a Treatment by the APOE isoforms, particularly APOE4, causes little alteration of the gene expression pattern in LilrB3-null HMC3 cells. Shown here are the volcano maps of the RNA-seq results. b Only 4 of the 18 up-regulated genes in APOE4-treated LilrB3-null HMC3 cells display APOE isoform dependence. The only down-regulated gene shows no APOE isoform dependence. c Treatment by APOE2, APOE3, and APOE4 has relatively little impact on the gene expression patterns of LilrB3-null HMC3 cells. Tabulated here are the heat maps for 95 and 72 genes that are either up- or down-regulated in APOE4-treated, WT LilrB3 HMC3 cells. d Results of qPCR of 13 representative genes in pro-inflammatory pathways in LilrB3-null HMC3 cells. Shown here are the results of qPCR from three independent experiments (means ± SD).
These RNA-seq results were confirmed by qPCR analysis on 13 representative up-regulated genes, 1 down-regulated gene, and 2 microglia marker genes (Fig. 7d). The expression levels for IFITM3, BST2, MX1, and ISG15 in the APOE4-treated WT HMC3 cells are 2.4-, 9.0-, 10.0-, and 4.3-fold higher, respectively, compared to the PBS-treated control (Fig. 6f). In contrast, the gene expression FC values for these genes have been markedly reduced in the LilrB3-null cells. The down-regulated gene TNFRSF10D displays 32% expression level in APOE4-treated WT HMC3 cells compared to the PBS-treated control (Fig. 6f). In contrast, TNFRSF10D is largely unaffected by APOE4 treatment in the LilrB3-null cells. Validating this analysis, the expression of two microglia marker genes IBA1 and P2RY12 remains indifferent to APOE4 treatment in both the WT and LilrB3-null HMC3 cells (Figs. 6f, 7d). Taken together, our experimental data demonstrate that the ability of APOE4 to activate the HMC3 cells is severely damaged in the absence of LilrB3 expression.
To further examine LilrB3-dependent microglia activation, we generated three APOE4 mutants, W39A, R114A, and W39A/R114A, each of which exhibits crippled ability to bind LilrB3 (Fig. 3b; Supplementary information, Fig. S9a). Upon treatment of HMC3 cells by each of the APOE4 variants, we analyzed the expression of six representative APOE4-responsive genes by qPCR. Compared to WT APOE4 treatment, the expression is markedly reduced in HMC3 cells treated by APOE4 single mutant W39A or R114A (Supplementary information, Fig. S9b). The expression is nearly abolished in cells treated by the APOE double mutant W39A/R114A.
Discussion
AD, characterized by amyloid plaques in patient brain, accounts for the vast majority of dementia in elderly population.79,80 Greater than 95% of all AD cases are classified as late onset AD (LOAD).81 APOE is the strongest known genetic risk factor for LOAD.38,39 Compared to APOE3, APOE4 confers a much higher AD risk with an earlier age of onset38 whereas APOE2 confers a lower AD risk.40 APOE was reported to display isoform-dependent binding to β-amyloid,41,42,43 suggesting a role in β-amyloid pathogenesis. Indeed, APOE4 carriers are usually associated with more severe β-amyloid deposition.82 The cellular uptake of APOE and β-amyloid is mediated by binding to APOE receptors, such as LDLR, LRP1 and HSPG. In this study, we identify the checkpoint protein LilrB3 as an APOE4-specific immune cell surface receptor. Because LilrB3 is abundantly expressed in lymphocytes, it may play a role in cellular clearance of β-amyloid. Activation of the ISG genes triggered by LilrB3–APOE4 interaction may ultimately impede the phagocytosis function of microglia, thus contributing to the β-amyloid deposition.
The specific recognition of APOE4 by LilrB3 has been revealed by our cryo-EM reconstruction of the unanticipated 2:2 complex between APOE4-NTD and LilrB3-ECD (Fig. 5). Two isolated molecules of APOE4-NTD each interacts with two molecules of LilrB3-ECD, but have no direct contacts with each other. Such a ligand–receptor binding mode is rare but has been previously documented, as exemplified by the interaction between the immunoregulatory cytokine IL-6 and the signaling receptor gp13083,84 (Supplementary information, Fig. S10). Similar to IL-6, APOE4-NTD mainly comprises a four-helix bundle. The overall spatial arrangement of the 2:2 complex between APOE4 and LilrB3 is reminiscent of that between IL-6 and gp130. Remarkably, the C-termini of the two gp130 molecules are separated by a distance of ~73 Å, compared to ~99 Å spanned by two LilrB3 molecules. The binding of monomeric IL-6 to gp130 is preceded by IL-6 engagement with its non-signaling α-receptor IL-6Rα. In contrast, APOE4 already exists in a lipid-containing particle prior to engagement with LilrB3.
Among the known APOE-binding receptors on the cell surface of microglia, the TREM family proteins are particularly attractive, because they contain the immunoreceptor tyrosine-based activating motifs (ITAMs) on the intracellular side. The ITAMs are thought to recruit downstream kinases, triggering microglia activation.85 Unfortunately, TREM2, which was reported to bind APOE, shows no APOE isoform specificity.62,86 This observation effectively rules out TREM2 as the mediator of different AD risks by APOE isoforms. This is also true for a number of other cell surface receptors.1,62,86,87
In contrast to TREM2, the LilrB/PirB family receptors contain the ITIMs on the intracellular side, binding of two APOE4 molecules to two LilrB3 receptors effectively brings their respective ITIMs to close proximity. It is conceivable that the signaling kinase and/or phosphatase recruited to one LilrB3 molecule can now be trans-regulated by that on the other LilrB3 molecule. However, the ITIMs on the intracellular side are thought to inhibit downstream signaling.88 How these ITIMs trigger downstream signaling events remains to be determined.
Notably, unlike most other APOE–receptor complexes,89 the binding affinity of 22 μM between APOE4 and LilrB3 is quite modest. This low-affinity binding constitutes a challenge for accurate measurement because it is difficult to achieve saturation. Nonetheless, this affinity, measured in vitro using recombinant proteins, is likely to be enhanced on the cell surface due to abundant expression of LilrB3 and high local concentration as a result of two-dimensional membrane environment.59 In addition, we cannot rule out the possibility of non-signaling co-receptors for APOE4, which may greatly strengthen its binding to LilrB3.
As a lipid-binding protein, APOE derived from APOE-expressing mice is known to form lipoprotein particles, with sizes ranging between 7 nm and 17 nm.68 These APOE particles are thought to exhibit preference for some lipids over others.90,91 In our study, the APOE proteins are derived from HEK293F cells and thus may show variation in lipid composition. Notably, however, the sizes of our HEK293F-derived APOE particles are similar to those isolated from immortalized astrocytes.68 In addition, application of recombinant APOE4 or isogenic knock-in of APOE4 are both able to activate microglia.92,93,94 Moreover, the NTD of APOE, which does not bind lipid, is responsible for receptor binding.15,95 Therefore, neither the size of the APOE particle nor its lipid composition may constitute a major concern for the conclusion of this study.
The identification of LilrB3 as an APOE4-specific immune cell surface receptor bears important ramifications for understanding APOE isoform-dependent functions and diseases, in particular AD. Microglia are known to play a key role in AD development.96,97 The transcriptional profile of microglia is altered in AD, switching from a homeostatic pattern to a disease-associated microglial (DAM) phenotype or microglia neurodegenerative (MGnD) phenotype.55,98,99 APOE ablation in AD model mice blocks the microglial transition from homeostasis to DAM or MGnD.55,99 Notably, activated interferon response is frequently associated with microglia that are derived from AD model mice and AD patients.73,100,101,102,103 Consistent with a direct role in AD development, type I interferon signaling promotes microglia dysfunction and memory loss.73,102,103,104 APOE4, either endogenously expressed or recombinantly applied, is associated with elevated pro-inflammatory cytokine production in mouse and human.2,105,106,107 Expression of APOE4, but not APOE2, is associated with elevated pro-inflammatory state in microglia, which in turn releases neurotoxic cytokine and eliminates synapses, ultimately resulting in AD.58,92,93,107,108 These lines of evidence point to an attractive model to explain elevated AD risks by APOE4: through microglia activation into a pro-inflammatory state.
This model has a critical gap: how does APOE4, but not APOE2, activate microglia? This gap might be filled by LilrB3 as a specific receptor for APOE4, but not APOE2. LilrB3 only recognizes the full-length APOE4, but not APOE2, both in solution and on cell surface (Figs. 1, 2). Consistent with existing knowledge, treatment by APOE4, but not APOE2, activates the HMC3 cells into a pro-inflammatory state that exhibits clear features of type I interferon response73,102,103,104 (Fig. 6). Importantly, LilrB3 ablation renders HMC3 cells no longer responsive to APOE4 treatment.
In summary, LilrB3 has been identified as a specific cell surface receptor for APOE4, but not APOE2. The structural basis for this specific ligand–receptor recognition has been elucidated. LilrB3 binding by APOE4, but not by APOE2, triggers activation of the microglia HMC3 cells that is characterized by activated interferon responses. These experimental findings reveal clues to APOE isoform-dependent functions and diseases.
Materials and methods
Plasmids and constructs
The cDNA clones for LilrB1 (GenBank ID: BC015731.1) and LilrB5 (GenBank ID: BC025704.1) were provided as a gift from Professor Jiahuai Han of Xiamen University. The cDNA clones for APOE3 (GenBank ID: NM_000041.2), LilrB2 (GenBank ID: BC036827), LilrB3 (GenBank ID: XM_006726314.4), LilrB4 (GenBank ID: NM_001278426.3), and PirB (GenBank ID: U96689.1) were purchased from SinoBiological Inc. The cDNA clones for APOE2 and APOE4 were generated by introducing the missense mutations R158C and C112R, respectively, based on the cDNA clone of APOE3. The clones for all missense mutants of APOE, the NTD of APOE, and the ECD of LilrB/PirB were generated using PCR-based methods. All plasmids, constructs, and primers are summarized and listed (Supplementary information, Tables S1, S2).
For the AlphaLISA assays, LilrB/PirB and APOE were Flag- and Strep-tagged, respectively, for binding the anti-Flag donor beads and Streptavidin acceptor beads. For Flag-tagged proteins, the cDNA sequences for the ECDs of LilrB1 (residues 1–438), LilrB2 (residues 1–440), LilrB3 (residues 1–420), LilrB4 (residues 1–238), LilrB5 (residues 1–435), and PirB (residues 1–618) were amplified using PCR. Each of the LilrB/PirB ECD was subcloned into the modified pCAG vector between the KpnI and XhoI sites using Gibson Assembly (E2611, NEB). The constructs for the Strep-tagged, full-length APOE isoforms were similarly generated as described above except that the C-terminal Flag was replaced by a Strep-tag. For the ITC assays, the cDNA for the NTD (residues 1–167) of each APOE isoform was subcloned into the pET-21b vector for bacterial expression in E. coli strain BL21(DE3). For the cell-based binding assays, the C-termini of LilrB/PirB and APOE isoforms were fused with GFP and RFP, respectively, for confocal microscopy and flow cytometry analysis. For the domain/fragment swapping experiments, we generated chimeric constructs by swapping LilrB3 domains/fragments with those from LilrB2 using the PCR-based quick-change method.
Expression and purification of LilrB/PirB ECDs and APOE isoforms
Following transfection of the HEK293F cells, each ECD was expressed in the culture medium 293TI (SinoBiological Inc.). The culture medium was harvested and loaded into the Ni-NTA affinity column. The Ni-NTA affinity column was eluted using 10 mL elution buffer (300 mM imidazole, 25 mM Tris, pH 8.0, 50 mM NaCl). The eluted protein was further fractionated using Source-15Q anion exchange column (GE Healthcare). The peak fractions were collected, visualized on SDS-PAGE gels through Coomassie blue staining, concentrated and subjected to size exclusive chromatography (SEC) using a Superdex-200 column (GE Healthcare) (Supplementary information, Fig. S1a). The peak fractions were collected and visualized on SDS-PAGE gels through Coomassie blue staining (Supplementary information, Fig. S1b). Protein concentration was determined using the BCA assay (Pierce). The full-length APOE isoforms were similarly expressed and purified (Supplementary information, Fig. S1b), except that the Source-15Q anion exchange column was substituted by a Heparin column as commonly practiced in other published studies.109,110
Examination of APOE isoforms by negative staining electron microscopy (EM)
The full-length APOE2, APOE3 and APOE4 proteins were individually diluted to 0.018 mg/mL and absorbed on Cu grids supported by a thin layer of carbon film (Zhongjingkeji Technology Co. Ltd). The proteins were stained using 3% (w/v) uranyl acetate. The images were taken using FEI Tecnai Spirit with iCorr D1319 at 120 kV accelerating voltage (Supplementary information, Fig. S1c).
Expression and purification of APOE-NTD
Each plasmid for a specific APOE-NTD isoform was transformed into the E. coli strain BL21 (DE3). A single colony was inoculated into a 5-mL starter culture in LB for growth at 37 °C. After 12 h, the starter culture was inoculated into 1 L LB culture. At an optical density (OD) of 0.6 at 595 nm, IPTG was added for induction of protein expression. The culture was then air-cooled to 18 °C over a 45-min period and maintained at 18 °C overnight. Cells were collected through centrifugation, resuspended in the TBS buffer, and lysed through sonication. Following centrifugation at 13,000 rpm using the F14-14×50cy rotor (Thermo Fisher Scientific), the supernatant was loaded into a Ni-NTA affinity column. The eluted protein was incubated with the SUMO protease overnight at 4 °C to remove the SUMO tag. The cleaved APOE-NTD was fractionated using a Superdex-200 column (GE Healthcare). The peak fractions were visualized on an SDS-PAGE gel through Coomassie blue staining (Supplementary information, Fig. S1b).
AlphaLISA luminescence assay
The AlphaLISA assay for measuring protein–protein interactions was performed using the EnSpire reader (PerkinElmer). The C-terminally Flag-tagged LilrB/PirB protein is bound to the anti-Flag donor beads (AS103D, PerkinElmer). The Strep-tagged, full-length APOE is bound to the Streptavidin acceptor beads (AL125C, PerkinElmer). The donor and acceptor beads are brought into close proximity through potential interactions between LilrB/PirB and APOE. Upon illumination at 680 nm, the donor beads with phthalocyanine convert ambient oxygen into singlet oxygen that activates thioxene derivatives in the acceptor beads, which then release photons at 615 nm. The experiments were conducted using 2.75 μM LilrB/PirB and 2.75 μM APOE in the presence of 5 μg/mL donor and acceptor beads in a buffer of 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, 1 mM DTT and 0.1 mg/mL bovine serum albumin. For the titration experiments, the concentration for each of the three APOE isoforms was 2.75 μM. The concentrations of LilrB3 were 5 μM, 4 μM, 2 μM, 1 μM, 500 nM, 250 nM, 125 nM, 62.5 nM, 31.3 nM, 15.6 nM, 7.8 nM, and 3.9 nM. Each experiment was independently repeated three times. The results are highly reproducible, with each measurement typically within 10% of the average. The result is presented as means ± SD.
Cell-based binding assay
HEK293F cells were transfected using the pCAG-LilrB/PirB-GFP plasmid, which expresses a full-length GFP-tagged LilrB or PirB cell surface receptor, or the pCAG-APOE-RFP plasmid, which expresses a full-length RFP-APOE isoform. The supernatant of the culture medium for the cells transfected by APOE-expressing plasmids was collected. The secreted APOE protein in the supernatant was quantified and diluted using fresh medium to a concentration of 10 μg/mL based on the dual criteria of RFP fluorescence signal and SDS-PAGE quantification. 0.5 mL of the APOE-containing medium was incubated with 0.5 mL of the cells transfected by LilrB/PirB-expressing plasmids. The final mixture was analyzed by flow cytometry and confocal laser scanning microscopy. The experimental data were analyzed using the FlowJo 10.5.3 Software (FlowJo LLC).
ITC
All ITC experiments were performed using the MicroCal iTC200 system. All proteins were buffer-exchanged into PBS using SEC. All three APOE-NTDs were first individually concentrated and then diluted to 765 μM. In order to get a good signal, each APOE-NTD loaded in the syringe was injected into the cell in a series of 2-μL aliquots with 100-s intervals and the LilrB3-ECD concentration in the cell was 35 μM. An initial injection of 0.5 μL ligand was made and discarded during data analysis. The full-length APOE proteins were individually concentrated and diluted to 413 μM. The titrations comprised 19 injections of 2-μL each of the full-length APOE protein into LilrB3-ECD of 31 μM. Under these conditions, titration of APOE2 or APOE3 into LilrB3 yielded noisy and unreliable signals. Therefore, we raised APOE2 and APOE3 concentration to 542 μM and 586 μM, respectively, which are close to their solubility limits. We also adjusted the concentration of LilrB3-ECD to 52 μM to increase the signal. Under these conditions, titration of APOE3 into LilrB3 yielded a nicely reproducible signal; however, titration of APOE2 still gave rise to undetectable signal. The raw ITC data were fitted to a single binding site model using the ITC200 Origin software (MicroCal LLC) provided by the manufacturer.
In vitro binding assay
Potential interaction between APOE4-NTD and each LilrB/PirB ECD was examined using SEC (Supplementary information, Fig. S2). Briefly, 3.6 mg APOE4-NTD and 1.2 mg LilrB/PirB ECD were incubated and applied to a Superdex-200 column (GE Healthcare) under the running buffer (25 mM Tris, pH 8.0, 150 mM NaCl). The elution profile was compared to those for 3.6 mg APOE4-NTD alone and 1.2 mg LilrB/PirB ECD alone. The appearance of a new peak with a smaller elution volume demonstrates stable interaction between the two proteins. The peak fractions were visualized on SDS-PAGE gels through Coomassie blue staining.
Assembly of the APOE4-NTD/LilrB3-ECD complex for cryo-EM analysis
To assemble the APOE4-NTD/LilrB3-ECD complex for cryo-EM analysis, ~20 mg purified APOE4-NTD were incubated with ~20 mg purified LilrB3-ECD in 1 mL gel filtration running buffer (1× PBS, Beyotime). The mixture was applied to SEC (Superdex-200, GE Healthcare) in the running buffer. The peak fraction for the APOE4-NTD/LilrB3-ECD complex was used for cryo-EM sample preparation.
Cryo-EM sample preparation and data acquisition
An aliquot of 3-μL sample was applied to a glow-discharged holey carbon grid (Quantifoil Au 300 mesh, R1.2/1.3), blotted for 3 s, and rapidly plunged into liquid ethane using Vitrobot Mark IV (Thermo Fisher Scientific) operating at 8 °C and 100% humidity. The samples were imaged on a 300-kV Titan Krios electron microscope (Thermo Fisher Scientific) using a nominal magnification of 64,000× (Supplementary information, Fig. S5a). Movies were recorded using a Gatan K3 detector (Thermo Fisher Scientific) equipped with a GIF Quantum energy filter (slit width 20 eV) at the super-resolution mode. Each stack was exposed for 2.56 s, with a dose rate of ~23 counts/s/physical-pixel (~19.5 e−/s/Å2) for each frame. All 32 frames in each stack were aligned and summed using MotionCor2111 and binned to a pixel size of 1.0979 Å. The defocus value for each image varied from 1.5 μm to 1.8 μm and was determined using Gctf.112
Cryo-EM data processing
Data processing of the APOE4-NTD/LilrB3-ECD complex involves 5,029 movie stacks (Supplementary information, Fig. S5a). 1,996,689 particles were picked using Gautomatch (developed by Kai Zhang, https://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch/). After particle extraction with a binned box size of 128 pixels (which is twice that in the original micrographs), three rounds of 2D classification were performed using cryoSPARC113 (Supplementary information, Fig. S5b), which gave rise to a final data set containing 1,299,413 good particles. This data set was then used for generation of an initial model (Supplementary information, Fig. S5c). The ab initial 3D model was generated using C1 symmetry, with the number of classes set to three. One class with 948,680 particles stood out with well-defined features for secondary structural elements. This subset was then subjected to non-uniform refinement with C1 symmetry. The reconstruction at this stage revealed apparent C2 symmetry. The particles were re-extracted using a box size of 256 pixels (which is the same as that in the original micrographs). Through several rounds of heterogeneous refinement and non-uniform refinement, a final reconstruction of the APOE4-NTD/LilrB3-ECD complex with C2 symmetry was obtained at an average resolution of 3.0 Å from 462,565 particles (Supplementary information, Figs. S5c–S7).
Reported resolution limits were calculated on the basis of the FSC 0.143 criterion with a high-resolution noise substitution method114 (Supplementary information, Fig. S6a, b). The final reconstruction exhibited good angular distribution (Supplementary information, Fig. S6c). Prior to visualization, all EM maps were sharpened by applying a negative B-factor that was estimated using automated procedure.115 Local resolution variations were estimated using ResMap.116
Model building and refinement
The atomic model of the APOE4-NTD/LilrB3-ECD complex was de novo built on the basis of our 3.0-Å EM maps (Supplementary information, Fig. S7). First, the predicted structures of APOE4-NTD and LilrB3-ECD from the AlphaFold database71 were fitted into the EM maps using Chimera117 and manually adjusted using COOT.118 Then, the linker sequences were adjusted, and de novo built on the basis of clear features of bulky amino acid residues. The final atomic model of the APOE4-NTD/LilrB3-ECD complex was refined against the EM maps using PHENIX119 in real space, with secondary structure restraints. Overfitting of the model was monitored by refining the model in one of the two independent maps and testing the refined model against the other120 (Supplementary information, Fig. S6d). The structures were validated through examination of the MolProbity scores and statistics of the Ramachandran plots (Supplementary information, Table S3). MolProbity scores were calculated as described.121
Ablation of LilrB3 in HMC3 cells
On the basis of the genomic sequences of LilrB3 from the NCBI database, the sgRNA plasmids were designed to target exon 3 (Supplementary information, Fig. S8a and Table S2). Using the CRISPR/Cas9 gene editing technique, Cas9 and sgRNA plasmids were co-transfected into HMC3 cells via electroporation. Monoclonal cells with CRISPR/Cas9-mediated LilrB3 deletion were screened and obtained using PCR. Ablation of both LilrB3 alleles was confirmed through DNA sequencing (Supplementary information, Fig. S8b), western blot (Supplementary information, Fig. S8c), and mass spectrometric analysis (Supplementary information, Fig. S8d).
Western blot
WT and LilrB3-null HMC3 cell samples were harvested using the RIPA Buffer (Pierce). Target protein was detected using a monoclonal antibody (11978-MM03, SinoBiological Inc.) against LilrB3 (residues 1–443). The blots were scanned by ChemiDoc XRS + and the band intensity was analyzed using Image Lab Software (Supplementary information, Fig. S8c).
Protein identification by mass spectrometry
Proteins from different cell lines were separated on SDS-PAGE gels. The gel bands of interest (~70 kDa) were excised, reduced using 5 mM dithiothreitol, and alkylated using 11 mM iodoacetamide. The gel was digested using sequencing grade modified trypsin in 50 mM ammonium bicarbonate at 37 °C overnight. The peptides were extracted twice using 0.1% trifluoroacetic acid (TFA) in 50% acetonitrile aqueous solution for 30 min. Extracts were then centrifuged in a SpeedVac (Thermo Fisher) to reduce the volume. Tryptic peptides were redissolved in 20 μL 0.1% TFA and analyzed using LC-MS/MS.
For LC-MS/MS analysis, the peptides were separated using 85-min gradient elution at a flow rate of 0.30 μL/min and a Thermo-Dionex Ultimate 3000 HPLC system, which was directly interfaced with a Thermo Scientific Q Exactive mass spectrometer. The analytical column was a fused silica capillary column (75 μm ID, 150 mm length; Upchurch, Oak Harbor, WA, USA) packed with C18 resin (300 Å, 5 μm, Varian, Lexington, MA, USA). The mobile phase A consists of 0.1% formic acid. The mobile phase B consists of 80% acetonitrile and 0.1% formic acid. The Q Exactive mass spectrometer was in the data-dependent acquisition mode using Xcalibur 2.2 software. A single full-scan mass spectrum in the orbitrap (300–1800 m/z, 70,000 resolution) was followed by 20 data-dependent MS/MS scans at 27% normalized collision energy (HCD).
The MS/MS spectra from each LC-MS/MS run were searched against the above LilrB3 sequence using an in-house Proteome Discoverer (Version PD1.4, Thermo-Fisher Scientific, USA). The search criteria were as follows: full tryptic specificity required; two missed cleavages allowed; carbamidomethyl (C) set as the fixed modification; the oxidation (M) set as the variable modification; precursor ion mass tolerances set at 20 ppm for all MS acquired in an orbitrap mass analyzer; and the fragment ion mass tolerance set at 0.02 Da for all MS2 spectra. The peptide FDR was calculated using Percolator provided by PD. When the q value was smaller than 1%, the peptide spectrum match (PSM) was considered to be correct.
RNA preparation and RNA-seq library construction
WT and LilrB3-null HMC3 cells, recovered from frozen stock vials on the same day, were cultured in DMEM (Cat# 11960051, Thermo Fisher) supplied with NEAA (Cat# 11140050, Thermo Fisher), Glumax (Cat# 35050061, Thermo Fisher) and 10% FBS (Cat# 10099-141 C, Gibco). Each of the three full-length APOE proteins purified from the same batch under identical conditions was concentrated and diluted to 0.24 mg/mL (~7 μM) using PBS. The APOE proteins and the PBS control were 10-fold diluted using the culture medium to make the APOE treatment solution and the PBS control treatment solution. The final APOE concentration was 24 μg/mL (~0.7 μM). Each treatment by APOE or PBS control was applied to three culture plates (as three replicates). 36 h after the treatment, the cells were harvested using TRIzol® and the total RNA was extracted following the manufacturer’s instructions. Roughly 20 μg total RNA was extracted from each replicate. The RNA was subjected to RNA-seq analysis using MGI-2000 system (Beijing Genomics Institute, China). In short, mRNA was enriched using poly(T) oligo-attached magnetic beads and reverse-transcribed to double-stranded cDNA, which was then subjected to adaptor ligation for library construction. Sequencing was performed on the completed library.122
RT-PCR and qPCR
Total RNAs were extracted as described in RNA preparation. RT-PCR was performed using the PrimeScript™ RT reagent Kit with gDNA Eraser (Takara) according to the manufacturer’s instructions. Briefly, 100 ng of RNA was used in each reaction for reverse transcription. qPCR analysis was performed using a SYBR Select Master Mix kit on ABi7500 real-time PCR system (Life Technologies). Transcript levels were normalized to ACTIN for gene expression. Primer sequences are listed in Supplementary information, Table S2. The qPCR results were analyzed using the comparative threshold cycle (ΔΔCt) method.123
Processing of RNA-seq data
The quality of the RNA-seq library was evaluated using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/); the adapters were removed using Cutadapt.124 Then, clean reads were mapped to the rRNA database (from NCBI RefSeq database) using Bowtie2.125 The unmapped reads were then mapped to the human genome sequences (UCSC genome build hg38) using STAR.126 Finally, a gene count matrix was generated using featureCounts with the options “-M –p –t exon –g gene_id”.127
We used edgeR128 to identify DEGs between the treatment and control. The significance cutoff was set at the FC value >1.5 and FDR < 0.05. When defining dose-dependent genes, we first selected DEGs treated with APOE4, and then imposed the criteria that the FC values by APOE4 and APOE2 treatments are the largest and the smallest, respectively.
Data availability
The atomic coordinates of the APOE4–LilrB3 complex have been deposited in the Protein Data Bank with the accession code 8GRX. The EM map for the APOE4–LilrB3 complex has been deposited in the EMDB with the accession code EMD-34216.
References
Holtzman, D. M., Herz, J. & Bu, G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring. Harb. Perspect. Med. 2, a006312 (2012).
Colton, C. A., Brown, C. M. & Vitek, M. P. Sex steroids, APOE genotype and the innate immune system. Neurobiol. Aging 26, 363–372 (2005).
Huang, Y. & Mahley, R. W. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 72 Pt A, 3–12 (2014).
Weisgraber, K. H., Rall, S. C. Jr. & Mahley, R. W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 256, 9077–9083 (1981).
Zannis, V. I. et al. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J. Lipid Res. 23, 911–914 (1982).
Huang, Y. Mechanisms linking apolipoprotein E isoforms with cardiovascular and neurological diseases. Curr. Opin. Lipidol. 21, 337–345 (2010).
Chen, Y., Strickland, M. R., Soranno, A. & Holtzman, D. M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 109, 205–221 (2021).
Arnon, R., Sehayek, E., Vogel, T. & Eisenberg, S. Effects of exogenous apo E-3 and of cholesterol-enriched meals on the cellular metabolism of human chylomicrons and their remnants. Biochim. Biophys. Acta 1085, 336–342 (1991).
Krimbou, L. et al. Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J. Lipid Res. 45, 839–848 (2004).
Avila, E. M., Holdsworth, G., Sasaki, N., Jackson, R. L. & Harmony, J. A. Apoprotein E suppresses phytohemagglutinin-activated phospholipid turnover in peripheral blood mononuclear cells. J. Biol. Chem. 257, 5900–5909 (1982).
Kowal, R. C., Herz, J., Goldstein, J. L., Esser, V. & Brown, M. S. Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc. Natl. Acad. Sci. USA 86, 5810–5814 (1989).
Sehayek, E. & Eisenberg, S. Mechanisms of inhibition by apolipoprotein C of apolipoprotein E-dependent cellular metabolism of human triglyceride-rich lipoproteins through the low density lipoprotein receptor pathway. J. Biol. Chem. 266, 18259–18267 (1991).
Fagan, A. M., Bu, G., Sun, Y., Daugherty, A. & Holtzman, D. M. Apolipoprotein E-containing high density lipoprotein promotes neurite outgrowth and is a ligand for the low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 30121–30125 (1996).
Li, X., Kypreos, K., Zanni, E. E. & Zannis, V. Domains of apoE required for binding to apoE receptor 2 and to phospholipids: implications for the functions of apoE in the brain. Biochemistry 42, 10406–10417 (2003).
Guttman, M., Prieto, J. H., Croy, J. E. & Komives, E. A. Decoding of lipoprotein-receptor interactions: properties of ligand binding modules governing interactions with apolipoprotein E. Biochemistry 49, 1207–1216 (2010).
Ji, Z. S., Dichek, H. L., Miranda, R. D. & Mahley, R. W. Heparan sulfate proteoglycans participate in hepatic lipaseand apolipoprotein E-mediated binding and uptake of plasma lipoproteins, including high density lipoproteins. J. Biol. Chem. 272, 31285–31292 (1997).
Burgess, J. W., Gould, D. R. & Marcel, Y. L. The HepG2 extracellular matrix contains separate heparinase- and lipid-releasable pools of ApoE. Implications for hepatic lipoprotein metabolism. J. Biol. Chem. 273, 5645–5654 (1998).
Gonzales, J. C., Gordts, P. L., Foley, E. M. & Esko, J. D. Apolipoproteins E and AV mediate lipoprotein clearance by hepatic proteoglycans. J. Clin. Invest. 123, 2742–2751 (2013).
Weisgraber, K. H., Innerarity, T. L. & Mahley, R. W. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J. Biol. Chem. 257, 2518–2521 (1982).
Innerarity, T. L., Weisgraber, K. H., Arnold, K. S., Rall, S. C. Jr. & Mahley, R. W. Normalization of receptor binding of apolipoprotein E2. Evidence for modulation of the binding site conformation. J. Biol. Chem. 259, 7261–7267 (1984).
Mahley, R. W., Huang, Y. & Rall, S. C. Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J. Lipid Res. 40, 1933–1949 (1999).
Schumacher, K., Maerker-Alzer, G. & Wehmer, U. A lymphocyte-inhibiting factor isolated from normal human liver. Nature 251, 655–656 (1974).
Fujii, D. K. & Edgington, T. S. Direct suppression of lymphocyte induction by the immunoregulatory human serum low density lipoprotein, LDL-In. J. Immunol. 124, 156–160 (1980).
Hui, D. Y. & Harmony, J. A. Phosphatidylinositol turnover in mitogen-activated lymphocytes. Suppression by low-density lipoproteins. Biochem. J. 192, 91–98 (1980).
Terkeltaub, R. A., Dyer, C. A., Martin, J. & Curtiss, L. K. Apolipoprotein (apo) E inhibits the capacity of monosodium urate crystals to stimulate neutrophils. Characterization of intraarticular apo E and demonstration of apo E binding to urate crystals in vivo. J. Clin. Invest. 87, 20–26 (1991).
Heun, R. et al. Apolipoprotein E allele 4 is not a sufficient or a necessary predictor of the development of Mild Cognitive Impairment. Eur. Psychiatry 25, 15–18 (2010).
Barger, S. W. & Harmon, A. D. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388, 878–881 (1997).
Colton, C. A., Brown, C. M., Czapiga, M. & Vitek, M. P. Apolipoprotein-E allele-specific regulation of nitric oxide production. Ann. N. Y. Acad. Sci. 962, 212–225 (2002).
Colton, C. A. et al. APOE and the regulation of microglial nitric oxide production: a link between genetic risk and oxidative stress. Neurobiol. Aging 23, 777–785 (2002).
van den Elzen, P. et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 437, 906–910 (2005).
Borg, N. A. et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 448, 44–49 (2007).
Ophir, G. et al. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-kappaB signaling cascade. Neurobiol. Dis. 20, 709–718 (2005).
Enzinger, C. et al. Accelerated evolution of brain atrophy and “black holes” in MS patients with APOE-epsilon 4. Ann. Neurol. 55, 563–569 (2004).
Shi, J., Zhao, C. B., Vollmer, T. L., Tyry, T. M. & Kuniyoshi, S. M. APOE epsilon 4 allele is associated with cognitive impairment in patients with multiple sclerosis. Neurology 70, 185–190 (2008).
Savettieri, G. et al. Gender-related effect of clinical and genetic variables on the cognitive impairment in multiple sclerosis. J. Neurol. 251, 1208–1214 (2004).
Shi, Y. & Holtzman, D. M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772 (2018).
Mahley, R. W. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 94, 739–746 (2016).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993).
Strittmatter, W. J. et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 1977–1981 (1993).
Corder, E. H. et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7, 180–184 (1994).
Yang, D. S., Smith, J. D., Zhou, Z., Gandy, S. E. & Martins, R. N. Characterization of the binding of amyloid-beta peptide to cell culture-derived native apolipoprotein E2, E3, and E4 isoforms and to isoforms from human plasma. J. Neurochem. 68, 721–725 (1997).
Castellano, J. M. et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 3, 89ra57 (2011).
Deane, R. et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013 (2008).
Verghese, P. B. et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. USA 110, E1807–E1816 (2013).
Rogers, J., Luber-Narod, J., Styren, S. D. & Civin, W. H. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 9, 339–349 (1988).
Griffin, W. S. et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 86, 7611–7615 (1989).
Combs, C. K., Johnson, D. E., Karlo, J. C., Cannady, S. B. & Landreth, G. E. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 20, 558–567 (2000).
Hickman, S. E., Allison, E. K. & El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360 (2008).
Marsh, S. E. et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl. Acad. Sci. USA 113, E1316–E1325 (2016).
Heppner, F. L., Ransohoff, R. M. & Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372 (2015).
Paolicelli, R. C. et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Wang, C. et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 367, 688–694 (2020).
Bartels, T., De Schepper, S. & Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 370, 66–69 (2020).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017).
Lucas, S. M., Rothwell, N. J. & Gibson, R. M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147, S232–S240 (2006).
Guillot-Sestier, M. V. et al. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 85, 534–548 (2015).
Shi, Y. et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017).
Gosselin, D. et al. An environment-dependent transcriptional network specifies human microglia identity. Science 356 (2017).
Deng, M. et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 562, 605–609 (2018).
Bailey, C. C., DeVaux, L. B. & Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem. 290, 26033–26042 (2015).
Atagi, Y. et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050 (2015).
Deczkowska, A., Amit, I. & Schwartz, M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 21, 779–786 (2018).
Perry, V. H. & Gordon, S. Macrophages and microglia in the nervous system. Trends Neurosci. 11, 273–277 (1988).
Barkal, A. A. et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 19, 76–84 (2018).
Djurisic, M. et al. PirB regulates a structural substrate for cortical plasticity. Proc. Natl. Acad. Sci. USA 110, 20771–20776 (2013).
Kim, T. et al. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341, 1399–1404 (2013).
Morikawa, M. et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol. Dis. 19, 66–76 (2005).
Bielefeld-Sevigny, M. AlphaLISA immunoassay platform- the “no-wash” high-throughput alternative to ELISA. Assay Drug Dev. Technol. 7, 90–92 (2009).
Chen, J., Li, Q. & Wang, J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. USA 108, 14813–14818 (2011).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Mathys, H. et al. Temporal Tracking Of Microglia Activation In Neurodegeneration At Single-cell Resolution. Cell Rep. 21, 366–380 (2017).
Gillespie, M. et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 50, D687–D692 (2022).
Smith, S., Weston, S., Kellam, P. & Marsh, M. IFITM proteins-cellular inhibitors of viral entry. Curr. Opin. Virol. 4, 71–77 (2014).
Iaccarino, H. F. et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235 (2016).
Perng, Y. C. & Lenschow, D. J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 16, 423–439 (2018).
Butturini, E., Boriero, D., Carcereri de Prati, A. & Mariotto, S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 669, 22–30 (2019).
Alzheimer, A. Uber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin. 64, 146–148 (1907).
Cahill, S. WHO’s global action plan on the public health response to dementia: some challenges and opportunities. Aging Ment. Health 24, 197–199 (2020).
(2022), A. s. A. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement 18, 700-789 (2022).
Polvikoski, T. et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N. Engl. J. Med. 333, 1242–1247 (1995).
Chow, D., He, X., Snow, A. L., Rose-John, S. & Garcia, K. C. Structure of an extracellular gp130 cytokine receptor signaling complex. Science 291, 2150–2155 (2001).
Boulanger, M. J., Chow, D. C., Brevnova, E. E. & Garcia, K. C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 300, 2101–2104 (2003).
Klesney-Tait, J., Turnbull, I. R. & Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 7, 1266–1273 (2006).
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C. & Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 91, 328–340 (2016).
Lane-Donovan, C. & Herz, J. ApoE, ApoE receptors, and the synapse in Alzheimer’s Disease. Trends Endocrinol. Metab. 28, 273–284 (2017).
Deng, M. et al. Leukocyte immunoglobulin-like receptor subfamily B: therapeutic targets in cancer. Antib. Ther. 4, 16–33 (2021).
Ruiz, J. et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 46, 1721–1731 (2005).
Fitz, N. F. et al. Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat. Commun. 12, 3416 (2021).
Hatters, D. M., Budamagunta, M. S., Voss, J. C. & Weisgraber, K. H. Modulation of apolipoprotein E structure by domain interaction: differences in lipid-bound and lipid-free forms. J. Biol. Chem. 280, 34288–34295 (2005).
Iannucci, J., Sen, A. & Grammas, P. Isoform-specific effects of apolipoprotein E on markers of inflammation and toxicity in brain glia and neuronal cells in vitro. Curr. Issues Mol. Biol. 43, 215–225 (2021).
Chen, S. et al. Isoform-specific effects of apolipoprotein E on secretion of inflammatory mediators in adult rat microglia. J. Alzheimers Dis. 7, 25–35 (2005).
Lin, Y. T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1294 (2018).
Wilson, C., Wardell, M. R., Weisgraber, K. H., Mahley, R. W. & Agard, D. A. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 252, 1817–1822 (1991).
DEL RIO-HORTEGA, P. El tercer elemento de los centros nerviosos. I. La microglia en estado normal II. Intervencion de la microglia en los procesos patologicos. HI. Naturaleza probable de la microglia. Boll Socieded. Esp. Biol. 9, 69–120 (1919).
McGeer, P. L., Itagaki, S., Tago, H. & McGeer, E. G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 79, 195–200 (1987).
Deczkowska, A. et al. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081 (2018).
Krasemann, S. et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017).
Friedman, B. A. et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847 (2018).
Akiyama, H., Ikeda, K., Katoh, M., McGeer, E. G. & McGeer, P. L. Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J. Neuroimmunol. 50, 195–201 (1994).
Roy, E. R. et al. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid beta plaques. Immunity 55, 879–894.e6 (2022).
Jin, M. et al. Type-I-interferon signaling drives microglial dysfunction and senescence in human iPSC models of Down syndrome and Alzheimer’s disease. Cell Stem Cell 29, 1135–1153.e8 (2022).
Mathur, V. et al. Activation of the STING-dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron 96, 1290–1302.e6 (2017).
Guo, L., LaDu, M. J. & Van Eldik, L. J. A dual role for apolipoprotein e in neuroinflammation: anti- and pro-inflammatory activity. J. Mol. Neurosci. 23, 205–212 (2004).
Brown, C. M. et al. Apolipoprotein E isoform mediated regulation of nitric oxide release. Free Radic. Biol. Med. 32, 1071–1075 (2002).
Vitek, M. P., Brown, C. M. & Colton, C. A. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360 (2009).
Zhu, Y. et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60, 559–569 (2012).
Roses, A. D. & Saunders, A. M. APOE is a major susceptibility gene for Alzheimer’s disease. Curr. Opin. Biotechnol. 5, 663–667 (1994).
Westerlund, J. A. & Weisgraber, K. H. Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J. Biol. Chem. 268, 15745–15750 (1993).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Chen, S. X. et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Swint-Kruse, L. & Brown, C. S. Resmap: automated representation of macromolecular interfaces as two-dimensional networks. Bioinformatics 21, 3327–3328 (2005).
Pettersen, E. F. et al. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Amunts, A. et al. Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–1489 (2014).
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 6, 1–9 (2017).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Acknowledgements
We thank Li Huang, Xiaojuan Wang, and Zhipeng Jiang of the Westlake University Cryo-EM facility for technical assistance, the High-Performance Computing Center, Mass Spectrometry & Metabolomics Core Facility of Westlake University for support and Meng Han of Tsinghua Mass Spectrometry center for technical assistance. This work was supported by funds from the National Key R&D Program of China (2020YFA0509300 to Y.S.), the National Natural Science Foundation of China (81920108015 to Y.S.), the Key R&D Program of Zhejiang Province (2020C04001 to Y.S.), and Start-up funds from Westlake University.
Author information
Authors and Affiliations
Contributions
J.Z. and Y.S. conceived the project. J.Z., Y.W., M.Y., C.J., D.J., and K.J. performed the experiments. J.Z., Y.W., G.H, C.J., Y.Z., and Y.S. analyzed the data. J.Z., Y.W., and G.H. prepared the figures. J.Z., G.H., and Y.S. wrote the manuscript. Y.S. guided the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhou, J., Wang, Y., Huang, G. et al. LilrB3 is a putative cell surface receptor of APOE4. Cell Res 33, 116–130 (2023). https://doi.org/10.1038/s41422-022-00759-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-022-00759-y
This article is cited by
-
Roles of ApoE4 on the Pathogenesis in Alzheimer’s Disease and the Potential Therapeutic Approaches
Cellular and Molecular Neurobiology (2023)
