
Abstract
The presence of SARS-CoV-2 mutants, including the emerging variant B.1.1.7, has raised great concerns in terms of pathogenesis, transmission, and immune escape. Characterizing SARS-CoV-2 mutations, evolution, and effects on infectivity and pathogenicity is crucial to the design of antibody therapies and surveillance strategies. Here, we analyzed 454,443 SARS-CoV-2 spike genes/proteins and 14,427 whole-genome sequences. We demonstrated that the early variant B.1.1.7 may not have evolved spontaneously in the United Kingdom or within human populations. Our extensive analyses suggested that Canidae, Mustelidae or Felidae, especially the Canidae family (for example, dog) could be a possible host of the direct progenitor of variant B.1.1.7. An alternative hypothesis is that the variant was simply yet to be sampled. Notably, the SARS-CoV-2 whole-genome represents a large number of potential co-mutations. In addition, we used an experimental SARS-CoV-2 reporter replicon system to introduce the dominant co-mutations NSP12_c14408t, 5′UTR_c241t, and NSP3_c3037t into the viral genome, and to monitor the effect of the mutations on viral replication. Our experimental results demonstrated that the co-mutations significantly attenuated the viral replication. The study provides valuable clues for discovering the transmission chains of variant B.1.1.7 and understanding the evolutionary process of SARS-CoV-2.
Similar content being viewed by others


Introduction
Since the outbreak in December 2019, COVID-19 has been pandemic in over 200 countries. Cases of infection and mortalities have been surging and are an ongoing threat to public health1,2. COVID-19 is caused by infection with the novel coronavirus SARS-CoV-23,4,5. Although as a coronavirus, SARS-CoV-2 has genetic proofreading mechanisms6,7,8, the persistent natural selection pressure in the population drives the virus to gradually accumulate favorable mutations6,9,10,11. Much attention has been paid to the mutations and evolution of SARS-CoV-212,13,14,15,16,17, since mutations are related to the infectivity and pathogenicity of viruses14,18,19,20,21,22. Beneficial mutants of the virus can better evolve and adapt to the host9, either strengthening or weakening the infectivity and pathogenicity. In addition, certain variants may generate drug resistance and reduce the efficacy of vaccines and therapeutics23,24,25,26,27,28. In short, studying mutations and evolution in detail is vital to understand the transformations of viral properties and to control the pandemic.
A new variant of SARS-CoV-2 named VOC-202012/01 (Variant of Concern 202012/01) or lineage B.1.1.7 was first detected in the United Kingdom last December29. It appears to be substantially more transmissible than other variants30. The variant has been growing exponentially in the United Kingdom and rapidly spreading to other countries31,32. However, it is not yet clear whether it evolved spontaneously in the United Kingdom or was imported from other countries. Studying how the variant B.1.1.7 mutates will enable researchers to track its spread over time and to understand the evolution of SARS-CoV-2.
In this study, large-scale SARS-CoV-2 sequences, consisting of more than 454,000 spike genes/proteins and 14,000 whole-genome sequences were analyzed. Our extensive sequence analysis showed that many mutations always co-occur not only in the spike protein of B.1.1.7, but in the whole genome of SARS-CoV-2. The mutation trajectories of the spike protein indicate that the early variant B.1.1.7 did not evolve spontaneously in the United Kingdom or even within human populations. We also investigated possible SARS-CoV-2 transmission chains of the variant B.1.1.7 based on the mutation analysis of large-scale spike proteins and the cluster analysis of spike genes. Over the whole genome, the top 25 high-frequency mutations of SARS-CoV-2 converged into several potential co-mutation patterns, each of which showed a strong correlation. The potential co-mutations depicted the evolutionary trajectory of SARS-CoV-2 virus in the population, shaping variable replication of SARS-CoV-2. In addition, we further explored the effect of the dominant (co-)mutations 5′UTR_c241t, NSP3_c3037t, and NSP12_c14408t on viral replication using a SARS-CoV-2 replicon based on a four-plasmid in vitro ligation system. The results suggest that such mutations significantly attenuate the replication of SARS-CoV-2.
Results
Evolutionary trajectories of variant B.1.1.7
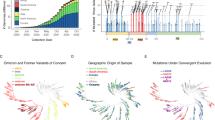
The variant B.1.1.7 was generally defined by multiple amino acid changes including three deletions (69–70 del and 145 del) and seven mutations (N501Y, A570D, D614G, P681H, T716I, S982A, and D1118H) in the spike protein31. The number of non-adjacent co-occurrent changes indicates that they resulted from accumulated mutations. We therefore explored the evolutionary trajectories of B.1.1.7 by tracing the incremental mutations (Fig. 1a). All routes along the directions of the arrows are possible evolutionary trajectories of lineage B.1.1.7. Among all the mutation routes, the green one was the most probable mutation trajectory based on the number of variant strains. However, it was unlikely that the earliest variant B.1.1.7 (GISAID: EPI_ISL_601443, 2020-09-20, England) with nine mutations evolved from the existing variants with 3–8 mutations, because the former arose much earlier than the latter. More than 454,000 SARS-CoV-2 strains have been collected and extensive sequenced from infected humans without finding intermediate variants with 3–9 mutations. It is therefore unlikely that the intermediate variants with 3–8 mutations have infected humans. Thus, the early variant B.1.1.7 might not have arisen spontaneously in the United Kingdom or within human populations. An alternative hypothesis is that spillover likely occurred from susceptible animals.

a Incremental mutations of variant B.1.1.7. The digits in the upper-right-corner rectangle with dotted line indicate the labels of mutations. For simplicity, the 69–70 deletions were labeled as “1”, and the other mutations “2”–“9”, respectively. The bottom nodes (rectangles) represent the variants with one mutation and the top one was the early variant B.1.1.7. Each rectangle with solid line consists of lineage (e.g., B.1.243), number of strains (e.g., N:2382), mutation sites (e.g., M:----56---), the earliest collection date (e.g., 20-03-29), and collection location (e.g., USA). In the labels of the mutation sites, sign “-” indicated the corresponding site did not mutate. All routes along the directions of the arrows are possible evolutionary trajectories of lineage B.1.1.7, where the green one was the most probable mutation trajectory. Large-scale SARS-CoV-2 analysis demonstrates that the early variant B.1.1.7 might not have arisen spontaneously in the United Kingdom or within human hosts. b Co-appearances of variant B.1.1.7 mutations. At least five mutations form a potential co-mutation pattern (coappearance rate >95%). c Possible transmission chains of variant B.1.1.7. Canidae, Mustelidae or Felidae, especially the Canidae family (e.g., dog) could be a possible host of the direct progenitor of variant B.1.1.7.
The coappearance rates (see Materials and methods) of all nine mutations are shown in Fig. 1b. We found that at least five mutations (145 del, A570D, T716I, S982A, and D1118H) of variant B.1.1.7 significantly co-occurred (rate > 95%), which indicates a potential co-mutation pattern in the spike protein, causing us to wonder what selection pressure drove such co-occurrences of mutations and rapid evolution in the population of SARS-CoV-2. Note that coronaviruses generally tend to exhibit rapid evolution when they jump to a different species33. We therefore analyzed the key spike genes and proteins of existing SARS-CoV-2 strains collected from animals to find a possible direct progenitor of variant B.1.1.7. The variant with mutations “56” (labeled by “*” in Fig. 1a, termed star variant) had the minimum phylogenetic distance with EPI_ISL_699508, which was collected from a dog on 2020-07-28 (Fig. 2) using MEGA34,35 (see Materials and methods). The strains collected from tigers, minks, and cats were also close to the star variant. Our extensive analyses including mutations, phylogeny (Fig. 2), collection date/location and the number of sequences (Supplementary Tables S1–S3) suggested that Canidae, Mustelidae or Felidae, especially the Canidae family (e.g., dog) could be a possible host of the direct progenitor of variant B.1.1.7. The possible transmission chains of variant B.1.1.7 are shown in Fig. 1c. This star variant strains in humans could not have evolved into the early variant B.1.1.7, but they might have infected high-density yet susceptible animals (such as dogs) and adapted to these species through rapid mutation. Such progenitor variants comprised most or all of the mutations of the early variant B.1.1.7 within the Canidae family populations, and they may have spilled back to humans after the rapid mutation period.

The digits on the left of the figure indicate the labels of mutations, which correspond with the mutation labels in Fig. 1a. The strains shown in the center of the figure contain at least one spike mutation of variant B.1.1.7. Moreover, these strain examples cover all existing SARS-CoV-2 viruses that collected from animal hosts. The strain labeled by orange star corresponds with the star variant in Fig. 1a. The strain with orange solid-round label was collected from a dog on 2020-07-28. Such two strains share the same mutations “56” and have the minimum phylogenetic distance by MEGA tool. Canidae, Mustelidae or Felidae, especially the Canidae family (e.g., dog) could be a possible host of the direct progenitor of variant B.1.1.7 based on the existing stains collected before the end of 2021-01.
High-frequency mutations converge into potential co-mutations
Based on sequence alignment and mutation analysis, we found that 7441 nucleotide alterations in the viral 29,903-letter RNA code occurred at least once in the samples from COVID-19 patients. These mutations were dispersed in the 14,427 SARS-CoV-2 strains collected from all around the world. As shown in the heatmap of the top 1% high-frequency mutations (Supplementary Table S4), some sites show very similar mutation rates on most days in samples isolated globally (Supplementary Fig. S1), including 8898 and 815 samples isolated from the U.S. (Supplementary Fig. S2) and Australia (Supplementary Fig. S3). Therefore, these mutations shown in Supplementary Fig. S4a were selected and clustered into co-occurrences, which we called potential co-mutation patterns. From the landscape of the mutation rates (Supplementary Fig. S4a), 25 nucleotide sites were clearly clustered into several potential co-mutation patterns. Among these patterns, there was one consisting of the top 4 high-frequency mutations (i.e., 5′UTR_c241t, NSP3_c3037t, NSP12_c14408t, and S_a23403g), which converged into a dominant potential co-mutation pattern. Such co-occurrence lineage has been found in almost all sequenced samples of SARS-CoV-2. Within this co-occurrence pattern, mutation S_ a23403g resulted in the amino acid change (D614G) that apparently enhances viral infectivity6,20, albeit debate exists18. Notably, there were three successive sites at the 28881st to 28883rd positions of the virus (N_g28881a, N_g28882a, and N_g28883c) that strictly co-occurred. Comparing Supplementary Fig. S4a–c and Table S4, we found that the top 14 high-frequency mutations formed five common co-occurrence patterns.
To assess the above co-occurrence patterns, we analyzed the correlations and statistical significance levels of the high-frequency co-occurrence mutations. The heatmap of the paired Pearson-correlation-coefficients (PCC, Fig. 3a) shows that the top 25 high-frequency mutations clearly cluster into several potential co-mutation groups/patterns with very strong correlation (≥0.8). By regression analyses, the above co-occurrence patterns have statistical significance levels with P-values < 10−44 (Fig. 3b). The detailed mutation transitions (Fig. 3c–f, g–i, j–k, l–n and Supplementary Figs. S5–S7) provide further evidence that the above mutations form co-mutation patterns.

a The correlation heatmap of the top 25 mutations. These mutations could be grouped into several clusters with high Pearson-correlation-coefficient (PCC). b Regression analysis of mutations shows that eight clusters all denote the statistical significance level: ***P-value < E-44. c–f, g–i, j–k, and l–n show the transitions of the high-frequency mutations. The sky-blue represents the rate per day of initial residue in population and the golden represents the rate per day of substitution/mutant. These mutation transitions provide further evidence that the above mutations potentially form co-mutation patterns.
Dominant mutations attenuate viral replication
We further explored the effect of the dominant mutations 5′UTR_c241t, NSP3_c3037t, and NSP12_c14408t on viral replication using a SARS-CoV-2 replicon based on a four-plasmid in vitro ligation system. This replicon is devoid of the viral structural proteins while undergoing viral replication, and the viral replication is sensitive to the antiviral agent remdesivir36. The 5′UTR_c241t mutation resides in a highly conserved region in the 5′UTR (Fig. 4a). The NSP3_c3037t mutation is synonymous. The NSP12_c14408t mutation is nonsynonymous with an amino acid change of a conserved amino acid P323 in the viral RNA-dependent RNA polymerase (Fig. 4b). We introduced the NSP12_c14408t mutation or the NSP12_c14408t mutation with the other two mutations 5′UTR_c241t and NSP3_c3037t into the replicon plasmids. The fragments were released from the plasmids by BsaI digestion, and then assembled by in vitro ligation with T4 ligase (Fig. 4c). Replicon RNA transcribed from the ligation products was co-transfected with nucleocapsid (N) mRNA into Huh7 cells. RNA replication was monitored by measuring the secreted Gaussia luciferase activity in the supernatants. Enzymatic dead mutants (759-SAA-761) of the RNA-dependent RNA polymerase (RdRp) NSP12 were introduced, and the mutated replicon served as a non-replication control. As shown in Fig. 4d, transfection of wild type replicon RNA resulted in an obvious increase of luciferase activity, and dead mutant SAA RNA did not replicate. Introduction of NSP12_c14408t mutation resulted in a significant reduction of viral replication. The combination of NSP12_c14408t mutation with the other two mutations further reduced viral replication. These results demonstrate that the P323L mutation in the viral RdRp reduces viral replication, and the synonymous mutations may further attenuate viral replication.

a Predicted RNA structure of the SARS-CoV-2 5′UTR. RNA structure of the 400-nt 5′UTR was predicted by “RNAstructure” (http://rna.urmc.rochester.edu/RNAstructureWeb). The start codon for nsp1 is gray, the TRS-L is orange, and the mutated nucleotides are red. The bottom panel shows the alignment of the 5′UTR of SARS-CoV-2 with 5′UTRs of related viruses, with c241 highlighted. b Structure of SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) or RNA complex. The structure of SARS-CoV-2 RdRp/RNA complex (PDB, 6X2G) was visualized by Chimera (UCSF). The P323 mutation is highlighted in red, with the alignment of the amino acid sequences of SARS-CoV-2 and related viruses near the P323 position. c Schematic of the in vitro ligation system for SARS-CoV-2 replicon. Four plasmids encompassing the viral genome were digested by BsaI to release the four fragments. After gel purification, the fragments were ligated by T4 ligase. The ligation products were purified and used as template for RNA in vitro transcription. sGluc, secreted Gaussia luciferase; 2A, foot-and-mouth disease virus (FMDV) 2A peptide; BSD, blasticidin. d Huh7 cells were co-transfected with in vitro transcribed replicon RNA (wild type (WT) or the indicated mutants) and an mRNA encoding the SARS-CoV-2 N protein. The luciferase activity in the supernatants was measured at the time points indicated. Medium was changed at 8 h post-transfection. Data are shown as mean ± SEM (n = 8). SAA, the NSP12 polymerase active-site mutant. Unpaired Student’s t-test was performed between the mutants and WT and between the mutants as indicated. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
A well-resolved phylogeny of variant B.1.1.7 spike genes provides an opportunity to understand the evolutionary process and transmission chains of variant B.1.1.7. Our incremental mutation and phylogenetic analyses on large-scale SARS-CoV-2 spike proteins/genes revealed that the early variant B.1.1.7 might not have evolved spontaneously in the United Kingdom or within human populations. In this case, the spillover likely occurred from susceptible animals. Current evidence37,38,39 indicates that SARS-CoV-2 can effectively infect both domestic animals (for example, dog, cat, pig, and bovine) and wild animals (e.g., mink, rabbit, and fox) by binding host angiotensin converting enzyme 2 (ACE2). Our further analyses including mutations, phylogeny, collection date/location and the number of sequences suggested that the earliest variant B.1.1.7 possibly originated from Canidae, Mustelidae or Felidae, especially the Canidae family (e.g., dog). The cases40 that the variant B.1.1.7 can easily infect dogs and cats indicated that both are susceptible to B.1.1.7. Still, due to the limited information available to date, an alternative hypothesis is that the direct progenitor of variant B.1.1.7 is yet to be sampled. In addition to variant B.1.1.7, as a future topic we will work on the analysis of other lineages such as P.1, B.1.351, B.1.427, and B.1.42, when sufficient numbers of their sequences are available.
By tracing the mutation trajectories, we found that at least five mutations of the spike proteins always co-occurred, and a large number of potential co-mutations appeared in the top 1% high-frequency mutations of SARS-CoV-2 whole genome. It has been documented that the mutation S_a23403g results in the amino acid change of the spike protein D614G and enhances viral infectivity14,41,42,43,44. Here, by using a SARS-CoV-2 reporter replicon system, we demonstrated that one of the dominant co-mutations NSP12_c14408t significantly reduced viral replication and combination of NSP12_c14408t mutation with the other two synonymous mutations 5′UTR_c241t and NSP3_c3037t further reduced viral replication further. As the 5′UTR plays an important role in regulating viral replication, the synonymous mutations 5′UTR_c241t may attenuate viral replication by change RNA secondary structure45. These findings imply that SARS-CoV-2 undergoes an evolution toward enhancing viral infectivity while attenuating viral replication. SARS-CoV-2 has exhibited significant mutations and co-mutations. We evaluated the replication of a co-mutation pattern including three dominant mutations. Whether other mutations act similarly on the viral replication needs to be verified. These results can be further explored for efficient vaccine design by combining the information of the associated network biomarkers46,47, dynamic network biomarkers48,49,50 or conserved k-mers51,52,53 in our future work. In summary, this study provides insights into the transmission chains of variant B.1.1.7 and the effect of viral dominant mutations on viral evolution.
Materials and methods
Data selection and pre-processing
The 454,443 spike gene/protein sequences of SARS-CoV-2 were obtained at https://www.gisaid.org/. The NCBI website at https://www.ncbi.nlm.nih.gov/sars-cov-2/ has released >1.7 thousand sequences of SARS-CoV-2 viruses before July 31, 2020. We selected 14,427 sequences that satisfied two criteria: (1) having specific collection dates; (2) sequence-lengths being no less than 29,305 nt (29903*0.98). It is inevitable that some sites of sequences are equivocal owing to the limitation of sequencing depth. For instance, many sites were labeled as letter N in genome sequences. The noise of indeterminate nucleic acids was taken into consideration in our experiments so as to boost accuracy. The co-mutation rate of multi-site co-mutations was calculated by \({\mathrm{co}} {\hbox{-}} {\mathrm{mutation}}\;{\mathrm{rate}} = \frac{{{\mathrm{number}}\;{\mathrm{of}}\;{\mathrm{sequences}}\;{\mathrm{containing}}\;{\mathrm{co}}{\hbox{-}} {\mathrm{mutations}}}}{{{\mathrm{number}}\;{\mathrm{of}}\;{\mathrm{all}}\;{\mathrm{sequences}}}}\). Moreover, the co-appearance rate of a mutation in B.1.1.7 variant was defined by \({\mathrm{co}} {\hbox{-}} {\mathrm{appearance}}\;{\mathrm{rate}} = \frac{{{\mathrm{number}}\;{\mathrm{of}}\;{\mathrm{B}}.{\mathrm{1}}.{\mathrm{1}}.{\mathrm{7}}\;{\mathrm{sequences}}}}{{{\mathrm{number}}\;{\mathrm{of}}\;{\mathrm{sequences}}\;{\mathrm{containing}}\;{\mathrm{a}}\;{\mathrm{mutation}}}}\).
Analyses on the possible animal hosts
In addition to the phylogenetic analysis, we further explored the possible animal hosts of the direct progenitor of variant B.1.1.7 by mutations, collection time/space of strains, the number of sequences and the edit distance54,55 of mutations (Supplementary Tables S1, S2). Owing to the late lockdown policies of some governmental agencies, the spread of SARS-CoV-2 has not been prevented well in Europe, America, and Australia. We could ignore the impact of policies for studying the origin of variant B.1.1.7. We quantified the multiple impact factors of viral transmission as shown in Supplementary Table S3 based on the criterion that the smaller the value, the more similar to the star variant. The results still supported that the Canidae family is a possible host of the direct progenitor of variant B.1.1.7.
MEGA version and parameter settings
Version: MEGA-X
Statistical method: maximum likelihood
Test of phylogeny: none
Model/method: Jones-Taylor-Thornton (JTT) model
Rates among sites: uniform rates
Gaps/missing data treatment: use all sites
ML Heuristic method: nearest-neighbor-interchange (NNT)
Initial tree for ML: make initial tree automatically (Default-NJ/BioNJ)
Branch swap filter: none
Number of threads: 7.
Statistical analysis
PCC is a classic statistic that measures linear correlation between two variables. Its value ranges from –1.0 to 1.0. Normally, the two variables meet a strong correlation or a very strong correlation when the absolutes value of PCC is between 0.6 and 0.8 or between 0.8 and 1.0. Linear regression is a linear approach to model the relationship between a scalar response and one or more variables. We used PCC and significance level (P-value) of regression analysis to evaluate the relationships of the co-occurrence mutations in large-scale SARS-CoV-2 examples.
Plasmids
Four plasmids encompassing the viral genome (pLC-nCoV-A-BsaI, pLC-nCoV-B-BsaI, pLC-nCoV-C-BsaI, and pnCoV-D-sGluc-BsaI) were described previously36. The 5′UTR_c-241-t and NSP3_c-3037-t mutations were introduced into the pLC-nCoV-A-BsaI by fusion Polymerase Chain Reaction (PCR). The NSP12_c-14408-t mutation was introduced into the pLC-nCoV-B-BsaI by fusion PCR.
Cell lines
The human hepatoma cells Huh 7 were purchased from the Cell Bank of the Chinese Academy of Sciences (www.cellbank.org.cn) and routinely maintained in Dulbecco’s modified medium supplemented with 10% FBS (Gibco) and 25 mM HEPES (Gibco).
In vitro ligation
BsaI digested fragments were gel purified using Gel Extraction Kit (OMEGA) and ligated with T4 ligase (New England Biolabs) at room temperature for 1 h. The ligation products were phenol/chloroform extracted, precipitated by absolute ethanol, and resuspended in nuclease-free water, quantified by determining the A260 absorbance.
In vitro transcription
Purified in vitro ligated product was used as template for the in vitro transcription by mMESSAGE mMACHINE T7 Transcription Kit (Ambion) according to the manufacturer’s protocol. For N mRNA production, we amplified the N protein-coding region by PCR (sense: GGC ACA CCC CTT TGG CTC T; antisense: TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TCT AGG CCT GAG TTG AGT CAG CAC) with phCMV-N as template. Then the purified PCR product was used as a template for in vitro transcription by mMESSAGE mMACHINE T7 Transcription Kit as described above. RNA was purified by RNeasy mini Elute (Qiagen), eluted in nuclease-free water, and quantified by UV absorbance (260 nm).
Transfection
Cells were seeded onto 48-well plates at a density of 7.5 × 104 per well and then transfected with 0.3 µg in vitro transcribed RNA using a TransIT-mRNA transfection kit (Mirus) according to the manufacturer’s protocol.
Luciferase activity
Supernatants were taken from cell medium and mixed with equal volumes of 2× lysis buffer (Promega). Luciferase activity was measured with Renilla luciferase substrate (Promega) according to the manufacturer’s protocol.
Data availability
The raw sequence data reported in this paper have been deposited in the GISAID and NCBI websites at https://www.gisaid.org/ and https://www.ncbi.nlm.nih.gov/sars-cov-2/, respectively. Code is available from the corresponding author on reasonable request.
References
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Chinazzi, M. et al. The effect of travel restrictions on the spread of the 2019 novel coronavirus (COVID-19) outbreak. Science 368, 395–400 (2020).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Chen, L. et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 9, 313–319 (2020).
Korber, B. et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827 (2020).
Smith, E. C., Blanc, H., Vignuzzi, M. & Denison, M. R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 9, e1003565 (2013).
Sevajol, M., Subissi, L., Decroly, E., Canard, B. & Imbert, I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 194, 90–99 (2014).
Vignuzzi, M., Stone, J. K., Arnold, J. J., Cameron, C. E. & Andino, R. J. N. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 (2006).
Garvin, M. R. et al. Potentially adaptive SARS-CoV-2 mutations discovered with novel spatiotemporal and explainable AI models. Genome Biol. 21, 304 (2020).
Yao, H. P. et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 6, 76 (2020).
Tang, X. et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci. Rev. 7, 1012–1023 (2020).
Acter, T. et al. Evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as coronavirus disease 2019 (COVID-19) pandemic: A global health emergency. Sci. Total Environ. 730, 138996 (2020).
Plante, J. A. et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592, 116–121 (2020).
Zohar, T. et al. Compromised humoral functional evolution tracks with SARS-CoV-2 mortality. Cell 183, 1508–1519.e1512 (2020).
Li, T. et al. The use of SARS-CoV-2-related coronaviruses from bats and pangolins to polarize mutations in SARS-Cov-2. Sci. China Life Sci. 63, 1608–1611 (2020).
Lu, B. et al. Integrated characterization of SARS-CoV-2 genome, microbiome, antibiotic resistance and host response from single throat swabs. Cell Discov. 7, 19 (2021).
Grubaugh, N. D., Hanage, W. P. & Rasmussen, A. L. Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell 182, 794–795 (2020).
Liu, Z. et al. Identification of common deletions in the spike protein of severe acute respiratory syndrome coronavirus 2. J. Virol. 94, e00790–20 (2020).
Li, Q. Q. et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182, 1284 (2020).
Blanco, J. D., Hernandez-Alias, X., Cianferoni, D. & Serrano, L. In silico mutagenesis of human ACE2 with S protein and translational efficiency explain SARS-CoV-2 infectivity in different species. PLoS Comput Biol. 16, e1008450 (2020).
Zhang, L. Z. et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 11, 6013 (2020).
Baum, A. et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 369, 1014–1018 (2020).
Hansen, J. et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369, 1010–1014 (2020).
Sheahan, T. P. et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 12, eabb5883 (2020).
Nunes-Santos, C. J., Kuehn, H. S. & Rosenzweig, S. D. N-glycan modification in Covid-19 pathophysiology: in vitro structural changes with limited functional effects. J. Clin. Immunol. 41, 335–344 (2020).
Lo, M. K. et al. Remdesivir targets a structurally analogous region of the Ebola virus and SARS-CoV-2 polymerases. Proc. Natl Acad. Sci. USA 117, 26946–26954 (2020).
Zhou, Y. D. et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 6, 14 (2020).
Editorial. Evolution goes viral. Nat. Ecol. Evolution 5, 143–143 (2021).
Editorial. COVID-19 vaccines: acting on the evidence. Nat. Med. https://doi.org/10.1038/s41591-021-01261-5 (2021).
Muik, A. et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine–elicited human sera. Science 371, 1152–1153 (2021).
Rice, B. L. et al. Variation in SARS-CoV-2 outbreaks across sub-Saharan Africa. Nat. Med. https://doi.org/10.1038/s41591-021-01234-8 (2021).
Zhou, P. & Shi, Z.-L. SARS-CoV-2 spillover events. Science 371, 120–122 (2021).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evolution 35, 1547–1549 (2018).
Tamura, K. & Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-dna in humans and chimpanzees. Mol. Biol. Evolution 10, 512–526 (1993).
Zhang, Y., Song, W., Chen, S., Yuan, Z. & Yi, Z. A bacterial artificial chromosome (BAC)-vectored noninfectious replicon of SARS-CoV-2. Antiviral Res. 185, 104974 (2020).
Shi, J. et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science 368, 1016–1020 (2020).
Wu, L. et al. Broad host range of SARS-CoV-2 and the molecular basis for SARS-CoV-2 binding to cat ACE2. Cell Discov. 6, 68 (2020).
Patterson, E. I. et al. Evidence of exposure to SARS-CoV-2 in cats and dogs from households in Italy. Nat. Commun. 11, 6231 (2020).
Ferasin, L. et al. Myocarditis in naturally infected pets with the British variant of COVID-19. bioRxiv https://doi.org/10.1101/2021.03.18.435945 (2021).
Raghav, S. et al. Analysis of Indian SARS-CoV-2 genomes reveals prevalence of d614g mutation in spike protein predicting an increase in interaction with TMPRSS2 and virus infectivity. Front. Microbiol. 11, 594928 (2020).
Johnson, M. C. et al. Optimized pseudotyping conditions for the SARS-COV-2 spike glycoprotein. J. Virol. 94, e01062–20 (2020).
Jiang, X. Y. et al. Bimodular effects of D614G mutation on the spike glycoprotein of SARS-CoV-2 enhance protein processing, membrane fusion, and viral infectivity. Signal Transduct. Tar. 5, 268 (2020).
Fernandez, A. Structural impact of mutation D614G in SARS-CoV-2 spike protein: enhanced infectivity and therapeutic opportunity. ACS Medicinal Chem. Lett. 11, 1667–1670 (2020).
Sun, L. et al. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell 184, 1865 (2021).
Liu, X. P., Wang, Y. T., Ji, H. B., Aihara, K. & Chen, L. N. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 44, e164 (2016).
Zhang, W. W., Zeng, T., Liu, X. P. & Chen, L. N. Diagnosing phenotypes of single-sample individuals by edge biomarkers. J. Mol. Cell Biol. 7, 231–241 (2015).
Chen, L. N., Liu, R., Liu, Z. P., Li, M. Y. & Aihara, K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep.-UK 2, 342 (2012).
Yang, B. W. et al. Dynamic network biomarker indicates pulmonary metastasis at the tipping point of hepatocellular carcinoma. Nat. Commun. 9, 678 (2018).
Liu, X. P. et al. Detection for disease tipping points by landscape dynamic network biomarkers. Natl Sci. Rev. 6, 775–785 (2019).
Zhang, J. S., Wang, Y. L. & Yang, D. Y. CCSpan: Mining closed contiguous sequential patterns. Knowl.-Based Syst. 89, 1–13 (2015).
Zhang, J. S., Wang, Y. L., Zhang, C. & Shi, Y. Y. Mining contiguous sequential generators in biological sequences. IEEE ACM Trans. Comput. Biol. Bioinform. 13, 855–867 (2016).
Zhang, J. S. et al. Efficient mining multi-mers in a variety of biological sequences. IEEE ACM Trans. Comput. Biol. Bioinform. 17, 949–958 (2020).
Ristad, E. S. & Yianilos, P. N. Learning string-edit distance. IEEE Trans. Pattern Anal. Mach. Intell. 20, 522–532 (1998).
Bille, P. A survey on tree edit distance and related problems. Pattern Anal. Appl. 337, 217–239 (2005).
Acknowledgements
We thank associate Prof. Tao Zeng, Dr. Hao Dai, and Dr. Shutao He for useful comments on the manuscript. This work is supported by the National Key Research and Development Program of China (2017YFA0505500 to L.C., 2017YFC0909502 to J.Z.); the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB38040400 to L.C.); National Science Foundation of China (31771476, 31930022, and 12026608 to L.C., 61602460 and 11701379 to J.Z.); Shanghai Municipal Science and Technology Major Project (2017SHZDZX01 to L.C.); National Science and Technology Major Project of China (2017ZX10103009 to Z.Y.); Emergency Project of Shanghai Science and Technology Committee (20411950103 to Z.Y.); National Postdoctoral Program for Innovative Talent (BX20180331 to J.K.); and China Postdoctoral Science Foundation (2018M642018 to J.K.).
Author information
Authors and Affiliations
Contributions
L.C. and J.Z. designed the study. Z.Y. and J.Z. designed the experiments. J.Z. analyzed data. Y.Z. performed the experiments of viral replication. J.Z., Z.Y., and J.-Y.K. designed the figures. S.C. repeated and checked the experiments of viral replication. B.H. checked the computational analyses. J.Z. and Z.Y. wrote the manuscript. Y.H., M.-F.L., L. Lu, and L. Li polished the manuscript. All authors participated in result interpretation and discussion.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Zhang, Y., Kang, JY. et al. Potential transmission chains of variant B.1.1.7 and co-mutations of SARS-CoV-2. Cell Discov 7, 44 (2021). https://doi.org/10.1038/s41421-021-00282-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-021-00282-1
