
Abstract
Error-free mitosis depends on accurate chromosome attachment to spindle microtubules, powered congression of those chromosomes, their segregation in anaphase, and assembly of a spindle midzone at mitotic exit. The centromere-associated kinesin motor CENP-E, whose binding partner is BubR1, has been implicated in congression of misaligned chromosomes and the transition from lateral kinetochore-microtubule association to end-on capture. Although previously proposed to be a pseudokinase, here we report the structure of the kinase domain of Drosophila melanogaster BubR1, revealing its folding into a conformation predicted to be catalytically active. BubR1 is shown to be a bona fide kinase whose phosphorylation of CENP-E switches it from a laterally attached microtubule motor to a plus-end microtubule tip tracker. Computational modeling is used to identify bubristatin as a selective BubR1 kinase antagonist that targets the αN1 helix of N-terminal extension and αC helix of the BubR1 kinase domain. Inhibition of CENP-E phosphorylation is shown to prevent proper microtubule capture at kinetochores and, surprisingly, proper assembly of the central spindle at mitotic exit. Thus, BubR1-mediated CENP-E phosphorylation produces a temporal switch that enables transition from lateral to end-on microtubule capture and organization of microtubules into stable midzone arrays.
Similar content being viewed by others


Introduction
Accurate chromosome segregation in mitosis depends on the spindle assembly checkpoint (SAC) that prevents premature exit from mitosis.1 The SAC plays a vital role in maintaining chromosome stability and avoiding aneuploidy as it inhibits advance to anaphase until proper MT-kinetochore attachment is established at every kinetochore. The SAC produces a four-protein inhibitory complex consisting of Bub3, Mad2, BubR1 and Cdc20 which binds to the APC/CCdc20 E3 ubiquitin ligase and inhibits its ubiquitination of cyclin B and securin.2
BubR1 shares significant similarity in both primary sequence and protein domain architecture with Bub1 including a C-terminal kinase domain.2,3 BubR1 kinase activity has been reported to be important for the SAC in both Xenopus laevis egg extracts and human cells.4,5 Replacing endogenous BubR1 with a kinase dead mutant form in Drosophila melanogaster,6 Xenopus laevis egg extracts,7 or human cells8 results in chromosome misalignment, which is consistent with early findings that BubR1 is essential for stable kinetochore-microtubule attachments via interacting with a plus end-directed kinetochore motor CENP-E.9,10,11 The mitotic kinesin CENP-E has also been reported to be an activator of BubR1 kinase, and CENP-E-dependent BubR1 autophosphorylation in response to spindle microtubule capture acts to enhance chromosome alignment and the SAC.5,12 During the prometaphase-metaphase transition, the motility of CENP-E motor has been reported to convert from a lateral mode into an end-on mode, and to maintain its association with both the assembling and disassembling microtubule plus ends during chromosome oscillation.13 CENP-E exhibits a dynamic distribution from kinetochore to the midzone of central spindle during metaphase-anaphase transition.14 However, the mechanism underlying the switch of CENP-E from lateral to end-on attachment to spindle microtubule and its relationship to BubR1 remain unknown.
Despite prior experimental evidence that BubR1 has kinase activity, this has been highly controversial, as a widely held view is that BubR1 is an unusual pseudokinase containing modules to interact with Bub1, Bub3, PP2A-B56 and KNL.15,16,17,18,19 To better understand the regulatory mechanism of the BubR1-CENP-E signaling pathway and to explore new BubR1-specific chemical modulators, we solved the crystal structures of Drosophila melanogaster BubR1 kinase domain in apo- and ADP-bound states, which reveal an active conformation capable of catalyzing phosphotransfer. Based on the structure, we discovered a novel chemical inhibitor of the BubR1 kinase, bubristatin, which harnesses an interaction between αN1 of N-terminal extension and αC of BubR1 kinase domain. We then used bubristatin as a small-molecule tool to probe BubR1-CENP-E signaling and identified CENP-E as a bona fide substrate of BubR1 in mitosis. Remarkably, the BubR1-elicited phosphorylation of CENP-E converted it (from lateral motility) to a plus-end tracker. Importantly, the phosphorylation facilitates the interaction between CENP-E and PRC1 to establish stable midzone arrays in metaphase-anaphase transition. Thus, phosphorylation of CENP-E by BubR1 provides a spatiotemporal cue for central spindle assembly.
Results
The structure of the BubR1 kinase
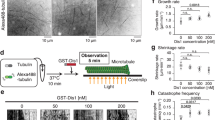
To obtain structural insights into BubR1 actions in mitotic regulation, a C-terminal region of Drosophila melanogaster BubR1 (DmBubR1c; aa 1124–1460, based on sequence alignment with Bub1, Supplementary information, Fig. S1a) that includes the kinase domain was crystalized and its structure was solved in apo- (1.85 Å) and 2Mg2+•ADP-complexed (1.95 Å) states, respectively (Fig. 1a; Supplementary information, Table S1). The overall architecture of DmBubR1c exhibits a canonical kinase fold with an unusual N-terminal extension (aa 1124–1171) attached at the flank, structurally similar to the configuration of human Bub1 C-terminal fragment (HsBub1c, PDB entry: 4R8Q).20 Like other Ser/Thr protein kinases, the kinase core of DmBubR1c is comprised of two parts, an N-lobe and a C-lobe, between which a deep cleft functions as the ATP binding site. An unusual N-terminal extension (yellow in Fig. 1a) folds into two tandem α-helices (αN1 and αN2) followed by two β-strands (βN1 and βN2), wrapping around the kinase domain N-lobe with its N-terminus anchored near the activation segment of kinase domain C-lobe. The anchoring is based on hydrophobic interactions between the aromatic ring of Phe1129 and the hydrophobic pocket formed by residues in the activation segment, which further enhances the interactions between the kinase domain and N-terminal extension and stabilizes the conformation of the activation segment (Supplementary information, Fig. S1b).

Both fly and human BubR1 kinase domains are active in phosphotransfer. a Cartoon presentation of the overall structure of DmBubR1c in complex with ADP and Mg2+. N-terminal extension (yellow) is highlighted. Bound ADP is shown in stick presentation. Mg2+ ions (green) are shown in spheres. N-lobe and C-lobe are labeled accordingly. b Detailed binding mode of ADP in the nucleotide-binding pocket of DmBubR1c. The nucleotides and essential residues involved are shown as sticks. Hydrogen bonds and salt bridges are shown in dashed lines. Coodinated waters are shown in red spheres. c The interactions between key residues Lys1204, Glu1213 and Asp1326. Left: apo state; Right: ADP-bound state. d, e The kinetics curves of DmBubR1 (d) and BubR1 (e) kinase activity. Purified DmBubR1 (d) or BubR1 (e) wild type and mutants were subjected to in vitro kinase assay using casein as substrate. Qualities and loading of recombinant BubR1 proteins were judged by Coomassie Brilliant Blue (CBB) staining as shown in insets. Data represent mean ± SEM from three independent experiments. f SAR study guided by molecule modeling of BRT-1-BubR1 interfaces (both BubR1 and DmBubR1 kinases). BubR1 wild type or mutants were subjected to in vitro phosphorylation using casein as substrate. Qualities and loading of recombinant BubR1 proteins were judged by CBB staining from same gels as shown in insets. Approximately, 50 ng BubR1 kinase (wild type or mutants) incubated with 1 µg casein were resolved and visualized in this SDS-PAGE gel. g Statistical analyses of kinase activity of BubR1 wild type or mutants in (f). Data represent mean ± SEM from three independent experiments. Statistical significance was tested by two-sided t-test (**p < 0.01). See also Supplementary information, Fig. S1 and Table S1
To study conformational changes of DmBubR1c induced by ligand binding, we co-crystallized it with ADP, in the presence of Mg2+. Structure determination led to clear electron densities of an ADP molecule captured in the binding cleft, as well as two Mg2+ ions coordinated to the ADP (Fig. 1b). As expected, the binding mode of ADP to DmBubR1c is the same as that in other Ser/Thr kinases such as Bub1 (Fig. 1b), while the conformational changes induced by ADP binding are, unexpectedly, limited (r.m.s. deviation of 331 Cα is only 0.36 Å). A notable difference involves Lys1204 and Glu1213, between which a hydrogen bond shrinks from 3.3 Å to 2.7 Å, indicating an enhancement of the interaction between those two residues (Fig. 1c). Actually, strong interaction between the counterparts of Lys1204 and Glu1213 in Ser/Thr kinase family will restrain the side chain of the lysine residue at a proper position to increase the affinity for ADP/ATP. In addition, other conserved residues involved such as Glu1251, Asp1305, Asn1306 and Asp1326 are also essential to the binding of ADP or its coordinated Mg2+ (Fig. 1b, c).
Careful comparison between DmBubR1 and other classical S/T kinase structures revealed that DmBubR1 adopts an active conformation (Supplementary information, Fig. S1c). The salt bridge formed between two highly conserved residues, the Lys at β3 and the Glu at αC helix, is thought to be a signature of an active kinase.21,22 Similarly, the corresponding amino acids Lys1204 and Glu1213 in DmBubR1c form a salt bridge, so that Lys1204 points to and stabilizes ATP through interaction with the α- and β-phosphates. In addition, in the DFG motif in the activation loop of DmBubR1, the conserved H-bond forms between the carbonyl of Phe1327 and the amide of Ser1330 to help maintain the catalysis-related residue Asp1326 at a favorable position for Mg2+ binding. Importantly, like other active kinases, the four hydrophobic residues (F1327, I1236, C1217 and H1299) stack in a spatially linear order and form a conserved “regulatory spine”, which stabilizes the kinase core. Furthermore, the activation segment of DmBubR1c adopts an ordered conformation which is characteristic of active kinases21 (Supplementary information, Fig. S1c). Thus, all these features suggest that DmBubR1 adopts an active conformation capable of catalyzing phosphotransfer.
Consistent with structural analyses, enzymatic assays confirmed that DmBubR1 is an active kinase and that Lys1204 and Asp1326 are critical for its kinase activity (Fig. 1d). The catalytic activities of DmBubR1K1204A and DmBubR1D1326A were lower than DmBubR1WT (kcat/Km value for DmBubR1WT is 5.6-fold higher than DmBubR1K1204A) (Supplementary information, Fig. S1d). Although we failed in efforts to crystallize human BubR1, mutagenesis studies were performed on Lys795 and Asp911 of HsBubR1, the counterparts of Lys1204 and Asp1326, suggesting their functional conservation. Indeed, human BubR1WT was found to have catalytic activity and our enzymatic assays confirmed that Lys795 and Asp911 are critical for that activity (Fig. 1e). The catalytic activities of BubR1K795A and BubR1D911A were much lower than that of BubR1WT (kcat/Km value for BubR1WT is 5.1-fold higher than BubR1K795A and 10-fold higher than BubR1D911A) (Supplementary information, Fig. S1e). Consistently, in vitro phosphorylation experiments confirmed that mutations of the ATP-binding sites in DmBubR1 and human BubR1 disrupted their kinase activities (Fig. 1f, g; **p < 0.01), while protein stability was not altered (as judged by the Commassie Brilliant Blue (CBB) staining). Thus, we conclude that both DmBubR1 and human BubR1 actively catalyze phosphate transfer to substrates.
To explore functional significance of the phosphotransfer activity of BubR1 in chromosome segregation, we used live cell imaging of HeLa cells coexpressing BubR1 siRNA, mCherry-H2B, together with RNAi-resistant forms of BubR1 wild type and mutants. Suppression of BubR1 via siRNA resulted in premature anaphase with lagging chromosomes (Supplementary information, Fig. S1f). Expression of wild-type BubR1 but not ATP-binding deficient mutants (BubR1K795A and BubR1D911A) rescued the aberrant metaphase-anaphase transition (Supplementary information, Fig. S1f). Thus, human BubR1 possesses all structural and enzymatic features of an active Ser/Thr kinase which are essential for accurate cell division.
Discovery of the BubR1 kinase inhibitor bubristatin
We identified small molecules that specifically inhibit BubR1 kinase activity by performing a virtual screening of a mitotic inhibitor library consisting of 153 diterpene compounds that elicit chromosome misalignment phenotype. The selected diterpene compounds were also characterized by the chemical property of diterpenoid ring in perturbation of hydrophobic interaction.23 Our use of this strategy previously identified syntelin as a selective inhibitor of CENP-E, a key kinetochore kinesin.24 Similar, computational molecular docking analyses identified one kaurene diterpenoid, which we named bubristatin-1 (or BRT-1; Fig. 2a) after its inhibition of BubR1 activity. Multiple attempts to generate co-crystals of Hs/DmBubR1 with BRT-1 did not succeed. Nevertheless, modeling of BubR1c (residues 720–1050) using DmBubR1c structure as a template produced a computational structural model highly similar to that of DmBubR1c (rmsd = 0.72 Å) (Supplementary information, Fig. S2a).

BRT-1 inhibits BubR1 kinase activity in an ATP non-competitive fashion. a Chemical structure of bubristatin-1 (BRT-1). b Cartoon representation of BRT-1 (yellow and red for carbon and oxygen, respectively) and ADP docked onto the BubR1c model. Potential residues for BRT-1 and ADP binding are shown as sticks. The αN1 helix (cyan) and the αC helix (green) are labeled. c BubR1 kinase was subjected to in vitro phosphorylation using casein as substrate in the presence of BRT-1 at different concentration. Upper panel is SDS-PAGE gel stained by CBB and lower panel is 32P autoradiograph from the same reaction and the same gel. d Statistical analyses of dose-responsive curve of BubR1 kinase activity towards BRT-1 in (c). Data represent mean ± SEM from three independent experiments. e Kinetics of BubR1 kinase activity in response to increased concentration of BRT-1 using casein as substrate. Data represent mean ± SEM from three independent experiments. f Kinetics of BubR1 kinase activity in the presence of BRT-1 with increased ATP concentration. Data represent mean ± SEM from three independent experiments. g Aliquots of BubR1 wild type and mutants were purified and subjected to in vitro phosphorylation using casein as substrate. Qualities and loading of recombinant BubR1 proteins were judged by CBB staining of a representative gel from the same reaction. Approximately, 50 ng BubR1 kinase (wild type or mutants) incubated with 1 µg casein were resolved and visualized in this SDS-PAGE gel. h Aliquots of BubR1 wild type and mutants were purified and subjected to in vitro phosphorylation in the presence of BRT-1 (3 μM). Qualities and loading of recombinant BubR1 proteins from the same gel were judged by CBB staining. Approximately, 50 ng BubR1 kinase (wild type or mutants) incubated with 1 µg casein were resolved and visualized in this SDS-PAGE gel. See also Supplementary information, Fig. S2 and Table S2
Computational molecular docking analyses suggested that BRT-1 would bind in the hydrophobic cavity between the αN1 of N-terminal extension and the so-called αC of BubR1c (Fig. 2b). Unlike other kinase inhibitors which function by competing with ATP, the BRT-1-binding site is far away from the ATP binding pocket (Fig. 2b). The clustering analyses showed that BRT-1 exhibits selectivity to BubR1 compared with Bub1 (Supplementary information, Fig. S2b, c) despite sequence and structural similarities (Supplementary information, Fig. S2a). In vitro phosphorylation assays and quantitative analysis demonstrated that BRT-1 inhibited BubR1 kinase activity in a dose-dependent manner with an IC50 of 1.2 µM (Fig. 2c, d). BRT-1 is relatively selective as a BubR1 inhibitor, as it exhibits no apparent inhibition of other mitotic kinases, including Bub1 (even at concentrations over 5 µM) in in vitro kinase assays (Supplementary information, Table S2). Use of phosphorylation of the CDK1 substrate RCC1 as a readout, immunoblotting analyses confirmed the BRT-1 did not inhibit CDK1 (Supplementary information, Fig. S2d).
We then carried out enzymatic assays and measured the kinetic parameters of recombinant BubR1 kinase toward two substrates, casein and ATP. The maximal velocity of phosphorylation by BubR1 was inhibited by BRT-1 in a dose-dependent manner (Fig. 2e, f). BRT-1 not only reduced the catalytic activity of BubR1, but also decreased the affinity between BubR1 and its substrates. kcat/Km of BubR1 was dramatically reduced in a dose-dependent manner by BRT-1 (Supplementary information, Fig. S2e, f). Thus, BRT-1 reduces BubR1 activity to catalyze phosphate transfer to its substrates.
To identify the binding sites of the substrate ATP and BRT-1 inhibitor, we carried out mutational analyses of human BubR1 followed by in vitro phosphorylation assays (Fig. 2g, h). Molecular modeling suggested residues which might be involved in BRT-1 binding. These were divided into three groups (marked in Supplementary information, Fig. S1a): (i) Trp722, Ser724, Arg727, Arg728 and Leu731 in αN1 helix; (ii) Gln799, Pro800, Val801, Pro802 and Trp803 in αC helix; (iii) Tyr914 next to the DFG motif. Three residues around the active site, Lys795, Asp882 and Asp911, were determined to be critical for BubR1 kinase activity: conversion of any of these to alanine diminished 32P incorporation into casein as a substrate or BubR1 auto-phosphorylation, both in the absence (Fig. 2g, lanes 6, 12 and 13) or presence of BRT-1 (Fig. 2h, lanes 6, 12 and 13).
Our molecular docking analyses also suggested that Arg727 and Arg728 from the BubR1 αN1 helix contribute their side chain guanidino groups for capture of the hydroxyl groups of BRT-1, while Val801, Pro802 and Trp803 from αC helix may form a hydrophobic cavity for BRT-1 binding (Fig. 2b). While converting Arg727, Arg728, Val801, Pro802 or Trp803 to alanine did not affect ATP binding and BubR1 kinase activity (Fig. 2g, lanes 4, 9-11), each of these substitutions abrogated the ability of BRT-1 to inhibit that activity (Fig. 2h, lanes 4, 9-11). Other residues surrounding potential inhibitor-binding sites did not affect either BubR1 kinase activity or BRT-1-dependent inhibition of that activity (Fig. 2h). Binding of BRT-1 for BubR1 was validated by incubation of soluble mitotic HeLa cell lysates with 10 μM biotinylated BRT-1, followed by binding to an avidin-affinity matrix and BRT-1 elution (illustrated in Supplementary information, Fig. S2g). This revealed that biotinylated BRT-1 bound cellular-derived BubR1, but not Bub1 (Supplementary information, Fig. S2h), and was released by addition of soluble BRT-1. Thus, BRT-1 is an inhibitor of BubR1, but not Bub1, through binding interactions within the αN1 and αC helices.
BubR1 function in mitosis identified with bubristatin
BubR1 has been implicated as an essential component for proper attachment of chromosomes to the spindle.25 To define how inhibition of BubR1 kinase activity affected its function in mitosis, we treated asynchronized HeLa cells expressing mCherry-tubulin and GFP-H2B with 3 μM BRT-1 or with siRNA to BubR1 and followed mitotic progression using real-time imaging with deconvolution microscopy. Untreated cells took 29 ± 2 min to progress from prophase (nuclear envelope breakdown; NEBD) to anaphase onset (identified by sepration of sister chromatids) (Fig. 3a, top panel). Efficient suppression of BubR1 via siRNA (Supplementary information, Fig. S3a) resulted in premature anaphase with lagging chromosomes (Fig. 3a, middle panel), consistent with an essential role of BubR1 in establishing stable kinetochore-microtubule attachment.25 Importantly, suppression of BubR1 kinase activity with BRT-1 produced cells with chromosomes that remained unaligned even 120 min after mitotic entry (Fig. 3a, bottom panel). Similarly, expression of a BubR1 variant deleted in its kinase domain (BubR1-ΔKD; amino acids 1–717 of BubR1) delayed mitotic advance similarly to chemical inhibition of BubR1 kinase activity (Fig. 3b; Supplementary information, Fig. S3b). Statistical analyses showed that chemical inhibition of BubR1 resulted in a significant delay in mitotic progression and a dramatic increase in aberrant anaphase cells (Fig. 3b; Supplementary information, Fig. S3c), consistent with BubR1 kinase activity playing an important role in chromosome alignment at metaphase and the metaphase-anaphase transition.

BRT-1 selectively inhibits BubR1 and perturbs chromosome segregation in mitosis. a Representative phenotypes of live HeLa cells transfected with BubR1 siRNA or treated with BRT-1. Scale bar, 10 μm. b Quantification of mitotic duration of live HeLa cells treated with DMSO or BRT-1. In addition, HeLa cells with BubR1 depleted (siBubR1-UTR), and expression of wild-type kinase (siBubR1 + WT) or kinase domain-deleted BubR1 (siBubR1 + ΔKD) were also assessed. BRT-1 was used at 3 μM. NEBD, nuclear envelope breakdown. Data represent mean ± SEM from three independent experiments. Statistical significance was tested by two-sided t-test (***p < 0.001). SiControl, n = 32; siBubR1-UTR, n = 55; BRT-1, n = 31; siBubR1 + GFP-BubR1-ΔKD, n = 61; siBubR1 + GFP-BubR1-WT, n = 45. c Quantification of mitotic phenotypes in HeLa cells expressing various GFP-BubR1 mutants and wild type. The endogenous BubR1 was suppressed by siRNA-UTR as indicated. GFP, n = 27; WT, n = 32; K795A, n = 17; D911A, n = 33; R727A/R728A, n = 16; V801A, n = 24; P802A, n = 26; W803A, n = 15. d, e Representative phenotypes of live HeLa cells transfected with BubR1 siRNA and expressed wild-type and BRT-1-resistant BubR1 in the presence of BRT-1. BRT-1 was used at 3 μM. Scale bars, 10 μm. f Quantification of mitotic phenotypes of live HeLa cells transfected with BubR1 siRNA and expressed wild-type and BRT-1-resistant BubR1 in the presence of BRT-1. BRT-1 was used at 3 μM. WT, n = 31; R727A/R728A, n = 18; V801A, n = 27; P802A, n = 34; W803A, n = 26. BRT-1-binding deficient mutants such as R727A/R728A rescued the mitotic phenotypes of BRT-1-treated cells. HeLa cells were transfected with mCherry-H2B and GFP-BubR1 (wild type and mutants) together with BubR1 siRNA (3’-UTR) to suppress endogenous BubR1 protein. Real-time imaging was performed as described in (a). See also Supplementary information, Figs. S3 and S4
BubR1 mutants deficient in BRT-1-binding were expressed in HeLa cells by transient transfection along with BubR1 siRNA (targeted to 3’-UTR) and a gene encoding mCherry-H2B. BubR1 knockdown efficiency was determined by immunoblotting (Supplementary information, Fig. S3a). Each of the BRT-1-binding deficient mutants accumulated to comparable levels judged by immunoblotting assay and each rescued the mitotic phenotypes in BubR1 siRNA transfected cells (Fig. 3c; Supplementary information, Fig. S3d, e). As predicted, BubR1 mutants whose kinase activity was not inhibited by BRT-1 rescued the mitotic phenotypes of BRT-1-treated cells, while wild-type BubR1 did not (Fig. 3d–f; Supplementary information, Fig. S3f, g). Thus, we conclude that the mitotic phenotypes in response to BRT-1 result from specific inhibition of BubR1 and not from any effect on any other mitotic kinase.
To test whether BubR1 kinase inhibiton by BRT-1 was reversible, HeLa cells were arrested in prometaphase by addition of monastrol and BRT-1. Following BRT-1 and monastrol removal with three rapid washes, chromosomes aligned to a metaphase plate within ~24 min and then progressed into anaphase(Supplementary information, Fig. S4a) with timing indistinguishable from that of cells released from monastrol alone, demonstrating that mitotic arrest due to BRT-1 treatment is also reversible. In addition, BRT-1 neither altered microtubule polymerization in vitro (Supplementary information, Fig. S4b) nor inhibited phospohorylation in cells of substrates of Aurora A (pT210-PLK1) and Aurora B (pSer7-CENP-A) kinases (Supplementary information, Fig. S4c).
Mitotic kinesin CENP-E is a bona fide substrate of BubR1
CENP-E transports misaligned chromosomes to the spindle microtubule plus-ends,24,27,28 via interacting with mitotic kinases BubR1 and Mps1.5,11,29 To further delineate the role of BubR1 kinase activity in CENP-E function, we isolated CENP-E from mitotic HeLa cells treated with or without BRT-1. Mass spectrometric analyses (not shown) suggested Ser2639 of CENP-E as a potential site for phosphorylation by BubR1. To test whether Ser2639 of CENP-E is a substrate of BubR1, we performed in vitro phosphorylation assays using an MBP-CENP-E C-terminal part fusion protein (CENP-ETAIL; aa 2131–2701). Both wild-type and S2639-mutated CENP-ETAIL (2639A) migrated at the predicted position of 99 kDa (Fig. 4a, left panel). Incubation of the fusion proteins with [γ-32P]-ATP and BubR1 resulted in the incorporation of 32P into wild-type CENP-ETAIL but not the S2639A mutant (Fig. 4a, right panel, lanes 4 and 6). Notably, incubation of CENP-E with [γ-32P]-ATP in the presence of BRT-1 resulted in undetectable incorporation of radioactivity into wild-type CENP-ETAIL (Fig. 4a, lane 5). Using an antibody (pS2639) that we generated to selectively recognize CENP-E phosphorylated at Ser2639, we confirmed that CENP-E is phosphorylated at this site by BubR1, but not the kinase activity-deficient variants BubR1D911A or BubR1D882N (Fig. 4b, lanes 1–3). In vitro kinase assays demonstrated that the CENP-E C-terminal tail (CENP-EC100; aa 2602-2701) was a better substrate (i.e., more rapidly phosphorylated) than casein (Supplementary information, Fig. S5a). As expected, phosphorylation of CENP-E by BubR1 was inhibited by BRT-1 in vitro (Fig. 4b, lane 4). Thus, Ser2639 of CENP-E is a substrate for BubR1 in vitro.

CENP-E is a bona fide substrate of BubR1 in mitosis. a BubR1 was incubated with CENP-ETAIL or CENP-ETAIL/S2639A mutant for in vitro phosphorylation assay judged by 32P incorporation (right panel). CBB, Coomassie Brilliant Blue. b BubR1 was incubated with CENP-ETAIL for in vitro phosphorylation assay in the presence of BubR1 wild type and mutants. An aliquot of reaction also included BRT-1. CENP-E phosphorylation level was analyzed by anti-phospho-CENP-E Ser2639 (pS2639). Note that CENP-E is phosphorylated by wild-type BubR1 but not BubR1 kinase-deficient mutants and the phosphorylation of CENP-E was abolished by treatment of 3 μM BRT-1. c Nocodazole-arrested HeLa cells were treated with reversine (300 nM) or BRT-1 (3 μM), and the CENP-E phosphorylation level was assessed by Western blot. d HeLa cells were released from a double thymidine block and collected at indicated time points followed by Western blot analyses of pS2639-CENP-E and other indicated antibodies. e Temporal profile of cyclin B1 accumulation and phosphorylation levels of CENP-E and Aurora B. Data represent mean ± SEM from three independent experiments. f Microtubule binding assay of recombinant MBP-CENP-ETAIL. CENP-E (0.5 μM) and tubulin (0–20 μM) at varied molar ratios were incubated for 10 min in polymerization buffer and centrifuged. Equal volumes of supernatant (S) and pellet (P) fractions were resolved by SDS-PAGE and visualized by CBB staining. g CENP-ETAIL was incubated with BubR1 kinase before microtubule co-sedimentation assay as described in (f). See also Supplementary information, Fig. S5
To examine if Ser2639 is a physiological substrate of BubR1 in mitotic cells, nocodazole-arrested HeLa cells were examined (by immunoblotting cell extracts with the pS2639 antibody) for CENP-E phosphorylation after treatment with BRT-1 or reversine (an Mps1 inhibitor).30 CENP-E was phosphorlyated in untreated mitotic cells (Fig. 4c, lane 1, top panel). This phosphorylation was inhibited by BRT-1, but not reversine treatment (Fig. 4c, lanes 2 and 3, top panel). To determine the kinetics of Ser2639 phosphorylation across the cell cycle, synchronized HeLa cells were released from cell-cycle arrest at the G1/S boundary31 and CENP-E phosphorylation analyzed by immunoblotting with the pS2639 antibody (Fig. 4d, e). Cyclin B1 accumulation (6 h after release), marked advance into the G2 phase and was sustained until anaphase onset (~11 h after release). Aurora B phosphorylation (pT232) appeared in the early G2 phase (4 h after release), and was sustained until anaphase, consistent with previous studies.31 CENP-E Ser2639 phosphorylation appeared after the rise of cyclin B1 and pT232-Aurora B, and was sustained beyond anaphase entry when cyclin B1 level declined (11 h after release; Fig. 4d, e), demonstrating that the Ser2639 phosphorylation is temporally independent of CDK1 activity. CENP-E phosphorylation at Ser2639 was diminished after BRT-1 treatment or siRNA-mediated BubR1 knockdown (Supplementary information, Fig. S5b). CENP-E phosphorylation was present at metaphase kinetochores and the labeling was abolished after BRT-1 treatment (Supplementary information, Fig. S5c). Thus, CENP-E is a physiological substrate of BubR1 during mitosis.
Immunoprecipitations from cell lysates were used to determine that BubR1 co-precipitates with the C-terminal tail of CENP-E (Supplementary information, Fig. S5d, e). Conversely, CENP-E co-precipitates with the kinase domain of BubR1 (Supplementary information, Fig. S5f, g), consistent with other recent biochemical evidence for a BubR1-CENP-E interaction.19 In contrast, Cdc20 co-precipitated with full length BubR1, but not the kinase domain (Supplementary information, Fig. S5g).
Since CENP-ETAIL exhibits phosphorylation-regulated microtubule binding29,32 and Ser2639 lies in its C-terminal tail region, we examined if BubR1 phosphorylation modulated the microtubule-binding activity of CENP-E. Microtubule co-sedimentation assays in which the CENP-ETAIL concentration was held constant with varying tubulin concentrations demonstrated that the CENP-ETAIL bound to assembled microtubules in a dose-dependent manner (Fig. 4f). Significantly, the CENP-ETAIL was quantitatively mcrotubule bound after BubR1 and ATP addition (Fig. 4g; lane 11).
CENP-E phosphorylation enables a microtubule plus-end tracking activity

BubR1 phosphorylation elicits CENP-E tracking on microtubule plus-ends. a HeLa cells transfected with BubR1 siRNA or treated with BRT-1 (3 μM) were subjected to immunofluorescence assay. Magnified views of kinetochore (red)-microtubule (green) association were shown. Scale bars, 10 µm. b HeLa cells transfected with FLAG-CENP-ETAIL wild type (CENP-ETAIL/WT) or mutant (CENP-ETAIL/S2639A) or treated with BRT-1 were subjected to immunofluorescence assay. Scale bars, 10 μm. c Quantitative analyses of chromosome alignment phenotype in BRT-1-treated cells and cells expressing CENP-ETAIL/WT, CENP-ETAIL/S2639A or CENP-ETAIL/S2639D. d Quantification of syntelic kinetochores from various treatment. Data represent mean ± SEM from three independent experiments with more than 30 cells in each conditions. Statistical significance was tested by two-sided t-test. **p < 0.01; ***p < 0.001. e Cartoon drawing of microtubule plus-end-tracking assay for CENP-E using TIRFM. f Micorotubule plus-end-tracking activity of CENP-ETAIL in the presence or absence of BubR1. Scale bar, 5 µm. g Kymograph of CENP-ETAIL tracking behavior on dynamic microtubule plus-ends in the presence or absence of BubR1. Scale bar, 5 µm. h Statistical analyses of tracking velocity of CENP-ETAIL wild type and mutants in the absence or presence of BubR1 and BRT-1 (n = 9 for each group). Data represent mean ± SEM from three independent experiments. Statistical significance was tested by two-sided t-test. **p < 0.01; ***p < 0.001. i HeLa cells were transiently transfected to express GFP-CENP-ETAIL wild type and mutants followed by immunofluorescence microscopic analyses. Scale bar, 10 μm. See also Supplementary information, Fig. S6
To probe the precise function of BubR1-mediated phosphorylation of CENP-E in chromosome segregation, we examined the attachment of kinetochores to spindle microtubules in BRT-1-treated cells. As shown in Fig. 5a, kinetochore-fibers are firmly attached to kinetochores in DMSO-treated cells (top panel and inset), but aberrantly attached after BRT-1 treatment (bottom panel and insets). To examine if BubR1 phosphorylation of CENP-E at Ser2639 is critical for a stable kinetochore-microtubule attachment, we generated FLAG-CENP-ETAIL constructs containing phospho-mimicking (S2639D) and non-phosphorylatable (S2639A) mutations. Epitope-tagged CENP-ETAIL proteins were expressed in HeLa cells at comparable levels about 4-fold higher than endogenous CENP-E (Supplementary information, Fig. S6a). Cells overexpressing non-phosphorylatable FLAG-CENP-ETAIL/S2639A mutant or treated with BRT-1, but not the ones expressing wild-type CENP-ETAIL, exhibited lagging chromosomes (Fig. 5b). High magnification imaging revealed (Fig. 5b, insets) that aberrant (syntelic and monotelic) attachments were increased in either CENP-ETAIL/S2639A-expressing or BubR1-inhibited cells accompanied by aberrant chromosome alignment (Fig. 5c; Supplementary information, Fig. S6b). Syntelic kinetochore attachments were significantly increased (Fig. 5d) following BRT-1 inhibition of the BubR1 kinase (and the accompanying reduction in phosphorylation of CENP-E at Ser2639) (**p < 0.01; ***p < 0.001).
A recent study has reported that CENP-E tracks dynamic microtubule plus-ends.13 To delineate the molecular mechanism underlying the aberrant attachments seen in BubR1-inhibited cells, we used single-molecule TIRF microsocpy analyses33 to examine whether BubR1-phosphorylation modulates the microtubule tracking characteristics of CENP-E (Fig. 5e). We initially determined that GFP-CENP-ETAIL binds to the lattice of GMPCPP-seeded rhodamine-microtubules without apparent end tracking (Fig. 5f). Surprisingly, BubR1 addition produced an increased concentration of GFP-CENP-E at the growing-end of microtubules (Fig. 5f; right panel). Analyses of kymographs revealed that this BubR1-dependent tip-tracking activity of the CENP-ETAIL was seen only on growing microtubules (Fig. 5g; right panel). No apparent plus-end tracking activity was observed in the absence of BubR1 (Fig. 5g; left panel) or presence of BubR1 and BRT-1 (Fig. 5h). The microtubule plus-end associated velocity of CENP-E motion is a function of BubR1 kinase activity as chemical inhibition with BRT-1 or use of the non-phosphorylatable GFP-CENP-ES2639A diminished tracking activity of CENP-E (Fig. 5h). To test if BubR1-mediated phosphorylation of CENP-E in mitotic cells enables microtubule end-tracking as seen in the in vitro TIRF microscopy assay, HeLa cells were transiently transfected to express wild-type and a phosphomimetic CENP-ETAIL. As shown in Fig. 5i, GFP-CENP-ES2639D concentrated at the plus-ends of kinetochore microtubules (marked by ACA, bottom panel). The wild-type GFP-CENP-ETAIL distributed along spindle microtubules, in addition to at the kinetochores (Fig. 5i, top panel). By contrast, GFP-CENP-ES2639A was mainly distributed along the spindle microtubules (Fig. 5i, middle panel). Thus, BubR1-mediated phosphorylation enables conversion of CENP-E from lateral binding to plus-end tracking.
BubR1 is essential for accurate central spindle establishment and anaphase onset
The functional relevance of BubR1-CENP-E interaction in mitosis has been suggested.5,34 However, the role of BubR1-CENP-E interaction in metaphase-anaphase transition has never been directly examined due to the lack of analytical tools for temporal dissection. Our immuno-electron microscopic analyses demonstrated that CENP-E was found to be microtubule-associated only in the electron dense region with overlapping microtubule plus-ends.14 To explore the potential role of BubR1-CENP-E interaction in metaphase-anaphase transition, we synchronized HeLa cells at metaphase by addition of the proteosome inhibitor MG132. Following three washes, cells progressed into anaphase in the presence or absence of BRT-1 (illustrated in Fig. 6a). As shown in Fig. 6b, mock treated cells achieved metaphase chromosomal alignment within ~20 min after MG132 wash-out and subsequently progressed into anaphase onset. However, inhibition of BubR1 kinase activity by BRT-1 addition at metaphase perturbed chromosome movement after the metaphase-anaphase transition, with cells entering anaphase faster and with chromosome bridges, a characteristic phenotype of SAC defects (Fig. 6b, lower panel; arrow).
Careful examination of the microtubule network revealed that inhibition of BubR1 kinase activity beginning at metaphase perturbed the establishment of a stable central spindle, as BRT-1-treated cells exhibited fewer anti-parallel microtubules in anaphase onset (Fig. 6b, lower panel; arrowhead). The central spindle in BRT-1-treated cells was fragile and eventually fragmented (by 70 min), suggesting that anti-parallel microtubules may not be established and/or stabilized in the absence of BubR1 kinase activity. To assess the involvement of CENP-E in central spindle organization, we employed two chemically distinct CENP-E inhibitors and BRT-1 to probe the importance of BubR1 and CENP-E in central spindle integrity and function. As shown in Supplementary information, Fig. S7a–d, addition of CENP-E or BubR1 chemical inhibitor into synchronized metaphase HeLa cells perturbed central spindle establishment following the transition to anaphase.

BubR1 is essential for accurate metaphase-anaphase transition. a Schematic illustration of BRT-1 treatment in HeLa cells at metaphase-anaphase transition. b Representative phenotypes of metaphase-anaphase transition in HeLa cells treated with DMSO or BRT-1. c Schematic illustration of induction of BubR1 degradation in BubR1AID engineered DLD-1 cells. d Representative phenotypes of metaphase-anaphase transition in BubR1AID engineered DLD-1 cells with or without auxin treatment. e Statistical analyses of aberrant central spindle phenotypes elicited by BRT-1. DMSO, n = 42; BRT-1, n = 43; Reversine, n = 38. Data represent mean ± SEM from three independent experiments. **p < 0.01. f Statistical analyses of aberrant central spindle phenotypes elicited by auxin-induced BubR1 degradation. Control, n = 49; Auxin, n = 42. Data represent mean ± SEM from three independent experiments. Statistical significance was tested by two-sided t-test. **p < 0.01. See also Supplementary information, Fig. S7
To validate that the aberrant central spindle reflected a requirement for BubR1, we adopted a recently established auxin-inducible degron (AID) system to induce BubR1 degradation.35 Specifically, DLD-1 cells engineered with BubR1AID were synchronized at prometaphase by monastrol followed by addition of auxin for 25 min before release into monastrol-free media to enter metaphase (illustrated in Fig. 6c). Auxin addition resulted in a rapid loss of BubR1 protein within 25 min (92 ± 3%; Supplementary information, Fig. S7e). Rapid, acute loss of BubR1 resulted in a delay of anaphase onset followed by loss of an anti-parallel central spindle structure (Fig. 6d), which was evident by the appearance of tripolar spindles (lower panel; arrows). By contrast, without induced BubR1 degradation, control DLD-1 cells progressed into anaphase soon after release from monastrol block (Fig. 6d, upper panel). Quantitative analyses showed that the stable anti-parallel microtubule organization requires BubR1 kinase activity, as chemical inhibition with BRT-1 or AID-elicited BubR1 loss in metaphase cells increased the rate of formation of aberrant central spindles (Fig. 6e, f; **p < 0.01; ***p < 0.001). This evidence demonstrates that BubR1 (and its kinase activity) plays a previously uncharacterized mitotic role post-metaphase.
Phosphorylation of CENP-E by BubR1 at Ser2639 generates marking of the midzone
The phenotype of aberrant central spindles in BRT-1-treated and BubR1-deficient anaphase cells prompted us to examine the distribution of microtubule bundling proteins such as PRC1. Real-time imaging of GFP-PRC1- and mCherry-H2B-expressing HeLa cells revealed that inhibition of BubR1 activity by BRT-1 at metaphase prevented the accumulation of PRC1 onto the overlapped anti-parallel microtubules (Fig. 7a; b’), demonstrating a potential functional link between BubR1 activity and central spindle establishment.
To delineate the mechanism underlying BRT-1-elicited aberrant central spindle organization, we examined the interaction of CENP-E with PRC1, a microtubule-bundling protein essential for central spindle plasticity.36,37 Both endogenous PRC1 and exogenous mCherry-PRC1 were found to co-distribute with CENP-E into the midzone microtubule arrays (Supplementary information, Fig. S8a). To examine whether BubR1 kinase activity regulates the interaction between CENP-E and PRC1, metaphase HeLa cells (synchronized by thymidine block and a 10 h release) were treated with 3 µM BRT-1 (or DMSO as control) for 30 min in the presence of MG132. Immunoprecipitation of CENP-E from clarified cell lysates co-precipitated PRC1 and BubR1 (Fig. 7b, lane 3; middle two panels). PRC1 was barely detectable in CENP-E immunoprecipitates when cells were treated with BRT-1 (lane 4). Immnoblotting analyses with the anti-pS2639 CENP-E antibody confirmed the inhibition of BubR1 activity by BRT-1 (lane 4; bottom panel). Thus, the interaction between endogenously expressed CENP-E and PRC1 requires BubR1 kinase activity.
We further characterized CENP-E-PRC1 interaction in vitro using recombinant GST-PRC1 and MBP-CENP-E proteins purified from bacteria or GFP-CENP-ETAIL and full-length proteins isolated from 293T cell lysates (Fig. 7c, d). Pull-down assays indicated that CENP-E tail binds to the N-terminal PRC1 directly (Fig. 7c; lane 5 and d; lane 7). Consistent with the proposed role of BubR1 phosphorylation, phospho-mimicking CENP-E tail bound well to PRC1-N, while wild-type CENP-E barely bound (Fig. 7d; lane 5 and 6), suggesting that BubR1 phosphorylation may elicit a conformational change of CENP-E which enables the CENP-E-PRC1 interaction in metaphase-anaphase transition.

Phosphorylation of CENP-E by BubR1 at Ser2639 constitutes a temporal cue for midzone establishment. a Real-time imaging of GFP-PRC1- and mCherry-H2B-expressing cells with inhibition of BubR1 activity by BRT-1 at metaphase. Scale bar, 10 μm. b Immunoprecipitation of endogenous CENP-E from metaphase-synchronized HeLa cells with DMSO (lanes 1 and 3) or BRT-1 (3 μM) treatment (lanes 2 and 4). Extracts from mitotic HeLa cells were incubated with an anti-CENP-E rabbit antibody and immunoprecipitates were resolved by SDS-PAGE followed by Western blot analyses using indicated antibodies. Top panel, CENP-E; second panel, PRC1; third panel, BubR1; bottom panel, pS2639. c Recombinant GST-PRC11-303 (PRC1-N) and GST-PRC1304-620 (PRC1-C) were incubated with MBP-CENP-ETAIL for 4 h, and their interaction was assessed by CBB-stained SDS-PAGE gel. d Recombinant GFP-CENP-E full-length (wild type and S2639D mutant) and GFP-CENP-E tail were transfected into 293T cells for 24 h, and clarified cell lysates were incubated with GST-PRC11-303 (PRC1-N) for 2 h, and the interaction between CENP-E and PRC1 was assessed by pull-down assay and GFP Western blotting. Note that CENP-E S2639D mutant was efficiently isolated by PRC1-N affinity matrix. e Immunofluorescence staining of pS2639-CENP-E and PRC1 in DMSO or BRT-1-treated HeLa cells. Scale bar, 10 μm. f A working model shows that BubR1-elicited phosphorylation switches CENP-E from lateral motility to plus-end tracking that is implicated in kinetochore rotation to facilitate an accurate microtubule ‘search and capture’ process in metaphase alignment and initiate central spindle establishment via a temporally controlled CENP-E-PRC1 interaction. Statistical significance was tested by two-sided t-test. See also Supplementary information, Fig. S8
If BubR1 kinase-dependent CENP-E-PRC1 interaction is important for midzone anti-parallel microtubule organization, inhibition of BubR1 kinase activity with BRT-1 added to metaphase cells would result in suppression of pS2639 level, with subsequent perturbation of central spindle formation. Consistent with our hypothesis, BRT-1 treatment of metaphase cells abolished the signal of pS2639 (Fig. 7e; Supplementary information, Fig. S5c). Importantly, while the characteristic midzone was apparent in DMSO-treated cells (Fig. 7e, upper panel, arrows), a stable midzone was not established in BRT-1-treated cells (Fig. 7e; lower panel, arrows).
Discussion
A general principle of mitotic spindle assembly is described as “search and capture” model in which dynamic plus-ends of microtubules grow and shrink until they are captured by kinetochores.38 However, actual completion of mitosis in diploid human cells takes much less time than anticipated,39,40 suggesting that additional mechanisms may guide microtubule growth toward kinetochores39,41 and/or position chromosomes to the areas with high-density of microtubules.9,42,43 Our study shows that BubR1-mediated CENP-E phosphorylation elicits a critical switch from the lateral association to end-on capture of microtubules (Fig. 7f), which provides molecular insight into mechanisms underlying the switch from laterally attached kinetochore to an end-on stable configuration in mitosis.42
One outstanding question in mitosis field is whether BubR1 is an actual kinase. Our studies suggest that metazoans evolved an elaborate mitotic checkpoint machinery including BubR1, which was considered as an unusual pseudokinase as it contains the canonical catalytic triad but lacks essential features in several critical kinase motifs. Previous works have shown that BubR1 has undetectable kinase activity without modifications or binding to other activators (such as CENP-E).5,12,15 Our structural analyses show that DmBubR1 is an exceptional vertebrate BubR1 kinase that retains the characteristics of Bub1 kinase which is exemplified by the salt bridge formed between two highly conserved residues, the Lys at β3 and the Glu at αC helix. However, this type of signature of an active kinase is not apparently conserved in human BubR1. Early study suggested that the classical kinase-dead human BubR1 by mutating the residues at the ATP-binding pocket had altered protein stability and therefore affected BubR1 function in a catalysis-independent way.15 Our present study of BubR1 mutant D882N, a mutation at the catalytic loop without altered protein stability, demonstrated kinase activity of human BubR1 and the observed effects are not due to lack of structure integrity. Structural comparison between human BubR1 and Bub1c (PDB: 4R8Q) revealed that although the overall structures of human Bub1 and BubR1 are virtually similar (rmsd = 1.62 Å), the conformation of the activation segment and the orientation of the Gly-rich loop are different (Supplementary information, Fig. S2a). For example, human BubR1 structure P + 1 loop forms a 310 helix, which shows similar conformation with PKA and other S/T kinases in active state. On the contrary, the P + 1 loop of the human Bub1c adopts a hairpin-like structure that covers the catalytic loop and restricts substrate access to ATP. We reason that the aforementioned difference seen in their structures suggests the basis for functional specificity of Bub1 and BubR1 and their roles in metazoans evolution.
Our structural determination of DmBubR1 revealed an unusual N-terminal extension folds that wrap around the kinase domain and the activation segment of kinase domain. The hydrophobic interactions between the aromatic ring of Phe1129 and the hydrophobic pocket formed by residues in the activation segment are thought to stabilize the conformation of the activation segment (Supplementary information, Fig. S1b). However, it was intriguing that F1129A mutation exhibits no effect on DmBubR1 kinase activity but the equivalent W742A dramatically reduced Bub1 kinase activity despite the fact that both of them contain similar N-terminal extension parts. For Bub1c, its autophosphorylation of S969 is essential for regulating the conformation of the P + 1 loop required for its activation. For DmBubR1, the equivalent residue is D1339 which naturally mimics the phosphorylated state of Ser and stabilizes the active conformation. Thus, the kinase core of DmBubR1 is naturally stabilized and less dependent on the N-terminal extension than Bub1 which may account for no dramatic alteration on kinase activity seen in F1129A mutant. Given their minor structural roles in the hydrophobic interaction with αC, it was not surprised that I1134A/S1135A mutant also exhibited no major alteration on the kinase activity. In fact, these amino acids are divergent among other species and their counterpart residues in human BubR1 are R727 and R728. Together, these results suggest that DmBubR1 and Bub1 might use distinctly different mechanisms for their catalyses. The fact that BRT-1 reacts with DmBubR1 and BubR1 but not Bub1 supports this notion. It would be of great interest to visualize the respective conformational flexibility of Bub1 and DmBubR1 using single molecule analyses and relate the conformational flexibility to mechanism of action in live mitotic cells.
Although CENP-E localizes to midbody and central spindle during metaphase-anaphase, the mechanism of action underlying CENP-E function in central spindle assembly was never addressed despite the fact that an early microinjection experiment suggested a role of CENP-E in anaphase.28 Surprisingly, BubR1 kinase inhibition perturbed the establishment of stable central spindle as BRT-1-treated cells exhibited fewer anti-parallel microtubules in anaphase onset (Fig. 6b; 20–25 min frames). Our findings suggest phosphorylation-elicited CENP-E binding to PRC1 contributes to central spindle organization during anaphase onset. The spindle midzone is assembled by a complex network of anti-parallel nonkinetochore microtubules, which were built up between separating chromosomes.44 A subset of these spindle microtubules overlaps with anti-parallel orientation while other microtubules such as spindle microtubules terminate at the kinetochore. During metaphase-anaphase transition, CENP-E and PRC1 relocate to the central spindle. Phosphorylated CENP-E binds and recruits greater amounts of PRC1 to stabilize longer central spindle microtubules, thus promoting the assembly of midzone. Concurrently, stable association of PRC1 with microtubule plus-ends promotes cross-linking of the anti-parallel microtubules. Further study will be directed to understanding how microtubule plus-ends are tagged by CENP-E-PRC1 as they transit into midzone array and whether BubR1 phosphorylation provides a spatial cue for the selectivity.
The BubR1-elicited CENP-E-PRC1 interaction identified in this study suggests that phosphorylation of Ser2639 opens intra-molecular interaction of CENP-E. To directly examine if conformational change elicited by BubR1 phosphorylation exists and is sufficient for central spindle organization, we attempted to reconstitute the PRC1-CENP-E interaction using recombinant phospho-mimicking CENP-ES2639D. Surprisingly, exogenous overexpression of CENP-ES2639D often resulted in formation of CENP-ES2639D condensates in cytoplasm and perturbed its accurate localization/distribution. This indicated that the phosphorylation of CENP-E by BubR1 is a dynamic process in a timely regulated fashion as persistent phosphorylation prevented the accurate targeting of CENP-E to mitotic spindle and kinetochore in early mitosis. Given this limitation, our attempt to examine whether CENP-ES2639D is sufficient to build a central spindle in metaphase cells in the presence of BRT-1 was inefficient. Future study will need to build a reconstitution system with purified components and monitor the assembly of central spindle in real time with manipulated BubR1 activity. A similar approach has been recently succeeded in delineating the regulation of SAC kinetics by Mps1 kinase.45
Our early studies indicated that metazoans evolve an elaborate SAC machinery including ZW10, BubR1 and CENP-E to ensure faithful chromosome segregation in mitosis.46 It has been a hot debate as whether BubR1 exhibits active kinase activity and whether the kinase activity is essential for the SAC function. The structure-function analyses established here provide a definitive conclusion to this long-standing controversy. That BRT-1 treatment resulted in an initial mitotic arrest of cells with unaligned chromosomes strongly supports the view that BubR1 kinase activity is not essential for SAC function, which is consistent with the observation in Drosophila.6 Given the spatiotemporal dynamics of mitotic regulators such as kinase and kinesin, chemical tools were explored and used to delineate their signaling cascades in mitosis.24,26,30,47 The identification and characterization of BRT-1 as a chemical probe of BubR1 will facilitate the molecular delineation of BubR1 function in the spindle plasticity, SAC signaling, and mitotic timing as previously reported in Drosophila by Karess and colleagues.6 During the course of our study, Chen and colleagues have reported their identification of an active kaurene diterpenoid named Pharicin A that can inhibit leukemia cell division.48 Interestingly, Pharicin A exhibits similar chemical structure to BRT-1 and binds to BubR1, which suggests that kaurene diterpenoid compounds are rich sources of BubR1 modulators. It would be of great interest to pinpoint the binding sites of Pharicin A on BubR1 and examine whether any binding sites are overlapped with those of BRT-1. The respective binding interface of BRT-1 and Pharicin A on BubR1 together with their chemical structural information will enable us to optimize BRT-1’s potency in modulating BubR1 activity in cells and animals. Given the functional significance of BubR1 in cell fate decision and perturbation of BubR1 function underlying pathogenesis of diseases such as gastrointestinal cancers,49,50,51 a combination of disease-bearing BubR1 mutant gastrointestinal organoids with kaurene diterpenoid compounds could be used to screen and identify synthetic lethal effects, an exciting avenue for precision therapeutics.52
In sum, our study demonstrates that BRT-1 is a chemical inhibitor specifically inhibiting BubR1 in vitro and in live cells. BRT-1 is thus a promising tool to study context-dependent BubR1 function in cellular dynamics. Importantly, BRT-1 also inhibits DmBubR1 in vitro and in vivo (data not shown), which would enable the synergism between chemical biology and fly genetics in the powerful model system. The spatiotemporal regulation of CENP-E by BubR1 kinase revealed in this study may provide an insight into the classic view that phosphorylation directs two opposing kinetochore motors on isolated chromosomes.53 To coordinate metaphase-anaphase transition, this phosphorylation-dependent switch turns on the plus-end-directed tracking activity of CENP-E to enable the establishment of central spindle from spindle microtubules. The BubR1-elicited phosphorylation of CENP-E provides a spatial cue within the mitotic spindle to regulate CENP-E activity according to the position of chromosomes and temporal cue to facilitate CENP-E-PRC1 interaction for accurate anaphase midzone formation. Finally, the possibility of being able to chemically antagonize BubR1 may advance our understanding of its relevance in various contexts of health and disease.
Materials and methods
Plasmids
To generate GFP-tagged and Flag-tagged full-length human BubR1 and its deletion mutants, PCR-amplified BubR1 cDNA was cloned into the pEGFP-C1 vector (Clontech) and 3× FLAG-Myc-CMV-24 vector (Sigma) with KpnI and BamHI digestion. GFP-BubR1-ΔKD plasmid was generated by inserting PCR-amplified BubR1 (aa 1–717) fragment into pEGFP-C1 vector via KpnI and BamHI sites. FLAG-, GFP-, and MBP-CENP-ETAIL plasmids were generated by inserting PCR-amplified CENP-E (aa 2131–2701) fragments into the p3× FLAG-Myc-CMV-24, pEGFP-C1 and pMal-c2 (New England Biolabs) vectors via BamHI and SalI sites, respectively. GFP-tagged CENP-E full length and truncations of CENP-E1–768, CENP-E769–1571, and CENP-E1572–2701 were generated by inserting the corresponding PCR-amplified fragments into the pEGFP-C1 vector via SalI and BamHI. FLAG-tagged CENP-E full length were generated by subcloning CENP-E cDNA into 3× FLAG-Myc-CMV-24 via SalI/BamHI digestion. FLAG-tagged BubR1 truncations of BubR11–717 and BubR1718–1050 were generated by inserting the corresponding PCR-amplified fragments into 3× FLAG-Myc-CMV-24 vector with KpnI and BamHI digestion. The GST-PRC1-N and -C constructs were obtained by cloning a PCR fragment corresponding to aa 1–303 and 304–620 of human PRC1 into the pGEX-6p1 vector (GE Healthcare) via EcoRI and SalI, respectively. PCR-amplified CENP-E C-terminal tail (CENP-E-C100; aa 2602–2701) cDNA was cloned into pET-22b (+) vector (Novagen) via BamHI and SalI. Site-specific mutants of BubR1 and CENP-E were generated by PCR-based, site-directed mutagenesis kit (Vazyme) according to the manufacturer’s instructions. All plasmids used were verified by sequencing (Invitrogen).
Cell culture, synchronization and transfection
HeLa cells and HEK293T cells (American Type Culture Collection)were cultured in DMEM (Gibco) with 10% (vol/vol) FBS (Hyclone) and penicillin-streptomcyin (100 units/mL and 100 μg/mL, respectively; Gibco) at 37 °C in a humidified atmosphere with 8% CO2. For cell cycle synchronization, HeLa cells were first blocked in G1/S with 2.5 mM thymidine (Sigma) for 16 h and then released in fresh culture medium for 8 h to enrich mitotic cells. For some experiments, cells were exposed to 10 μM MG132 and 3 μM BRTs or DMSO for another 1 h before fixation. Plasmid transfections were performed with Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions.
Antibodies, short interfering RNA and chemical inhibitors
Anti-α-tubulin (mouse, FITC-DM1A, Sigma F2168) and ACA (anti-centromere antibody, from Immunovision HCT-0100) were used for immunofluorescence. To generate phospho-CENP-E (pSer2639) antibody, a synthetic peptide containing phosphorylated Ser2639 (C-ERNLQDPVPKE-pS-PK) was conjugated to rabbit albumin (Sigma) and immunized into rabbits as described.14,31 The serum was collected by a standard protocol and preabsorbed by non-phosphorylated CENP-E peptide (C-ERNLQDPVPKESPK) followed by affinity-purification using (C-ERNLQDPVPKE-pS-PK)-conjugated sulftone sepharose beads. The appropriate secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. Antibodies used for Western blots were anti-α-tubulin (mouse, DM1A, Sigma T9017), anti-Ezrin (mouse, BD Biosciences 610602), anti-Cyclin B1 (mouse, Cell Signaling Technology 4138S), anti-BubR1 (mouse, BD Biosciences 612503), anti-Bub1 (mouse, Abcam ab54893), anti-pS11-RCC1 (Cell Signaling Technology 5500), anti-RCC1 (Cell Signaling Technology 5134), anti-MBP-tag (Cell Signaling Technology 2396), anti-pS7-CENP-A (Cell Signaling Technology 2187), anti-CDC20 (Cell Signaling Technology 4823), anti-pT232-Aurora B (Rockland 600-401-677) and mouse anti-PLK1 pT210 (BD Biosciences 558400). Anti-FLAG-tag (M2, 1:2,000) and anti-GFP mAb (#G6539, 1:2000) antibodies were from Sigma. Anti-CENP-E antibody (HpX) and anti-CENP-E tail antibody were generated as previously.14,54 Anti-PRC1 antibody used in immunofluorescence and Western blot was generated as described.36
The short interfering RNA duplexes targeting human BubR1 (5’-AACGGGCAUUUGAAUAUGAAA-3’) and 3’-UTR (5′-GTCTCACAGATTGCTGCCT-3’) were purchased from Qiagen.
Nocodazole (100 ng/mL, ≥99%), monastrol (50 μM, ≥98%), MG132 (10 μM, ≥90%), puromycin (1 μg/mL, ≥98%), auxin (500 μM, ≥98%), roscovitine (20 μM, ≥98%), and reversine (300 nM, ≥98%) were from Sigma. ZM447439 (2 μM, >99%) was from Tocris Bioscience. BI2536 (100 nM, >99%) was from Selleckchem. Taxol (1 μM, >99.5%) was from Cytoskeleton Incorporation. The protease inhibitor cocktail was from Sigma.
Recombinant protein expression and purification
Fruitfly RNA was extracted, gene fragments encoding for human BubR1 (NCBI Gene ID: 701) or DmBubR1 (NCBI Gene ID: 35522) were cloned into pFastBac HTA according to the protocols of Bac-to-Bac system (Invitrogen). Proteins were expressed in Sf9 insect cells. Cells were infected with the baculovirus and cultured in SFM at 27 °C for 72 h post-infection. The cells were harvested, suspended and lysed by sonication in the PBS buffer. The supernatant was obtained by centrifugation at 16,000 rpm at 4 °C for 30 min. 6× His tag-fused proteins were purified by standard affinity chromatography. For crystallization, DmBubR1c protein was further cleaved by tobacco etch virus over night. After a second affinity chromatography step, DmBubR1c was further purified through gel filtration chromatography on a Superdex 200 column (GE Healthcare) in 20 mM Tris (pH 7.5), 500 mM NaCl and 5 mM MgCl2. The purified proteins were concentrated to 15 mg/mL, flash frozen in liquid nitrogen.
Human His-BubR1 WT/K795A/D911A and fly His-BubR1 WT/K1204A/D1326A were expressed in Sf9 cells using BAC-TO-BAC expression system (Invitrogen) and purified with Ni-NTA agarose (Qiagen). Protein samples for crystallization were further purified with size exclusion chromatography on a Superdex 200 column (GE Healthcare) in a solution containing Mg2+ ions. FLAG-tagged BubR1 wild type or mutants were transfected into HEK293T cells for 36 h. Cells were harvested and then solubilized by sonication in lysis buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 2 mM EGTA, 1 mM MgCl2, 1 mM DTT) containing 0.2% Triton X-100, 1 mM PMSF, and protease inhibitor mixture (Sigma). The lysates were clarified by centrifugation at 12,000 rpm for 20 min at 4 °C and the supernatant was incubated with FLAG-M2 resin. The resins were washed three times with the lysis buffer containing 0.2% Triton X-100 and 1 mM PMSF and three times with the lysis buffer alone.
MBP-CENP-ETAIL plasmid was transformed into E. coli strain Rosetta (DE3) and protein expression was induced with 0.5 mM IPTG at 16 °C for 20 h. Bacteria expressing maltose-binding protein (MBP)-tagged proteins were suspended, lysed in MBP column buffer (20 mM Tris-HCl, pH 7.4; 200 mM NaCl; 1 mM EDTA) and incubated with amylose resin (New England Biolabs) for 1.5 h. The resin was washed three times by column buffer, then proteins were eluted with 10 mM maltose in appropriate buffer for corresponding experiments. All purification procedures were performed at 4 °C, and protease inhibitor cocktail was added to prevent protein degradation.
GST-PRC1-NT (aa 1–303) and CT (aa 304–620) plasmids were transformed into E. coli strain Rosetta (DE3), and protein expression was induced with 0.5 mM IPTG at 16 °C for 20 h. Bacteria expressing GST-PRC1 were suspended and lysed by sonication in PBS buffer supplemented with 0.1% Triton X-100. The preparation was incubated with glutathione-Sepharose 4B (GE Healthcare) for 1.5 h at 4 °C. The resin was washed three times, and GST-PRC1 protein was eluted with 10 mM reduced glutathione.
Crystallization, X-ray diffraction, data collection, and structure solution
Apo DmBubR1c in Mg2+-contained buffer was crystallized using hanging-drop vapor-diffusion methods in a solution containing 3.5%-4% polyethylene glycol (PEG) 3000 and 0.1 M Bis-tris propane (pH 7.5) at 16 °C. The crystals of DmBubR1c-ADP-2Mg2+ complex were obtained from the same well solution using the protein co-crystallized with 5 mM ADP. The final cryoprotectant solution contained 30% (vol/vol) glycerol. Crystals were flash frozen in liquid nitrogen. Diffraction data were collected at home (apo form) and at beamline BL-17U1 of Shanghai Synchrotron. Radiation Facility (SSRF) (ADP-bound form), and subsequently indexed, scaled and merged by using HKL2000.55 The phasing problem was solved by molecular replacement method using Phenix.Phaser56 with the structure of HsBub1c (PDB entry: 4R8Q) as a search model. The final model was manually adjusted in COOT57 and refined by Phenix.Refine.56 The statistics of data collection and structure refinement are listed in Supplementary information, Table S1.
Modeling and docking
A computational structural model of human BubR1c was obtained using SWISS-MODEL workspace58 on the basis of DmBubR1c structure. BRT-1 was docked into human BubR1c structures by using AutoDock.59 Ligand molecules and all waters were removed from the original coordinate files before docking simulations. Each experiment consisted of 100 GA runs. The docking parameters were kept to their default values. The 100 independent GA runs were processed using the built-in clustering analysis with a 0.5-Å cutoff.
Kinase assay
The kinase reactions were performed in 40 μL kinase buffer (25 mM HEPES, pH 7.2, 50 mM NaCl, 2 mM EGTA, 5 mM MgSO4, 1 mM DTT, and 0.01% Brij35) with casein (2 μg) as substrate, purified BubR1 (100 ng) as kinase, 5 μCi of [γ-32P]-ATP and 500 μM ATP as described previously.31 Reaction mixtures were incubated at 30 °C for 30 min, then terminated by 5× SDS-PAGE loading buffer (10% SDS; 0.5% bromophenol blue; 50% glycerol; 100 mM DTT). After the samples were boiled at 100 °C for 2 min, 50% of the sample was resolved by SDS-PAGE and stained by Coomassie Brilliant Blue R250. For autoradiograms, CBB-stained SDS-PAGE gels were destained, scanned and then dried between sheets of cellulose for 4 h.60 The semi-dried gels were then placed between intensifier and X-ray film for 12–24 h in a −80 °C freezer, and the incorporation of 32P into casein proteins was quantified by PhosphorImager (Amersham Biosciences).
The kinetics of BubR1 was characterized by the AmpliteTM Universal Fluorimetric Kinase Assay Kit (AAT Bioquest) according to the manufacturer’s manual. 50 nM purified BubR1 (wide type or mutants) was incubated with indicated concentrations of casein in 20 μL kinetics assay buffer (60 mM HEPES, pH 7.5; 3 mM MgCl2; 3 mM MnCl2; 3 μM Na-orthovanadate) at 37 °C for 30 min. ADP sensor and sensor buffer were added into the preparations, which were incubated for another 15 min at room temperature. The amount of ADP produced from the kinase reaction was detected by monitoring fluorescence intensity at Ex/Em = 540/590 nm on TECAN Genios Plus. BubR1 autophosphorylation was determined by measuring the fluorescence intensity in the absence of substrate and subtracted from the experiments in the presence of substrate. Km and kcat values were calculated according to the Michaelis-Menten equation.
Immunoprecipitation and pull-down assays
For immunoprecipitation, cells were treated with DMSO or BRT-1 (3 μM) before being lysed in IP buffer (50 mM Tris-HCl, pH 8.0; 120 mM NaCl; 0.5% NP-40) supplemented with protease inhibitor cocktail. After pre-clearing with protein A/G resin (Thermo Fisher Scientific), the lysate was incubated with CENP-E antibody at 4 °C for 4 h with gentle rotation. Protein A/G resin was then added into the cell lysates to incubate for another 6 h followed by spun down and washed five times with lysis buffer. The immunoprecipitates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. For FLAG-tagged protein immunoprecipitation, the FLAG-M2 resin was added to the lysates and incubated for 2 h before washing.
For pull-down assays, GST-PRC1-bound Sepharose beads were incubated with HEK293T cell lysates containing ectopically-expressed FLAG-tagged CENP-E or with purified MBP-tagged CENP-ETAIL from bacteria in PBS containing 0.2% Triton X-100 at 4 °C for 4 h. The binding fraction was washed with PBS for 3 times and analyzed by CBB-stained SDS-PAGE gel.
BRT-1 affinity pull-down assay
Mitotic HeLa cell lysates were incubated with biotin-BRT-1 (10 μM) or DMSO for 2 h. Then the cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% (vol/vol) Triton X-100, 5 mM EDTA and protease inhibitors) and centrifuged at 16,000 × g for 20 min at 4 °C. The supernatant was incubated with Avidin Agarose (ThermoFisher Scientific) for 4 h. The beads were washed three times with lysis buffer and then resuspended in 50 μL lysis buffer with 10 μM BRT-1 to compete with biotin-BRT-1. After 30 min incubation for competition, supernatant was removed and SDS-Sample buffer was added into the beads, respectively. The samples were resolved by SDS-PAGE and immunoblotted with indicated antibodies.
Immunofluorescence microscopy
To examine cold-stable microtubules, HeLa cells growing on coverslips after transfection or drug treatment were incubated on ice for 10 min in L-15 medium (Gibco) with 20 mM Hepes, followed by fixation with 3.7% paraformaldehyde in PTEM (100 mM PIPES, pH 6.8, 10 mM EGTA, 1 mM MgCl2, and 0.2% Triton X-100) for 10 min at room temperature. In other cases, cells were extracted by pre-warmed PHEM (60 mM PIPES, 25 mM HEPES, pH 6.9, 10 mM EGTA, 2 mM MgCl2, and 4 M Glycerol), followed by 1 min of permeabilization with PHEM containing 0.1% Trition X-100. The extracted cells were then fixed with 3.7% paraformaldehyde in PHEM for 5 min After being washed three times with PBST (0.05% Tween-20 in PBS), cells were blocked with 1% BSA (Sigma) in PBST for 1 h, then incubated with primary antibodies for 1 h and secondary antibodies for 1 h at room temperature. DNA was stained with 4’,6-diamidino-2-phenylindole-dihydrochloride (DAPI, Sigma). Images were collected under a DeltaVision microscope (Applied Precision) with a 60× objective lens, NA = 1.42, with optical sections acquired 0.2 μm apart in the z axis. Deconvoluted images from each focal plane were projected into a single picture using softWoRx (Applied Precision). Images projected by softWoRx software were processed with Adobe Photoshop.
Live cell imaging
HeLa cells expressing indicated proteins were grown in glass-bottomed culture dish (MatTek, MA). During imaging, cells were maintained in CO2-independent medium (Gibco) containing 10% FBS (vol/vol) and 2 mM glutamine in a sealed chamber heated to 37 °C. Frames were acquired at 2 or 3 min intervals for mitotic process by a DeltaVision deconvolution microscope system built on an Olympus IX-71 inverted microscope base (Applied Precision). For imaging, a 60 × 1.42 NA lens was used, and optical sections were taken at intervals of 0.2 μm. Images for display were generated by projecting single optical sections as described previously.31 Images were prepared for publication using Adobe Photoshop software.
Microtubule co-sedimentation assay
The co-sedimentation assays were performed with taxol-stabilized microtubules as described previously.61 Purified recombinant MBP-CENP-ETAIL proteins (2 μM) or indicated drugs were incubated with different concentration of taxol-stabilized microtubules for 10 min followed by sedimentation for 10 min at 80,000 rpm in a TLA100 rotor at 25 °C. For visualization and quantification of CENP-E, pellets and supernatants were resolved by SDS-PAGE separately. Gels were stained with CBB. The fractions of CENP-ETAIL partitioned to the supernatant and the pellet were quantified by densitometry of the stained gel using ImageJ software (NIH). Binding curve fitting and apparent Kd calculation were performed with the Prism software (GraphPad).
In vitro microtubule polymerization assay
Tubulin 50 μM (Cytoskeleton, Inc.) was polymerized in BRB80 buffer (80 mM K-PIPES, pH 6.8, 1 mM MgCl2, 1 mM EGTA) containing 1 mM GTP and 1 mM DTT for 10 min at 37 °C in the presence of BRT-1 at indicated concentration. Taxol (Tax; 1 μM) and nocodazole (Noc; 1 μM) were used as polymerization and depolymerization controls, respectively. After sedimentation, the supernatant (S) and pellet (P) were analyzed by SDS-PAGE.
Total internal reflection fluorescence microscopy
The microtubule plus-end tracking experiment was performed as described recently.33,62 The GMPCPP microtubule seeds were prepared by polymerizing 30 μM tubulin (at a bovine tubulin-rhodamine/tubulin-biotin/tubulin ratio of 30:2:1) in the presence of 1 mM GMP-CPP (Jena Bioscience) at 37 °C for 40 min. The seeds were then centrifuged and resuspended in BRB80 buffer. These seeds were sheared with a 25-gauge needle to generate even-length seeds.
Flow chambers were prepared as described previously.33,62 Chambers were coated with 10% monoclonal anti-biotin antibody (Sigma) followed by blocking with 5% Pluronic F-127 (Sigma). After a brief wash, sheared MT seeds (125 nM) were added into the chamber. Tubulin polymerization mixture (30 μM tubulin in total containing 1:30 rhodamine-labeled bovine tubulin in BRB80, 50 mM KCl, 5 mM DTT, 1.25 mM Mg-GTP, 0.25 mg/mL κ-casein, 0.15% methylcellulose (Sigma), an oxygen-scavenging system, and +TIPs) was introduced into the chamber to initiate polymerization. Unless stated otherwise, the final concentrations of +TIPs were 100 nM GFP-CENP-ETAIL or BubR1-phosphorylated GFP-CENP-ETAIL. The temperature was kept at 25 °C. Images were collected with a super-resolution microscope configured on an ELYRA system (Carl Zeiss). The laser intensities were kept at a low level to avoid photobleaching. For +TIPs tracking assays, 1 frame was taken per second. Plus-end tracking of GFP-CENP-ETAIL, phospho-mimicking mutants and phosphorylated GFP-CENP-ETAIL was analyzed using kymographs in ZEN software (Carl Zeiss).
Application of auxin-inducible degron (AID) system for temporal depletion of BubR1
GFP-AID-BubR1 was introduced into Flp-In TRex-DLD-1, TIR1-9Myc stable cell line as previously described.35 The DLD-1 cells were maintained in DMEM containing 10% (vol/vol) FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine (Gibco) supplemented with 1 μg/mL puromycin at 37 °C in a 5% CO2 atmosphere. Monastrol was used at 50 μM to synchronize cells at prometaphase and MG132 was used at 10 μM to synchronize and maintain cells at metaphase. Auxin was used at 500 μM to induce BubR1 degradation as described.35
Statistical analysis
All statistics were described in the figure legends. Two-sided unpaired Student’s t-test was applied for experimental comparisons, using GraphPad Prism. All Western blot analyses were taken from three separated experiments. No statistical method was used to predetermine sample size. All data were expected to have normal distribution.
Data availability
The accession numbers for apo DmBubR1c and ADP-bound DmBubR1c structures reported in this paper are PDB: 6JKK and 6JKM, respectively.
References
Cleveland, D. W., Mao, Y. & Sullivan, K. F. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112, 407–421 (2003).
Sudakin, V., Chan, G. K. & Yen, T. J. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936 (2001).
Ditchfield, C. et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 161, 267–280 (2003).
Kops, G. J., Foltz, D. R. & Cleveland, D. W. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl Acad. Sci. USA 101, 8699–8704 (2004).
Mao, Y., Abrieu, A. & Cleveland, D. W. Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell 114, 87–98 (2003).
Rahmani, Z., Gagou, M. E., Lefebvre, C., Emre, D. & Karess, R. E. Separating the spindle, checkpoint, and timer functions of BubR1. J. Cell. Biol. 187, 597–605 (2009).
Zhang, J., Ahmad, S. & Mao, Y. BubR1 and APC/EB1 cooperate to maintain metaphase chromosome alignment. J. Cell Biol. 178, 773–784 (2007).
Matsumura, S., Toyoshima, F. & Nishida, E. Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J. Biol. Chem. 282, 15217–15227 (2007).
Kapoor, T. M. et al. Chromosomes can congress to the metaphase plate before biorientation. Science 311, 388–391 (2006).
Kim, Y., Holland, A. J., Lan, W. & Cleveland, D. W. Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 142, 444–455 (2010).
Yao, X., Abrieu, A., Zheng, Y., Sullivan, K. F. & Cleveland, D. W. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat. Cell Biol. 2, 484–491 (2000).
Guo, Y., Kim, C., Ahmad, S., Zhang, J. & Mao, Y. CENP-E–dependent BubR1 autophosphorylation enhances chromosome alignment and the mitotic checkpoint. J .Cell Biol. 198, 205–217 (2012).
Gudimchuk, N. et al. Kinetochore kinesin CENP-E is a processive bi-directional tracker of dynamic microtubule tips. Nat. Cell Biol. 15, 1079–1088 (2013).
Yao, X., Anderson, K. L. & Cleveland, D. W. The microtubule-dependent motor centromere-associated protein E (CENP-E) is an integral component of kinetochore corona fibers that link centromeres to spindle microtubules. J. Cell Biol. 139, 435–447 (1997).
Suijkerbuijk, S. J. et al. The vertebrate mitotic checkpoint protein BUBR1 is an unusual pseudokinase. Dev. Cell 22, 1321–1329 (2012).
Zhang, G., Mendez, B. L., Sedgwick, G. G. & Nilsson, J. Two functionally distinct kinetochore pools of BubR1 ensure accurate chromosome segregation. Nat. Commun. 7, 12256 (2016).
Overlack, K. et al. BubR1 promotes Bub3-dependent APC/C inhibition during spindle assembly checkpoint signaling. Curr. Biol. 27, 2915–2927 e7 (2017).
Di Fiore, B., Wurzenberger, C., Davey, N. E. & Pines, J. The mitotic checkpoint complex requires an evolutionary conserved cassette to bind and inhibit active APC/C. Mol. Cell 64, 1144–1153 (2016).
Ciossani, G. et al. The kinetochore proteins CENP-E and CENP-F directly and specifically interact with distinct BUB mitotic checkpoint Ser/Thr kinases. J. Biol. Chem. 293, 10084–10101 (2018).
Kang, J. et al. Structure and substrate recruitment of the human spindle checkpoint kinase Bub1. Mol. Cell 32, 394–405 (2008).
Jeffrey, P. D. et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320 (1995).
Kim, C., Xuong, N. H. & Taylor, S. S. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science 307, 690–696 (2005).
Wang, S. et al. Small-molecule modulation of Wnt signaling via modulating the Axin-LRP5/6 interaction. Nat. Chem. Biol. 9, 579–585 (2013).
Ding, X. et al. Probing CENP-E function in chromosome dynamics using small molecule inhibitor syntelin. Cell Res. 20, 1386–1389 (2010).
Lampson, M. A. & Kapoor, T. M. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat. Cell Biol. 7, 93–98 (2005).
Mayer, T. U. et al. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 286, 971–974 (1999).
Wood, K. W., Sakowicz, R., Goldstein, L. S. & Cleveland, D. W. CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment. Cell 91, 357–366 (1997).
Yen, T. J. et al. CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J. 10, 1245–1254 (1991).
Espeut, J. et al. Phosphorylation relieves autoinhibition of the kinetochore motor Cenp-E. Mol. Cell 29, 637–643 (2008).
Santaguida, S., Tighe, A., D’Alise, A. M., Taylor, S. S. & Musacchio, A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 190, 73–87 (2010).
Mo, F. et al. Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat. Chem. Biol. 12, 226–232 (2016).
Liao, H., Li, G. & Yen, T. J. Mitotic regulation of microtubule cross-linking activity of CENP-E kinetochore protein. Science 265, 394–398 (1994).
Ward, T. et al. Regulation of a dynamic interaction between two microtubule-binding proteins, EB1 and TIP150, by the mitotic p300/CBP-associated factor (PCAF) orchestrates kinetochore microtubule plasticity and chromosome stability during mitosis. J. Biol. Chem. 288, 15771–15785 (2013).
Chan, G. K., Jablonski, S. A., Sudakin, V., Hittle, J. C. & Yen, T. J. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 146, 941–954 (1999).
Holland, A. J., Fachinetti, D., Han, J. S. & Cleveland, D. W. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl Acad. Sci. USA 109, E3350–E3357 (2012).
Fu, C. et al. Mitotic phosphorylation of PRC1 at Thr470 is required for PRC1 oligomerization and proper central spindle organization. Cell Res. 17, 449–457 (2007).
Subramanian, R., Ti, S. C., Tan, L., Darst, S. A. & Kapoor, T. M. Marking and measuring single microtubules by PRC1 and kinesin-4. Cell 154, 377–390 (2013).
Kirschner, M. W. & Mitchison, T. Microtubule dynamics. Nature 324, 621 (1986).
Wollman, R. et al. Efficient chromosome capture requires a bias in the ‘search-and-capture’ process during mitotic-spindle assembly. Curr. Biol. 15, 828–832 (2005).
Yang, Z., Loncarek, J., Khodjakov, A. & Rieder, C. L. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat. Cell Biol. 10, 748–751 (2008).
O’Connell, C. B., Loncarek, J., Kalab, P. & Khodjakov, A. Relative contributions of chromatin and kinetochores to mitotic spindle assembly. J. Cell Biol. 187, 43–51 (2009).
Magidson, V. et al. The spatial arrangement of chromosomes during prometaphase facilitates spindle assembly. Cell 146, 555–567 (2011).
Paul, R. et al. Computer simulations predict that chromosome movements and rotations accelerate mitotic spindle assembly without compromising accuracy. Proc. Natl. Acad Sci. USA 106, 15708–15713 (2009).
Eggert, U. S., Mitchison, T. J. & Field, C. M. Animal cytokinesis: from parts list to mechanisms. Annu. Rev. Biochem. 75, 543–566 (2006).
Faesen, A. C. et al. Basis of catalytic assembly of the mitotic checkpoint complex. Nature 542, 498–502 (2017).
Wang, H. M. et al. Human Zwint-1 specifies localization of zeste white 10 to kinetochores and is essential for mitotic checkpoint signaling. J. Biol. Chem. 279, 54590–54598 (2004).
Wood, K. W. et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl Acad. Sci. USA 107, 5839–5844 (2010).
Xu, H. Z. et al. Pharicin A, a novel natural ent-kaurene diterpenoid, induces mitotic arrest and mitotic catastrophe of cancer cells by interfering with BubR1 function. Cell Cycle 9, 2897–2907 (2010).
Broderick, P. et al. Validation of recently proposed colorectal cancer susceptibility gene variants in an analysis of families and patients-a systematic review. Gastroenterology 152, 75–77 e4 (2017).
Cahill, D. P. et al. Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303 (1998).
Rio Frio, T. et al. Homozygous BUB1B mutation and susceptibility to gastrointestinal neoplasia. N. Engl. J. Med. 363, 2628–2637 (2010).
Yao, X. & Smolka, A. Cellular dynamics underlying gastric parietal cell physiology and microbial-induced pathobiology. Gastroenterology 156, 2158–2173 (2019).
Hyman, A. A. & Mitchison, T. J. Two different microtubule-based motor activities with opposite polarities in kinetochores. Nature 351, 206–211 (1991).
Schaar, B. T., Chan, G. K., Maddox, P., Salmon, E. D. & Yen, T. J. CENP-E function at kinetochores is essential for chromosome alignment. J. Cell Biol. 139, 1373–1382 (1997).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Bordoli, L. et al. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13 (2009).
Goodsell, D. S. & Olson, A. J. Automated docking of substrates to proteins by simulated annealing. Proteins 8, 195–202 (1990).
Yao, X. & et al. Stimulation of gastric acid secretion by cAMP in a novel α-toxin-permeabilized gland model. Am. J. Physiol. 271, C61–C73 (1996).
Huang, Y. et al. CENP-E kinesin interacts with SKAP protein to orchestrate accurate chromosome segregation in mitosis. J. Biol. Chem. 287, 1500–1509 (2012).
Xia, P. et al. EB1 acetylation by P300/CBP-associated factor (PCAF) ensures accurate kinetochore-microtubule interactions in mitosis. Proc. Natl Acad. Sci. USA 109, 16564–16569 (2012).
Acknowledgements
We are grateful to Prof. Yunyu Shi for support; to Dr Hongtao Yu for reagents and Drs Guo-Qiang Chen for input. We thank all the members of our laboratories for discussion and Qiaoqiao Zhang for help on several BubR1 mutant constructs. This work was initiated by the chemical biology grant PGX-2 from the ProteoGenomics Research Laboratory, and supported in part by the National Key Research and Development Program of China (2017YFA0503600, 2017YFA0504901, 2016YFA0100500), National Natural Science Foundation of China (31320103904, 31430054, 91313303, 31621002, 31301120, 91854203, 91853115, 91753000, 31671405, 31470792, 31601097, 21672201, 31600607, and B1661138004), ‘Strategic Priority Research Program’ of the Chinese Academy of Sciences (XDB19000000 and XDB08030102), MOE Innovative team (IRT_17R102).
Author information
Authors and Affiliations
Contributions
X. Y. and R. Z. conceived the project. Y. H., L. L., X. L. & S. Y. designed and performed most structure-activity relationship experiments. X. L., P. Y. Y., Y. Z., J. Z., J. Za., M. T., Z. W., and X. D. designed and performed chemical screening and synthesis of chemical derivative. Y. H., W. W., X. G., F. Y., J. L., Xu L., K.R., and X. D. performed in vitro reconstitution experiment and data analyses. Z. Y., Zhenye Y., L. Li and D. W. C. contributed to reagents. All authors contributed to the writing or editing of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Supplementary information
41422_2019_178_MOESM1_ESM.pdf
Supplementary information, Figure S1 Please replace supplementary figures S2, S3, S4, S5 with new pdf files named "Revised Figure S2/3/4/5-20190523" respectively.
Rights and permissions
About this article

Cite this article
Huang, Y., Lin, L., Liu, X. et al. BubR1 phosphorylates CENP-E as a switch enabling the transition from lateral association to end-on capture of spindle microtubules. Cell Res 29, 562–578 (2019). https://doi.org/10.1038/s41422-019-0178-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-019-0178-z
This article is cited by
-
CKAP5 stabilizes CENP-E at kinetochores by regulating microtubule-chromosome attachments
EMBO Reports (2024)
-
Kinesin-7 CENP-E mediates chromosome alignment and spindle assembly checkpoint in meiosis I
Chromosoma (2024)
-
Phase separation of EB1 guides microtubule plus-end dynamics
Nature Cell Biology (2023)
-
Structural insights into human CCAN complex assembled onto DNA
Cell Discovery (2022)
-
Physiological relevance of post-translational regulation of the spindle assembly checkpoint protein BubR1
Cell & Bioscience (2021)
