
Abstract
Heart failure (HF) has reached epidemic proportions in developed countries, affecting over 20 million people worldwide. Despite modern medical and device therapies, 60–70% of HF patients still die within 5 years of diagnosis as it relentlessly progresses through pervasive apoptotic loss of cardiomyocytes. Although fortilin, a 172-amino-acid anti-p53 molecule, is one of the most expressed proteins in the heart, its precise role there has remained unknown. Also unclear is how cardiomyocytes are protected against apoptosis. Here, we report that failing human hearts express less fortilin than do non-failing hearts. We also found that mice lacking fortilin in the heart (fortilinKO-heart) die by 9 weeks of age due to extensive cardiomyocyte apoptosis and severe HF, which suggests that fortilin sustains cardiomyocyte viability. The lack of fortilin is also associated with drastic upregulation of p53 target genes in the hearts. The heart-specific deletion of p53 in fortilinKO-heart mice extends their life spans from 9 to 18 weeks by mitigating cardiomyocyte apoptosis. Our data suggest that fortilin is a novel cardiac p53 inhibitor and that its inadequate expression in failing hearts and subsequent overactivation of the p53 apoptosis pathway in cardiomyocytes exacerbates HF.
Similar content being viewed by others

Introduction
Heart failure (HF)—a complex clinical syndrome secondary to structural and functional impairments of the heart muscle—has reached epidemic proportions in developed countries, currently affecting over 20 million people worldwide. Despite modern medical and device therapies, 30–40% of HF patients die within 1 year and 60–70% die within 5 years of diagnosis [1]. Although coronary artery disease and hypertension are the two most common causes of HF [2], HF progresses even after they are adequately treated, as biological changes initiated in failing human cardiomyocytes continue to expand [3]. These changes include myocyte hypertrophy, desensitization of β-adrenergic signaling, changes in excitation–contraction coupling, progressive loss of myofilaments within cardiomyocytes, and, most importantly, gradual and irreversible apoptotic loss of cardiomyocytes [3,4,5,6,7]. Although prevention of cardiomyocyte apoptosis could slow the progression of HF, how apoptosis is regulated in the heart is unclear.
Fortilin (also known as translationally controlled tumor protein, histamine-releasing factor, and TPT1) is a highly conserved, 172-amino-acid, 20 kDa protein that blocks apoptosis [8,9,10], and it is one of the most expressed genes in the heart [11]. Fortilin directly binds to and negatively regulates the tumor suppressor protein p53 [12, 13] and the endoplasmic reticulum (ER)-stress handling protein IRE1α [14], both of which promote loss of cardiomyocytes and subsequent HF [15,16,17]. Despite its abundance, the exact role of fortilin in the heart is poorly understood. Here we present evidence that fortilin is a major negative regulator of p53 in the heart and that its lack leads to massive apoptotic loss of cardiomyocytes and lethal HF.
Results
Human HF and fortilin
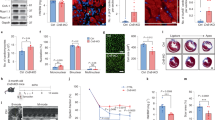
To evaluate the role of fortilin in the heart and in human HF, we subjected tissue lysates of human hearts from subjects with non-failing hearts (NFHs) and HF patients with non-ischemic cardiomyopathy (NICM) and ischemic cardiomyopathy (ICM) from the Duke Human Heart Repository [18] to an automated capillary-based quantitative Western blot analyses (JESS™, Protein Simple) [19, 20] (Fig. 1a). We calculated a fortilin expression index by dividing the area under the curve of a fortilin peak by the total proteins loaded in the same capillary. We found that fortilin expression was significantly lower in NICM and ICM hearts than in NFHs (Fig. 1b).

a, b Fortilin expression levels were lower in the hearts of patients with NICM and ICM than in those of NFH patients. c A strain of mice lacking fortilin in the cardiomyocytes (fortilinKO-heart) (3a) and its control (fortilinWT-heart) (3b) were generated by crossing fortilinflox/flox (1) and αMHC-Cre+/+ mice (2). d Fortilin protein levels were drastically lower in the hearts of fortilinKO-heart mice than in those of fortilinWT-heart mice. e All fortilinKO-heart mice died by 9 weeks of age. f, g Transthoracic echocardiography at 7 weeks of age revealed dilated, thinned, and severely dysfunctional left ventricle in fortilinKO-heart mice. h Sagittal heart sections of fortilinWT-heart and fortilinKO-heart mice. NFH non-failing hearts, NICM non-ischemic cardiomyopathy, ICM ischemic cardiomyopathy, JESS an automated capillary-based quantitative Western blot system with in-capillary total protein evaluation capability, A.U. arbitrary units, WT fortilinWT-heart (or αMHC-Cre−/−fortilinflox/flox) mice that express fortilin normally in the heart, KO fortilinKO-heart (or αMHC-Cre+/+fortilinflox/flox) mice that do not express fortilin in the heart, GAPDH glyceraldehyde 3-phosphate dehydrogenase, LVEF left ventricular (LV) ejection fraction, FS fractional shortening, LVIDs LV internal diameters in systole, LVIDd LV internal diameters in diastole, IVSd interventricular septum thickness in diastole, LVPWd LV posterior wall thickness in diastole. Scale bar = 500 µm; Error bars, means ± SD, statistical analyses performed using Student’s two-sample t-test, NS not statistically significant; *P < 0.05; ***P < 0.005; ****P < 0.001 (see also Fig. S1).
Heart-specific fortilin-knockout (KO) mice
To test the hypothesis that fortilin deficiency in the heart leads to HF, we first generated fortilinflox/flox mice using standard homologous recombination techniques [21]. We then generated heart-specific fortilin KO mice (αMHC-Cre+/+fortilinflox/flox, referred to here as fortilinKO-heart mice) by crossing fortilinflox/flox mice with mice over-expressing Cre-recombinase under the control of the cardiac-specific Myh6 promoter [22] (αMHC-Cre+/+, Jackson Laboratory) (Fig. 1c). In cardiomyocytes of fortilinKO-heart mice, the fortilin genomic sequence flanked by the LoxP sequences (Fig. S1a-1) is excised by the tissue-specifically expressed Cre-transgene (Figs. S1a-2, S1a-3a), while fortilin is normally expressed in all other tissue (Fig. S1a-3b). We used αMHC-Cre−/−fortilinflox/flox (fortilinWT-heart) mice as the control. As expected, fortilin was not detectable in the hearts of fortilinKO-heart mice, whereas it was normally expressed in all other tissues at both message (Fig. S1b) and protein (Fig. 1d) levels.
Although apparently normal at birth and fertile, fortilinKO-heart mice started to die as early as 6 weeks of age and all were dead by 9 weeks of age (Fig. 1e). We performed transthoracic echocardiography on the surviving mice at 7 weeks of age (Fig. 1f) and found that the hearts were (i) dilated (increased left ventricular (LV) internal diameter in systole (LVIDs) and increased LV internal diameter in diastole (LVIDd)), (ii) thinned (decreased interventricular septum thickness in diastole (IVSd) and decreased LV posterior wall thickness in diastole (LVPWd)), and (iii) severely dysfunctional (decreased LV ejection fraction (LVEF) and decreased fractional shortening (FS) [%]) (Fig. 1g), consistent with the gross examination of the hearts of fortilinWT-heart and fortilinKO-heart mice (Fig. 1h). In addition, the ratio of heart weights to body weights (HW/BW) and that of lung weights to body weights (LW/BW) of fortilinKO-heart mice were significantly greater than those of fortilinWT-heart mice at 8 weeks of age (Fig. S1c), suggesting the presence of cardiomegaly and lung congestion, respectively, in fortilinKO-heart mice. Further, RT-qPCR assays on RNAs from the hearts revealed the upregulation of heart-failure genes [23, 24] (Col1, Myh7, and Anf) in fortilinKO-heart mice, but not in fortilinWT-heart mice (Fig. S1d).
The size of cardiomyocytes did not differ between fortilinWT-heart and fortilinKO-heart mice (Fig. 2a), but both Masson and picrosirius red staining showed severe fibrosis in the hearts of fortilinKO-heart mice (Fig. 2b). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) revealed a drastically higher frequency of apoptotic cardiomyocytes in fortilinKO-heart hearts compared to fortilinWT-heart hearts (Fig. 2c). Additionally, the immunogenicity of the apoptosis inducer BAX was seven times greater in fortilinKO-hearts than in fortilinWT-hearts (Fig. 2d).

a The size of cardiomyocytes was similar in fortilinWT-heart and fortilinKO-heart mice, as shown in laminin staining. b Both Masson and picrosirius red staining showed severe fibrosis in the heart of fortilinKO-heart mice. c TUNEL staining revealed a significantly higher frequency of apoptotic cardiomyocytes in fortilinKO-heart hearts compared to fortilinWT-heart hearts. d BAX immunogenicity was drastically greater in fortilinKO-heart than in fortilinWT-heart mice. e The message levels of the p53-target, pro-apoptotic genes BAX, PUMA, and NOXA were significantly greater in the hearts of fortilinKO-heart mice than in those of fortilinWT-heart mice according to RT-qPCR assays of the heart RNAs. f The protein levels of BAX, PUMA, and NOXA were significantly greater in the hearts of fortilinKO-heart mice than in those of fortilinWT-heart mice, as shown by Western blot analyses of the heart lysates. A.U. arbitrary units, WT fortilinWT-heart (or αMHC-Cre−/−fortilinflox/flox) mice that express fortilin normally in the heart, KO fortilinKO-heart (or αMHC-Cre+/+fortilinflox/flox) mice, TUNEL terminal deoxynucleotidyl transferase dUTP nick-end labeling to identify apoptotic cells; scale bar = 100 µm; IB immunoblot, α-fortilin anti-fortilin antibody, GAPDH glyceraldehyde 3-phosphate dehydrogenase. Error bars means ± SD, statistical analyses performed using Student’s two-sample t-test: NS not statistically significant; *P < 0.05; **P < 0.01; ***P < 0.005, ****P < 0.001.
Chen et al. reported that fortilin binds to the sequence-specific DNA binding domain of p53, which blocks p53-induced transcriptional activation of BAX [12]. To test the status of activation of BAX, PUMA, and NOXA, which are all p53-target, pro-apoptotic genes, in the hearts of fortilinKO-heart mice, we subjected the hearts to both RT-qPCR and Western blot analyses. All three genes were significantly more expressed in the hearts of fortilinKO-heart mice than in those of fortilinWT-heart mice at both message (Fig. 2e) and protein (Fig. 2f) levels, suggesting that the lack of fortilin in cardiomyocytes may lead to p53-induced transcriptional activation of these genes, leading to cardiomyocyte apoptosis and myocardial fibrosis. The results of these processes are severe HF and death of fortilinKO-heart mice.
Lack of p53 improved survival
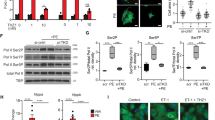
To test the hypothesis that the lack of fortilin causes the heart to fail through overactivation of the p53 apoptotic pathway, we knocked out p53 by crossing fortilinKO-heart mice with p53flox/flox mice [25] and generated fortilinKO-heartp53KO-heart and fortilinWT-heartp53KO-heart mice (Fig. S2a and S2b). Fortilin expression levels were significantly higher in fortilinWT-heartp53WT-heart mice (fortilinWTp53WT, hereafter) than in fortilinKOp53WT and fortilinKOp53KO mice (the latter two had comparable expression), both at the message (Fig. S2c, fortilin, a vs. b and a vs. c) and protein (Fig. 3a and b, fortilin, a vs. b and a vs. c) levels. In addition, total p53 expression levels were significantly higher in fortilinKOp53WT than in fortilinKOp53KO mice, both at the message (Fig. S2c, p53, b vs. c) and protein levels (Fig. 3a and b, p53, b vs. c; Fig. S3a, p53, b vs. c). p53 expression levels also were significantly higher in fortilinKOp53WT than in fortilinWTp53WT mice, both at the message (Fig. S2c, p53, a vs. b) and protein levels (Fig. 3b, p53, a vs. b; Fig. S3a, p53, a vs. b). On the other hand, phosphorylated p53, an active form of p53, was most abundant in the hearts of fortilinKOp53WT mice, followed by those of fortilinWTp53WT and then by those of fortilinKOp53KO (Fig. S3a, phospho-p53, b vs. a vs. c), suggesting that the lack of fortilin increases not only total p53 protein but also phosphorylated and activated p53 protein.

a, b Western blot analyses show that fortilin protein levels were significantly lower in fortilinKOp53WT and fortilinKOp53KO mice than in fortilinWTp53WT mice. p53 protein levels were significantly lower in fortilinKOp53KO mice than in fortilinKOp53WT mice. c The lack of p53 in the heart of fortilinKO-heart mice increased the M.S. from 8 to 18 weeks. d, e Echocardiography revealed that the hearts of fortilinKOp53KO mice had better overall heart function than those of fortilinKOp53WT mice, as evidenced by greater LVEF, less LVIDs (less cavity dilatation), and greater IVSd and LVPWd (less wall thinning). f Immunohistochemistry showed that the lack of p53 drastically reduced the expression levels of both BAX and cleaved lamin, an apoptosis marker. g Western blot analyses showed that the expression levels of BAX, PUMA, and NOXA were significantly decreased when p53 was deleted from fortilin-deficient hearts. a fortilinWTp53WT mice (αMHC-Cre−/−fortilinflox/floxp53flox/flox mice), b fortilinKOp53WT mice (αMHC-Cre+/+fortilinflox/floxp53WT/WT mice), c fortilinKOp53KO mice (αMHC-Cre+/+fortilinflox/floxp53flox/flox mice), IB immunoblot, α-fortilin anti-fortilin antibody, GAPDH glyceraldehyde 3-phosphate dehydrogenase, A.U. arbitrary unit, M.S. median survival, LVEF left ventricular (LV) ejection fraction, LVIDs LV internal diameters in systole, LVIDd LV internal diameters in diastole, LVPWd LV posterior wall thickness in diastole, IVSd interventricular septum thickness in diastole. Scale bar = 200 µm; Error bars, means ± SD, statistical analyses performed using ANOVA with Fisher’s multiple comparison except for the survival assay, for which the Log-Rank (Mantel–Cox) test was used: NS not statistically significant; *P < 0.05; **P < 0.01; ***P < 0.005, ****P < 0.001 (see also Figs. S2 and S3).
Because fortilin binds p53 and facilitates its proteasome-mediated degradation at the protein level [13], it is expected the protein levels of total p53 to be significantly higher in the heart of fortilinKOp53WT mice than in those of fortilinWTp53WT mice as we observed above. However, this process does not explain why the message levels of p53 were also significantly higher in the hearts of fortilinKOp53WT mice than in those of fortilinWTp53WT mice (Fig. S2c, p53, a vs. b).
Therefore, we tested the hypothesis that fortilin represses the transcription of p53. We first stably transduced H9C2 rat heart myoblast cells with a lentiviral construct containing both the p53 promoter sequence driving the expression of Gaussia luciferase (GLuc) and the secreted alkaline phosphatase (SEAP) gene under the control of the constitutional CMV promoter (H9C2p53-pomoter-GLuc/SEAP cells). We then transiently silenced fortilin by treating the cells with lentiviral vector containing shRNA against fortilin (sh-fortilin). These cells were irradiated with a moderate X-ray dose (8 Gy) and subjected to the GLuc-SEAP assay (Fig. S3b). Treatment with sh-fortilin effectively silenced fortilin in the cell (Fig. S3c), whereas X-ray irradiation nearly doubled the expression of p53 in the cells (Fig. S3d). In this system, we found that the knockdown of fortilin protein activated the p53 promoter with and without X-ray irradiation (Fig. S3e, columns 1 vs. 3; columns 2 vs. 4). Taken together, these data suggest that fortilin is a transcriptional repressor of p53 transcription in addition to facilitating degradation of p53 protein.
We also found that fortilinKOp53KO mice, which express less p53 in the heart than do fortilinKOp53WT mice (Fig. 3a and b), survived significantly longer that fortilinKOp53WT mice (Fig. 3c, median survival, fortilinKOp53WT vs. fortilinKOp53KO = 8.0 vs. 18.0 weeks, P < 0.0003 by Log-rank Mantel–Cox test). Echocardiography showed that the hearts of fortilinKOp53KO mice had better overall heart function (Fig. 3d), as evidenced by greater LVEF and FS, less LV dilatation, and less LV wall thinning (Figs. 3e and S2d). The expression of the αMHC-Cre transgene in the heart did not impact LV systolic function as measured by LVEF and FS (Fig. S2e).
Immunohistochemical staining of myocardium showed that the lack of p53 drastically reduced the expression levels of both BAX and cleaved lamin, which are apoptosis markers (Fig. 3f). Consistently, Western blots revealed that the protein levels of BAX, PUMA, and NOXA were drastically decreased when p53 was deleted from fortilin-deficient hearts (Fig. 3g, b vs. c). These data suggest that fortilin sustains heart function by negatively regulating p53 and that the lack of fortilin leads to inappropriate overactivation of the p53 pathway, apoptosis and loss of cardiomyocytes, dilated cardiomyopathy, HF, and death.
Survival benefit via IRE1α inhibition
Although the deletion of p53 in the heart of fortilinKO-heart mice increased their survival by about 10 weeks (Fig. 3c), it did not fully normalize their survival. Because the ER stress pathway is activated in a failing heart [26] and fortilin binds and negatively regulates IRE1α, a key component of the ER stress pathway [14], we assessed the activation status of IRE1α by immunostaining the heart tissue from fortilinWTp53WT, fortilinKOp53WT, and fortilinKOp53KO mice (Fig. 4a). We found that the phosphorylated and activated IRE1α (p-IRE1α) levels in the hearts of fortilinKOp53KO mice were significantly higher than those of fortilinWTp53WT mice, suggesting that even in the absence of p53, the lack of fortilin leads to the activation of the ER stress pathway (Fig. 4a, a vs. c).

a Activation of the endoplasmic reticulum (ER) stress pathway, as evidenced by phosphorylation of IRE1α, occurred in the hearts of fortilinKOp53WT mice. Its activation was not fully reversed by the deletion of p53 in fortilinKOp53KO mice. b Experimental scheme. Five study and five control mice were weighed, underwent echocardiography, and were sacrificed at week 10 of treatment; the rest of the mice (N = 5 each) were observed until their deaths. c KIRA6 treatment allowed fortilinKOp53KO mice to live significantly longer than did vehicle treatment. d Echocardiography showed no significant difference between fortilinKOp53KO mice treated with either KIRA6 or vehicle. e KIRA6 treatment led to less phosphorylation and activation of IRE1α compared with vehicle treatment. a fortilinWTp53WT mice (αMHC-Cre−/−fortilinflox/floxp53flox/flox mice), b fortilinKOp53WT mice (αMHC-Cre+/+fortilinflox/floxp53WT/WT mice), c fortilinKOp53KO mice (αMHC-Cre+/+fortilinflox/floxp53flox/flox mice), p-IRE1α phosphorylated inositol-requiring enzyme 1 alpha, A.U. arbitrary unit, KIRA6 IRE1α Kinase Inhibiting RNase Attenuator 6, DMSO dimethyl sulfoxide used as vehicle, i.p. intraperitoneal injection, M.S. median survival, LVEF left ventricular (LV) ejection fraction, FS fractional shortening, LVIDd LV internal diameters in diastole, LVIDs LV internal diameters in systole, IVSd interventricular septum thickness in diastole; LVPWd LV posterior wall thickness in diastole, IB immunoblot, α-p-IRE1α anti-phosphorylated IRE1α antibody, GAPDH glyceraldehyde 3-phosphate dehydrogenase, WES an automated capillary-based quantitative Western blot system without total protein evaluation capability; Scale bar = 200 µm; Error bars, means ± SD, statistical analyses performed using Student’s two-sample t-test, except for the survival assay, for which the Log-Rank (Mantel–Cox) test was used: NS not statistically significant, *P < 0.05; **P < 0.01 (see also Fig. S4).
To test the hypothesis that the lack of fortilin in the heart overactivates not only the p53 pathway but also IRE1α and that blockage of both p53 and ER stress pathways is required to normalize the survival of fortilinKO-heart mice, we treated fortilinKOp53KO mice with either KIRA6, a selective IRE1α inhibitor [27], or vehicle (Fig. 4b). We found that KIRA6 treatment allowed fortilinKOp53KO mice to live modestly longer compared to vehicle treatment (Fig. 4c, median survival of vehicle vs. KIRA6 = 17.86 vs. 18.71 weeks, P = 0.0342 by Log-rank Mantel-Cox test). At week 10 of treatment (about 18 weeks of age), both body weights and HW/BW ratios of KIRA6-treated fortilinKOp53KO mice were modestly but significantly greater than those of vehicle-treated fortilinKOp53KO mice (Fig. S4a and S4b), although echocardiography showed no significant difference between the two groups (Fig. 4d). The hearts of KIRA6-treated fortilinKOp53KO mice exhibited less p-IRE1α than those of vehicle-treated fortilinKOp53KO mice (Fig. 4e), suggesting that KIRA6 treatment effectively blocked the activation of IRE1α.
Discussion
Fortilin is abundant in the heart but its function has been unknown. Herein, we have demonstrated that fortilin sustains cardiomyocyte survival at least partly through inhibition of p53 and p53-dependent apoptosis. We found that the lack of fortilin in the heart led to a drastic increase in apoptotic cardiomyocytes, severely decreased heart function, and death and that deletion of p53 from cardiomyocytes nearly totally inhibited cardiomyocyte apoptosis and improved heart function and survival.
The activation of p53 plays a pivotal role in the regulation of cardiac tissue homeostasis under normal conditions [15] and the development of cardiomyocyte damage and HF under biomechanical stress [17, 28, 29]. Transaortic constriction (TAC) and resultant LV pressure overload lead to marked upregulation of P53, induction of BAX, and HF [17]. PUMA is a pro-apoptosis protein induced by P53, and Mandle et al. showed that its deletion from the heart protects mice against TAC-induced HF [28]. The p53 activator quinacrine accelerated TAC-induced HF [29], whereas global p53-deficient mice showed less HF than their wild-type control after TAC [17]. Despite the well-documented role of p53 in the heart, how p53 itself is regulated in the stressed heart has been unclear. Our current work presents evidence that fortilin is a critical negative regulator of p53 in the heart.
We now know that fortilin negatively regulates p53 in three distinct ways. First, fortilin binds to the sequence-specific DNA-binding domain of p53 and prevents it from transcriptionally activating p53-target genes [12]. Second, fortilin promotes the degradation of p53 by inhibiting mouse double minute 2 (MDM2) auto-ubiquitination and promoting MDM2-mediate ubiquitination and degradation of p53 [13]. Finally, fortilin represses the transcriptional activation of the p53 gene itself, as shown by the significantly higher p53 levels in fortilinKOp53WT mice compared to fortilinWTp53WT mice at the message (Fig. S2c, p53, a vs. b) level and by activation of the p53 promoter by knockdown of fortilin in the cardiomyocyte cell line (Fig. S3e).
KIRA6, a small molecular weight inhibitor of IRE1α [27], significantly and modestly prolonged the lifespan of fortilinKOp53KO mice (Fig. 4c). Although this result may be due to incomplete inhibition of IRE1α by KIRA6 (Fig. 4e), another possibility is that fortilin negatively regulates not only p53 and IRE1α but also unknown apoptosis pathways. This possibility requires further investigation.
There were p53 signals detectable in the Western blot of the total heart lysates from fortilinKOp53KO mice (Fig. 3a, α-p53, c). Because there are more endothelial cells and fibroblasts combined than cardiomyocytes in the murine hearts [30,31,32], it is possible that the p53 signals are from those non-cardiomyocyte cells. It is also possible that the Cre-transgene expressed under the αMHC promoter (Fig. S1a-2) was not capable of cleaving the floxed p53 gene in all cardiomyocytes of the fortilinKOp53KO heart. To evaluate the status of activated and functional p53 in the heart, we stained the tissue with anti-phospho-p53 antibody. We found that phosphorylated p53 were significantly less in fortilinKOp53KO mice than that in fortilinWTp53WT mice, suggesting that despite the similar total p53 signals between fortilinKOp53KO and fortilinWTp53WT hearts by both immunohistochemistry (Fig. S3a, p53, a vs. c) and Western blots (Fig. 3a, α-p53, a vs. c), functional p53 is significantly less in fortilinKOp53KO hearts than in fortilinWTp53WT hearts.
Because cardiomyocytes are not capable of proliferating in normal conditions, their continuous apoptotic loss leads to irreversible progression of HF. Protecting cardiomyocytes against apoptosis represents a new approach in HF gene therapy, distinct from the ones focusing on calcium metabolism and β-adrenergic receptor signal transduction [33]. The current work highlights fortilin as a viable molecular target of HF gene replenishment therapy and prepares us for further investigation of how fortilin expression is maintained in NFHs.
Methods
Reagents and materials
Kinase-Inhibiting RNase Attenuator 6 (KIRA6) [27], an inhibitor of IRE1α kinase, was obtained from EMB Millipore (Catalog #: 532281, Burlington, MA). 2,2,2-trichloroethanol (TCE) was purchased from Sigma (St. Louis, MO). Lentiviral particles that contained short-hairpin RNA (shRNA) against human fortilin (lentivirussh-fortilin) and random control sequence (lentivirussh-control) were purchased from Sigma (MISSION® shRNA lentiviral transduction particles). The lentivirussh-fortilin was experimentally shown to silence rat fortilin (Fig. S3c). Lentiviral particles that contained (a) the murine p53 promoter sequence fused to the GLuc cDNA, (b) the SEAP gene under the constitutional SV40 promoter, and (c) the puromycin resistance gene were obtained from GeneCopoeia (Rockville, MD) (Catalog #: LPP-MPRM33498-LvPG04-200; Lentivirusp53-promoter-GLuc/SEAP). The actual promoter sequence is as follows: GGATCTGTGGCTAGCTGGGGTTGGTCATCACCACCGCATGGCGGAGGCACCGGTTCAAAGTCTGTATTTTTCTCCGCTGGGGAACCTTGGGGTACCGGAGCTGGGGCCAGGTCAGGAGGGAGGCTATCCGGAGCTAAGAGTCGCTCCTCCGACGTCTTCATTCTGTAGAGTAAGCCCCCGGAAGGCAGAGGTCGGGCAAGTCTCGCTGAGCCGGCTACCAGCTGCCGAGGCTAGAGTGCATTACCGTTCCCAGGGATGCTCAGAGACCGGAGTCCGCTTTCCTCTTCCGGAAAATGTAAGCCGAACCTAAAGCAATCACCAGGGAACGAGTGTCCAAAGCCAAGCGCCTAGGGTCGCTAGGCGCCGCCAGGGCTTCTTGCTCTCGCGGGAGTCGGGCCACCTTCCGATAGGCTCTCCGCATCCTCCTCCGATTCCGAGCGGGAAGGCGGGAAGGAACGACTTTGCCTACACCTCAAGCGCTGGAGAATTCCTAGAGGTTTCTGGGAGTTGTAGTCTGAACTCTGGGCCTTGGCGAAAACTACACGAGCGCCCCCTACCGTCCCCTGGGGGTAATTCTTAAAGCGCCTATCCTCCCTGGCCTGCAGAGGGCGCATAATTTCTACAGTTTTTGCCCCTCTTGACTATCTTGTTTTGAATCCC
GTAACCTCAGGTTTCCTTTCTCCCCATCTCTCCCCCCTTCTTGTTCCTCTCTTTCCCTTTCTCCCCCGCCCTCCCTTCATTCATTCGACATTTATTATCAAGTTCTTACTGCCTAACCCAGGACTATACAAGGCATTGGGAAAAAAATAGCAATGTTTTCTAGTTCTTAATCTCCATAAAGTTTTCGTTGCTGTGCAATTAAAGGCTGTGAAAACAGTCTTTACAGAGAGTGATAAGGACTGTACAGGAAATTAAACACGGTGGTGCGATACCAAGTATCTCGGAGAACACGTTAGATTGAGATACTATGAAAAGCCTTTCTAAAGTGACATTTTAGCTAATGAGGGGAAAAAGAACTTAGGGGCCCGTGTTGGTTCATCCCTGTACTTGGAAGGCCTAAAGCAGGAAGACGGCCGCGAATTCCAGGCCAGCCTTGGCTACAAAGACTCTGTCTTAAAAATCCAAAAAGATGGCTATGACTATCTAGCTGGATAGGAAAGAGCACAGAGCTCAGAACAGTGGCGGTCCACTTACGATAAAAACTTAATTCTTTCCACTCTTTATACTTGACACAGAGGCAGGAGTCCTCCGAATCGGTTTCCACCCATTTTGCCCTCACAGCTCTATATCTTAGACGACTTTTCACAAAGCGTTCCTGCTGAGGGCAACATCTCAGGGAGAATCCTGACTCTGCAAGTCCCCGCCTCCATTTCTTGCCCTCAACCCACGGAAGGACTTGCCCTTACTTGTTATGGCGACTATCCAGCTTTGTGCCAGGAGTCTCGCGGGGGTTGCTGGGATTGGGACTTTCCCCTCCCACGTGCTCACCCTGGCTAAAGTTCTGTAGCTTCAGTTCATTGGGACCATCCTGGCTGTAGGTAGCGACTACAGTTAGGGGGCAC.
Cell culture
MycoFluor™ (ThermoFisher Scientific-Molecular Probe, Eugene, OR) was used to detect mycoplasma contamination when appropriate. The H9C2(2-1) (ATCC® CRL-1446™) cell line (H9C2 hereafter) was directly obtained from American Type Culture Collection (Manassas, VA, USA). Cells were maintained in Dulbecco’s modified Eagle’s medium (Corning, Corning, NY, Catalog #: 0-013-CV) with 10% fetal bovine serum (FBS) (Catalog #: 10082-147; ThermoFisher Science–Gibco, Waltham, MA) at 37 °C in an atmosphere containing 5% CO2. H9C2 cells stably harboring the lentiviral construct that contained both the p53 promoter sequence driving the expression of GLuc and the SEAP gene under the control of the constitutional SV40 promoter were generated by transducing the cells with Lentivirusp53-promoter-GLuc/SEAP and selecting them with puromycin (H9C2p53-promoter-GLuc/SEAP). They were maintained in puromycin-containing (0.3 µg/mL) media.
Dual-luciferase assay
On the first day, H9C2p53-promoter-GLuc/SEAP cells were transiently transduced by either (lentivirussh-fortilin) or (lentivirussh-control) at the multiplicity of infection (MOI) of 1. The next day, cells were irradiated using the RS-2000 X-ray irradiator (QuaStar, Buford, GA) at 8 Gy. Seventy-two hours after the irradiation, conditioned media were collected and subjected to both SEAP and luciferase assays using the Secrete-Pair Dual Luminescence Assay Kit according to the manufacturer’s instruction (GeneCopoeia). The degree of activation of the p53 promoter was assessed by dividing the luciferase activity units by the SEAP activity units and expressing it in arbitrary units (A.U.).
General veterinary care
Mice were given standard mouse chow (Lab Diet; Catalog #: 5L0D) and water ad libitum, maintained on a 12-h light–dark cycle and seen by a scientist daily to observe their behavior and health. Any animal that displayed substantive signs of distress anytime during the protocol, including but not limited to poor grooming, hunched back posture, or a loss in weight exceeding 10% of the weights at the initiation of the experiments, were identified and treated accordingly, including removal from the experiment and euthanasia. For mouse experiments where grouping was based on pharmacological treatments, mice of the same genotype were randomly assigned to treatment and control groups.
Generation of heart-specific fortilin KO (fortilinKO-heart) mice
Fortilinflox/flox mice, in which the fortilin gene was flanked by the LoxP sequence to allow tissue-specific deletion, were generated using the standard homologous recombination technique described previously: we micro-injected a mutant C57BL6 embryonic stem cell (ESC) line that contained two LoxP sequences flanking all six fortilin exons into C57BL6 blastocysts [14]. The resultant fortilinflox/flox mice were fully in the C57BL/6J genetic background from the beginning. Fortilinflox/flox mice were then crossed with a transgenic flipase strain to remove the neomycin resistance gene cassette. To generate fortilinKO-heart mice, fortilinfloxflox mice were crossed with C57BL/6J mice overexpressing the Cre transgene under the control of cardiac-specific α-myosin heavy chain (Myh6) promoter [22] (αMHC-Cre+/+, Jackson Laboratory, Ann Arbor, ME, Stock # 011038, already backcrossed to C57BL/6J for eight generations by the donating laboratory). Genotyping of fortilinKO-heart and fortilinWT-heart mice was performed on tail-derived genomic DNA using standard PCR-based methods. The presence of the αMHC-Cre transgene was detected using the following primer sets: 5′-ATGACAGAC AGATCCCTCCTATCTCC-3′ (M1) and 5′-CTCATCACTCGTTGCATCATCGAC-3′ (M2). αMHC-Cre+/− mice yielded a 300 bp amplified fragment and αMHC-Cre−/− mice yielded no amplicons. To determine the hemi- (αMHC-Cre+/−) or homo- (αMHC-Cre+/+) zygosity of the MHC-Cre transgene, RT-qPCR was performed using the primers and probe sets described previously [14] (see below). We used the 2–ΔΔCT method [34] to determine the expression levels of the αMHC-Cre transgene relative to the internal control levels in the sample, according to the Protocol 20627 of the Jackson Laboratory.
-
αMHC-Cre-transgene:
-
Forward primer (oIMR1084): 5’-GCGGTCTGGCAGTAAAAACTATC-3’
-
Reverse primer (oIMR1085): 5’-GTGAAACAGCATTGCTGTCACTT-3’
-
Probe (13593): 5’-FAM-AAACATGCTTCATCGTCGGTCCGG-IBFQ-3’
-
FAM, 6-carboyfluorescein; IBFQ, Iowa Black™ double-quenched probe (Integrated Data Technologies (IDT), Coralville, IA)
-
-
Internal control to calculate ΔCT for the αMHC-Cre-transgene:
-
Forward primer (oIMR1544): 5’-CACGTGGGCTCCAGCATT-3’
-
Reverse primer (oIMR3580): 5’-TCACCAGTCATTTCTGCCTTTG-3’
-
Probe (TmoIMR0105): 5’-JOE-CCAATGGTCGGGCACTGCTCAA-IBFQ-3’
-
The target gene is Apob (apolipoprotein B) [35].
-
The presence of the LoxP-fortilin-LoxP knock-in construct was verified using the following primer sets: 5′-TGGACCCTGACTTTCATCACCTC-3′ (F1) and 5′- GTCATCTAACCTTACCCCAGTAAGC-3′ (F2): fortilinflox/flox mice yielded a 405 bp fragment and wild-type fortilin mice (fortilinWT/WT mice) yielded a 280 bp fragment.
Generation of heart-specific fortilin p53 double KO (fortilinKOp53KO) mice
To generate a strain of C57BL/6J mice that lack both fortilin and p53 in the heart, fortilinKO-heart mice were crossed with p53flox/flox mice available from the Jackson Laboratory (Stock Number: 008462, p53LoxP). The status of the αMHC-Cre-transgene was evaluated by both PCR and RT-qPCR as described above, and the status of fortilin and p53 loci was determined using the following PCR-based genotyping strategies:
-
Fortilin locus:
-
Forward primer: 5’-TGGACCCTGACTTTCATCACCTC-3’
-
Reverse primer: 5’-GTCATCTAACCTTACCCCAGTAAGC-3’
-
Expected size of the mutant (floxed fortilin gene) allele: 405 bp
-
Expected size of the wildp-type allele: 280 bp
-
-
P53 locus:
-
Forward primer: 5’-GGTTAAACCCAGCTTGACCA-3’
-
Reverse primer: 5’-GGAGGCAGAGACAGTTGGAG-3’
-
Expected size of the mutant (floxed fortilin gene) allele: 390 bp
-
Expected size of the wild-type allele: 270 bp
-
KIRA6 rescue experiment
For the KIRA6 rescue experiment, 8-week-old male fortilinKOp53KO mice were daily and intraperitoneally injected with either 5 mg/kg of KIRA6 in solution (3% ethanol, 7% Tween-80, and 90% saline) or the same solution without KIRA6 as vehicle (a) for the subsequent 10 weeks for the sacrifice group (N = 5 each) or (b) until death for the survival analysis group (N = 5 each). Mice were examined daily for their behavior and health. Mice in the sacrifice group were weighed and underwent transthoracic echocardiography before they were sacrificed, and their hearts were harvested and processed as described elsewhere in “Methods” section.
Mouse echocardiography
Mouse transthoracic echocardiography was performed using a Vevo 2100 High-Resolution Imaging System (FUJIFILM VisualSonics, Toronto, ON, Canada) on mice anesthetized initially by 4% isoflurane in 1 L/min of O2 and then by 0.5–3.0% isoflurane and placed on a heated platform, as described previously [36]. We imaged the heart in the parasternal long-axis view in both 2D and M-modes. We obtained the following parameters using the software on the system: left ventricular ejection fraction (LVEF, %), fractional shortening (FS, %), left ventricular internal diameter in systole (LVIDs, mm), left ventricular internal diameter in end-diastole (LVIDd, mm), interventricular septum thickness in end diastole (IVSd, mm), left ventricular posterior wall thickness in end diastole (LVPWd, mm), and LV mass (mg). For the initial characterization of fortilinKO-heart mice, the baseline echo was obtained when they were 7 weeks of age.
Processing of the mouse hearts
After being weighed, mice were sacrificed by isoflurane inhalation until effective, followed by exsanguination. Both the heart and lungs were routinely harvested. The liver, kidney, and spleen were harvested, when appropriate, for protein and RNA analyses. The harvested hearts and lungs were rinsed with phosphate-buffered saline (PBS), drained on absorbent paper, and weighed. The ratio of heart weight to body weight (HW/BW ratio) as well as that of lung weight to body weight (LW/BW ratio) was then calculated. For the sagittal cross-sectional analysis of the heart, the entire heart was fixed in 4% paraformaldehyde and subjected to paraffin embedding, sectioning, and hematoxylin and eosin staining according to the standard protocol. The right ventricle and atria were removed from the left ventricle (LV). The LV was then divided into three sections of equal size (basal, mid-ventricular, and apical). The mid-ventricular part of the LV was fixed in 4% paraformaldehyde before being embedded in paraffin for immunohistochemistry. The apical portion of the LV was flash-frozen for subsequent protein extraction, and the basal portion of the LV was placed directly into Tri-Reagent (Molecular Research Center, Cincinnati, OH, USA) and frozen for subsequent RNA extraction.
RT-qPCR
RT-qPCR was performed as described previously [14]. We used the 2–ΔΔCT method [34] to calculate the expression levels of a gene in question relative to the 18S rRNA levels in the sample. Briefly, the hearts (and, when appropriate, other organs) of mice were harvested into Tri-Reagent. RNA was isolated following the manufacturer’s instructions and treated with DNAse (ABI, Foster City, CA, USA). RT-qPCR was performed in quadruplicate with exactly 50 ng of total RNA using the TaqMan® RT-PCR kit (Applied Biosystems [ABI] at Life Technologies, Grant Island, NY, USA) in the ABI Step One Plus Real-Time PCR system and the following primer and probe sets (Integrated DNA Technologies):
-
1.
Mouse Fortilin:
-
a.
Forward primer: 5′-TCCGACATCTACAAGATCCGG-3′,
-
b.
Reverse primer: 5′-ATCTTGCCCTCCACCTCCA-3′,
-
c.
Probe: 5′-FAM-AGATCGCGG/ZEN/ACGGGCTGTGC-IBFQ-3′
-
d.
ZEN™ = an internal quencher to enhance the quenching activity of the 3’ quencher IBFQ (IDT)
-
a.
-
2.
Mouse Col1:
-
a.
Forward primer: 5′-GAAACCCGAGGTATGCTTGA-3′,
-
b.
Reverse primer: 5′-GTTGGGACAGTCCAGTTCTT-3′,
-
c.
Probe: 5′-FAM-TGTGCGATGACGTGCAATGCAATG-IBFQ-3′
-
a.
-
3.
Mouse Myh7
-
a.
Forward primer: 5′-CCATCTCTGACAACGCCTATC-3′,
-
b.
Reverse primer: 5′-GGATGACCCTCTTAGTGTTGAC-3′,
-
c.
Probe: 5′-FAM-TCAGTCCATCCTCATCACCGGAGA-IBFQ-3′
-
a.
-
4.
Mouse ANF
-
a.
Forward primer: 5′-TCCGATAGATCTGCCCTCTT-3′,
-
b.
Reverse primer: 5′-CTCCAATCCTGTCAATCCTACC-3′,
-
c.
Probe: 5′-FAM-AAAGCAAAC/ZEN/TGAGGGCTCTGCTCG-IBFQ-3′
-
a.
-
5.
Mouse Acta1
-
a.
Forward primer: 5′-CTCCCTGGAGAAGAGCTATGA-3′,
-
b.
Reverse primer: 5′-CGATAAAGGAAGGCTGGAAGAG-3′,
-
c.
Probe: 5′-FAM-CATCGGCAATGAGCGTTTCCGTTG-IBFQ-3′
-
a.
-
6.
Mouse Serca2
-
a.
Forward primer: 5′-CATCAGTATGACGGGCTTGTAG-3′,
-
b.
Reverse primer: 5′-CTCGGTAGCTTCTCCAACTTTC-3′,
-
c.
Probe: 5′-FAM-AGCCACCATCTGTGCTCTGTGTAA-IBFQ-3′
-
a.
-
7.
Mouse p53
-
a.
Forward primer: 5′-CAGCTTTGAGGTTCGTGTTTG-3′,
-
b.
Reverse primer: 5′-AGTTCAGGGCAAAGGACTTC-3′,
-
c.
Probe: 5′-FAM-TCTTCTTCT/ZEN/GTACGGCGGTCTCTCC-IBFQ-3′
-
a.
-
8.
Mouse Bax
-
a.
Forward primer: 5′-TTGCTGATGGCAACTTCAACTGGG-3′,
-
b.
Reverse primer: 5′-TGTCCAGCCCATGATGGTTCTGAT-3′,
-
c.
Probe: 5′- FAM-TTTGCTAGC/ZEN/AAACTGGTGCTCAAGGC-IBFQ-3′
-
a.
-
9.
Mouse Puma
-
a.
Forward primer: 5′-ATGGCGGACGACCTCAAC-3′,
-
b.
Reverse primer: 5′-AGTCCCATGAAGAGATTGTACATGAC-3′,
-
c.
Probe: 5′-FAM-AGCAGCATC/ZEN/GACACCGACCCTCAC-IBFQ-3′
-
a.
-
10.
Mouse Noxa
-
a.
Forward primer: 5′-TGCACCGGACATAACTGTGGTTCT-3′,
-
b.
Reverse primer: 5′-TGAGCACACTCGTCCTTCAAGTCT-3′,
-
c.
Probe: 5′-FAM-AAAGAGCAG/ZEN/GATGAGGAGCCCAAGC-IBFQ-3′
-
a.
-
11.
Mouse 18S rRNA
-
a.
Forward primer: 5′- GCCGCTAGAGGTGAAATTCT-3′,
-
b.
Reverse primer: 5′-TCGGAACTACGACGGTATCT-3′,
-
c.
Probe: 5′-JOEN-ACCAGAGCG/ZEN/AAA GCATTTGCCAAG-IBFQ-3′
-
d.
JOEN = 6-carboxy-4′,5′-dichloro-2′,7′-dimethoxyfluorescein
-
a.
Western blot analyses
SDS–PAGE and Western blot analyses were performed as described previously [8, 37,38,39,40] on the lysates from the mouse organs. The following primary antibodies were used at the indicated dilutions/concentrations:
-
Anti-fortilin (Abnova, Taipei City, Taiwan; Clone 2C4, H00007178-M03; 1:1000 dilution)
-
Anti-p53 (Santa Cruz Biotechnology, Dallas, TX; sc-6243; 1:1000 dilution)
-
Anti-GAPDH (Santa Cruz Biotechnology; Clone 6C5, sc-32233; 1:10,000 dilution)
-
Anti-Bax (Santa Cruz Biotechnology; sc-493; 1:1000 dilution)
-
Anti-PUMA (Cell Signaling Technology, Danvers, MA; 7467; 1:1000 dilution)
-
Anti-NOXA (EMD Millipore, Billerica, MA; AB5761; 1:1000 dilution)
-
Anti-Tropomyosin (Santa Cruz Biotechnology; sc-28543; 1:1000 dilution)
All antibodies were used with appropriate IRDye680LT- or IRDye800CW-conjugated secondary antibodies (LI-COR, Lincoln, NE, USA). The signal intensities of protein bands were quantified using the Odyssey Infrared Imaging System (LI-COR) and normalized to the signal intensity of the loading control protein (GAPDH or tropomyosin) and expressed in A.U. To quantify p53 expression in response to X-ray irradiation, 0.5% (v/v) TCE was added to a polyacrylamide gel before polymerization. After standard SDS–PAGE, the gel was UV-irradiated on the Bio-Rad ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA) for 2 min. The image was electronically captured, and the cumulative band densities were calculated to assess loading conditions as previously described [41]. The signal intensity of Western blot bands was divided by that of the TCE bands to derive the p53 expression index. Results were expressed in A.U.
JESS/WES analysis
To evaluate the expression levels of fortilin in human hearts, we obtained de-identified tissue lysates of human hearts from patients with NFHs, non-ischemic cardiomyopathy (NICM), and ischemic cardiomyopathy (ICM) from the Duke Human Heart Repository (Duke University, Durham, NC). An automated capillary-based quantitative Western blot system called JESS™ (Protein Simple, San Jose, CA) was used to (a) detect fortilin (anti-fortilin antibody, MBL International Corporation, Woburn, MA; PM017; 1:10 dilution) and (b) visualize total proteins loaded as described previously [19, 20]. Compass Software (v3.1, Protein Simple) was used to calculate a fortilin expression index by dividing the area under the curve of a fortilin peak by the total proteins loaded in the same capillary (“in-capillary normalization”). The fortilin expression index was expressed in A.U.
To quantify phosphorylated IRE1α, the mouse heart lysates from fortilinKOp53KO mice that were treated with either vehicle or KIRA6 were subjected to WES™ (Protein Simple), an automated capillary-based quantitative Western blot system, to detect p-IRE1α (anti-phospho-IRE1α antibody, Novus Biologicals, Centennial, CO; NB100-2323; 1:10 dilution) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; anti-GAPDH antibody; Santa Cruz Biotechnology; Clone 6C5, sc-32233; 1:250 dilution); the latter served as the loading control. JESS™, but not WES™, can visualize the total proteins loaded for normalization. Compass Software (v3.1) was used to calculate a fortilin expression index by dividing the area under the curve of a fortilin peak by that of a GAPDH peak loaded in the same capillary (“in-capillary normalization”). The fortilin expression index was expressed in A.U.
Immunohistochemistry of mouse hearts
Mouse hearts were fixed in 4% paraformaldehyde and embedded in paraffin before they were sectioned at 5 µm thickness. Immunohistochemistry of mouse hearts was performed as described previously [42] using the primary antibodies listed below and 3,3’-diaminobenzidine (DAB) as the chromogen. Myocardial fibrosis was quantified by both Masson staining and picrosirius red staining as previously described [43, 44]. All immunostained sections were digitally imaged using the EasyScan Digital Slide Scanner (Motic, San Francisco, CA). Using the ImageJ software (National Institutes of Health, Bethesda, MD), expression indices were calculated by dividing the DAB-positive area (or signal-positive area for Masson and Picrosirius staining) by the region of interest (ROI), and results were expressed in A.U. For quantification of phospho-p53 positive cells in the heart, DAB-positive nuclei were counted in a randomly selected field on the DAB-only images generated by the color deconvolution function of ImageJ (version 1.53k; National Institutes of Health, Bethesda, MD). The process was repeated for 5 different fields per mouse for 5 mice.
-
Cleaved lamin A (Cell Signaling Technology; 2035; 1:100 dilution). The cleavage of lamin is a well-characterized event in apoptosis [45].
-
BAX (Santa Cruz Biotechnology; sc-493; 1:500 dilution)
-
p-IRE1α (Abcam, Cambridge, MA; ab48187; 1:4000 dilution)
-
p53 (Novocastra, Wetzlar, Germany; NCL-L-p53-CM5p; 1:250 dilution)
-
phosphorylated p53 (phospho-p53, Cell Signaling Technologies, Danvers, MA; #9284; 1:100 dilution)
TUNEL staining
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of heart tissue was performed on formalin-fixed, paraffin-embedded samples as previously described [12, 46] using the FragELTM DNA Fragmentation Detection Kit (EMD Millipore-Calbiochem) following the manufacturer’s instructions. TUNEL-positive cells within about 0.145 mm2 of the ROI were counted, and TUNEL indices were calculated as the number of TUNEL-positive cells per unit area (in mm2). Results are expressed in A.U.
Cardiomyocyte area
To assess the presence of cardiomyocyte hypertrophy, the heart tissue that had been formalin-fixed and paraffin-embedded was sectioned at 5 µm thickness and stained with laminin (ThermoScientific, Waltham, MA; RB082A; 1:100 dilution). After stained sections were digitally captured using the EasyScan Digital Slide Scanner, cardiomyocyte areas were measured using ImageJ and expressed in A.U. as described previously [47]. At least five distinct areas were quantified per mouse sample.
Ethics statement
This study was performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All experiments involving animals were approved by the Institutional Animal Care and Use Committees of our institution. Human tissue samples used for this study were procured from the Duke Human Heart Repository (DHHR), which is a Duke University Health System Institutional Review Board (DUHS IRB) approved tissue repository. Samples were procured by the DHHR using written informed consent or a waiver of consent for discarded tissues. No HIPAA information was provided with any of the samples used in this study. Human myocardium was acquired from the left ventricular free wall of explanted hearts following cardiac transplantation. Non-failing (NF) left ventricular tissue was acquired from donors whose hearts were not utilized for transplant, thus becoming available for research.
Statistical analysis
All measurements were taken from distinct samples (biological replicates) and the size of biological samples (N) is indicated in either figures or the main text. The degree of the spread of data was expressed by the standard deviation (±SD). The difference between the control and study groups was analyzed using unpaired two-tailed Student’s t-test for two groups or one-way analysis of variance (ANOVA) followed by Fisher’s pairwise multiple comparisons for multiple groups. P < 0.05 was considered to be statistically significant. For survival analyses, Kaplan–Meier survival curves were generated, and the Log-Rank (Mantel–Cox) test was used to compare the curves. The numbers of mice used in in vivo experiments were determined by (i) power analysis, assuming an α error rate of 0.05, a β error rate of 0.20, and an expected difference of 25%, in Minitab 17 (State College, PA) or (ii) our previous dataset and experience from similar experiments performed as part of past research. A similar variance was observed between the groups that were statistically compared. No data were excluded unless outliers were identified and verified by the outlier tests (Minitab 17). Although the scientists were not blinded to allocation during experiments and readouts evaluation, all readouts from the experiments were predetermined, highly objective, and obtained according to the validated protocols.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files. All relevant data are available from the authors upon request.
References
Mann DL. Getting pumped about heart failure. Cell Metab. 2014;19:896–7. https://doi.org/10.1016/j.cmet.2014.05.008.
Narula J, Haider N, Arbustini E, Chandrashekhar Y. Mechanisms of disease: apoptosis in heart failure-seeing hope in death. Nat Clin Pract Cardiovasc Med. 2006;3:681–8. 10.1038/ncpcardio0710
Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. 2017;14:30–8. https://doi.org/10.1038/nrcardio.2016.163.
Guerra S, Leri A, Wang X, Finato N, Di Loreto C, Beltrami CA, et al. Myocyte death in the failing human heart is gender dependent. Circ Res. 1999;85:856–66.
Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–41.
Saraste A, Pulkki K, Kallajoki M, Heikkila P, Laine P, Mattila S, et al. Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur J Clin Investig. 1999;29:380–6.
Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Investig. 2003;111:1497–504. https://doi.org/10.1172/JCI17664.
Li F, Zhang D, Fujise K. Characterization of fortilin, a novel anti-apoptotic protein. J Biol Chem. 2001;276:47542–9.
Tuynder M, Susini L, Prieur S, Besse S, Fiucci G, Amson R, et al. Biological models and genes of tumor reversion: cellular reprogramming through tpt1/TCTP and SIAH-1. Proc Natl Acad Sci USA. 2002;99:14976–81.
Yang Y, Yang F, Xiong Z, Yan Y, Wang X, Nishino M, et al. An N-terminal region of translationally controlled tumor protein is required for its antiapoptotic activity. Oncogene 2005;24:4778–88.
Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, et al. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–21. https://doi.org/10.1161/CIRCULATIONAHA.113.003863.
Chen Y, Fujita T, Zhang D, Doan H, Pinkaew D, Liu Z, et al. Physical and functional antagonism between tumor suppressor protein p53 and fortilin, an anti-apoptotic protein. J Biol Chem. 2011;286:32575–85. https://doi.org/10.1074/jbc.M110.217836.
Amson R, Pece S, Lespagnol A, Vyas R, Mazzarol G, Tosoni D, et al. Reciprocal repression between P53 and TCTP. Nat Med. 2011;18:91–9. https://doi.org/10.1038/nm.2546.
Pinkaew D, Chattopadhyay A, King MD, Chunhacha P, Liu Z, Stevenson HL, et al. Fortilin binds IRE1α and prevents ER stress from signaling apoptotic cell death. Nat Commun. 2017;8:1–16.
Mak TW, Hauck L, Grothe D, Billia F. p53 regulates the cardiac transcriptome. Proc Natl Acad Sci USA. 2017;114:2331–6. https://doi.org/10.1073/pnas.1621436114.
Nomura S, Satoh M, Fujita T, Higo T, Sumida T, Ko T, et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat Commun. 2018;9:4435 https://doi.org/10.1038/s41467-018-06639-7.
Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007;446:444–8. https://doi.org/10.1038/nature05602.
Schechter MA, Hsieh MK, Njoroge LW, Thompson JW, Soderblom EJ, Feger BJ, et al. Phosphoproteomic profiling of human myocardial tissues distinguishes ischemic from non-ischemic end stage heart failure. PLoS ONE. 2014;9:e104157 https://doi.org/10.1371/journal.pone.0104157.
Campos ML, Yoshida Y, Major IT, de Oliveira Ferreira D, Weraduwage SM, Froehlich JE, et al. Rewiring of jasmonate and phytochrome B signalling uncouples plant growth-defense tradeoffs. Nat Commun. 2016;7:12570 https://doi.org/10.1038/ncomms12570.
DeRamus ML, Stacks DA, Zhang Y, Huisingh CE, McGwin G, Pittler SJ. GARP2 accelerates retinal degeneration in rod cGMP-gated cation channel beta-subunit knockout mice. Sci Rep. 2017;7:42545 https://doi.org/10.1038/srep42545.
Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–71. https://doi.org/10.1016/j.cell.2013.08.032.
Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Investig. 1997;100:169–79. https://doi.org/10.1172/JCI119509.
Tan FL, Moravec CS, Li J, Apperson-Hansen C, McCarthy PM, Young JB, et al. The gene expression fingerprint of human heart failure. Proc Natl Acad Sci USA. 2002;99:11387–92. https://doi.org/10.1073/pnas.162370099.
Das S, Frisk C, Eriksson MJ, Walentinsson A, Corbascio M, Hage C, et al. Transcriptomics of cardiac biopsies reveals differences in patients with or without diagnostic parameters for heart failure with preserved ejection fraction. Sci Rep. 2019;9:3179 https://doi.org/10.1038/s41598-019-39445-2.
Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004.
Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004;110:705–12. https://doi.org/10.1161/01.CIR.0000137836.95625.D4.
Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–48. https://doi.org/10.1016/j.cell.2014.07.002.
Mandl A, Huong Pham L, Toth K, Zambetti G, Erhardt P. Puma deletion delays cardiac dysfunction in murine heart failure models through attenuation of apoptosis. Circulation 2011;124:31–9. https://doi.org/10.1161/CIRCULATIONAHA.110.988303.
Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci USA. 2005;102:17448–53. https://doi.org/10.1073/pnas.0508888102.
Chen SN, Lombardi R, Karmouch J, Tsai JY, Czernuszewicz G, Taylor MRG, et al. DNA Damage Response/TP53 pathway is activated and contributes to the pathogenesis of dilated cardiomyopathy associated with LMNA (Lamin A/C) mutations. Circ Res. 2019;124:856–73. https://doi.org/10.1161/CIRCRESAHA.118.314238.
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–9. https://doi.org/10.1161/CIRCRESAHA.115.307778.
Zhou P, Pu WT. Recounting cardiac cellular composition. Circ Res. 2016;118:368–70. https://doi.org/10.1161/CIRCRESAHA.116.308139.
Kieserman JM, Myers VD, Dubey P, Cheung JY, Feldman AM. Current landscape of heart failure gene therapy. J Am Heart Assoc. 2019;8:e012239 https://doi.org/10.1161/JAHA.119.012239.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001;25:402–8. https://doi.org/10.1006/meth.2001.1262.
Machado TS, Macabelli CH, Sangalli JR, Rodrigues TB, Smith LC, Meirelles FV, et al. Real-time PCR quantification of heteroplasmy in a mouse model with mitochondrial DNA of C57BL/6 and NZB/BINJ strains. PLoS ONE. 2015;10:e0133650 https://doi.org/10.1371/journal.pone.0133650.
Gao S, Ho D, Vatner DE, Vatner SF. Echocardiography in mice. Curr Protoc Mouse Biol. 2011;1:71–83. https://doi.org/10.1002/9780470942390.mo100130.
Zhang D, Li F, Weidner D, Mnjoyan ZH, Fujise K. Physical and functional interaction between MCL1 and fortilin. The potential role of MCL1 as a fortilin chaperone. J Biol Chem. 2002;277:37430–8.
Graidist P, Phongdara A, Fujise K. Antiapoptotic protein partners fortilin and MCL1 independently protect cells from 5-FU-induced cytotoxicity. J Biol Chem. 2004;279:40868–75.
Graidist P, Yazawa M, Tonganunt M, Nakatomi A, Lin C, Phongdara A, et al. Fortilin binds Ca2+ and blocks Ca2+-dependent apoptosis in vivo. Biochem J. 2007;408:181–91.
Fujita T, Felix K, Pinkaew D, Hutadilok-Towatana N, Liu Z, Fujise K. Human fortilin is a molecular target of dihydroartemisinin. FEBS Lett. 2008;582:1055–60.
Ladner CL, Edwards RA, Schriemer DC, Turner RJ. Identification of trichloroethanol visualized proteins from two-dimensional polyacrylamide gels by mass spectrometry. Anal Chem. 2006;78:2388–96. https://doi.org/10.1021/ac051851y.
Pinkaew D, Le RJ, Chen Y, Eltorky M, Teng BB, Fujise K. Fortilin reduces apoptosis in macrophages and promotes atherosclerosis. Am J Physiol Heart Circ Physiol. 2013. https://doi.org/10.1152/ajpheart.00570.2013.
Marcos-Garces V, Harvat M, Molina Aguilar P, Ferrandez Izquierdo A, Ruiz-Sauri A. Comparative measurement of collagen bundle orientation by Fourier analysis and semiquantitative evaluation: reliability and agreement in Masson’s trichrome, Picrosirius red and confocal microscopy techniques. J Microsc. 2017;267:130–42. https://doi.org/10.1111/jmi.12553.
Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979;11:447–55. https://doi.org/10.1007/BF01002772.
Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135:1441–55.
Sinthujaroen P, Wanachottrakul N, Pinkaew D, Petersen J, Phongdara A, Sheffield-Moore M, et al. Elevation of serum fortilin levels is specific for apoptosis and signifies cell death in vivo. BBA Clin. 2014;2:103–11. https://doi.org/10.1016/j.bbacli.2014.10.002
Tirziu D, Chorianopoulos E, Moodie KL, Palac RT, Zhuang ZW, Tjwa M, et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Investig. 2007;117:3188–97. https://doi.org/10.1172/JCI32024.
Acknowledgements
This manuscript is dedicated to Dr. James T. Willerson (1939–2020), a mentor of K.F. We thank all members of the Fujise laboratory for their collaborative work: Nong Gao He, Ph.D. and Minako Oda, Ph.D. for maintaining and improving the lab environment as Lab Managers; Dianne Reyes, Tu Dinh, and Rachael Korpi for research administration; Nicole Enger, a research scientist, for technical assistance; and Lynn Lauerman for scientific editing of the manuscript. The project was supported in part by grants from the National Heart, Blood, and Lung Institute within the National Institutes of Health (HL138992, HL152723, HL117247 to K.F.), the American Heart Association Established Investigator Award (0540054N to K.F.), and the American Heart Association Grant-in-Aid (7770000 to K.F.).
Author information
Authors and Affiliations
Contributions
K.F. conceived the general idea and framework of the project, designed the majority of the experiments, and oversaw the project to its completion. P.C. and D.P. contributed to the experimental design and performed most of the experiments. P.S. contributed to animal experiments. D.E.B. provided de-identified tissue lysates from human hearts to D.P. for protein analyses. P.C., D.P., and K.F. analyzed data and composed figures. P.C. and D.P. analyzed the immunohistochemistry slides. K.F. wrote the manuscript. K.F. and D.P. proofread the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chunhacha, P., Pinkaew, D., Sinthujaroen, P. et al. Fortilin inhibits p53, halts cardiomyocyte apoptosis, and protects the heart against heart failure. Cell Death Discov. 7, 310 (2021). https://doi.org/10.1038/s41420-021-00692-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-021-00692-w
