
Abstract
MVP17 encodes a mitochondrial inner-membrane protein, and mutation of human MVP17 can cause mitochondria DNA depletion syndrome (MDDS). However, the underlying function of mpv17 is still elusive. Here, we developed a new mutant with mpv17 knockout by using the CRISPR/Cas9 system. The mpv17−/− zebrafish showed developmental defects in muscles, liver, and energy supply. The mpv17−/− larvae hardly survived beyond a month, and they showed abnormal growth during the development stage. Abnormal swimming ability was also found in the mpv17−/− zebrafish. The transmission electron microscope (TEM) observation indicated that the mpv17−/− zebrafish underwent severe mitochondria dysfunction and the disorder of mitochondrial cristae. As an energy producer, the defects of mitochondria significantly reduced ATP content in mpv17−/− zebrafish, compared to wild-type zebrafish. We hypothesized that the disorder of mitochondria cristae was contributed to the dysfunction of muscle and liver in the mpv17−/− zebrafish. Moreover, the content of major energy depot triglycerides (TAG) was decreased dramatically. Interestingly, after rescued with normal exogenous mitochondria by microinjection, the genes involved in the TAG metabolism pathway were recovered to a normal level. Taken together, this is the first report of developmental defects in muscles, liver, and energy supply via mitochondria dysfunction, and reveals the functional mechanism of mpv17 in zebrafish.
Similar content being viewed by others


Introduction
MVP17 encodes proteins with four-transmembrane spanning in the mitochondrial inner membrane, but its function was not well understood. Different health problems were reported in multiple species. In mice, Mpv17 was identified early in 1990, and the defect of Mpv17 was generated by the insertional inactivation of recombinant retrovirus. The homozygotes of Mpv17−/− mice were found with health problems like nephrotic syndrome and chronic renal failure [1]. Subsequently, more works were reported on the mouse Mpv17 defect [2,3,4,5,6,7], implying that the function of the Mpv17 gene was involved in the ROS production and glomerulosclerosis. Due to the morphological degeneration of cochlea [5, 6], the abnormality of the mouse inner ear was also reported after the loss of Mpv17, which was similar to human Alport syndrome [3, 4]. Clozel et al. [7] revealed that Mpv17−/− mice developed significant systemic hypertension and tachycardia, and followed by polyuria and elevated natriuresis, indicating the key role of Mpv17 in the mouse.
In humans, MVP17 mutation caused mitochondria DNA depletion syndrome (MDDS) and led to death in infancy [8,9,10,11,12,13]. The clinical symptoms of MVP17 mutation were hepatopathy, peripheral neuropathy, and failure to thrive [14]. Wong et al. [15] reported that mutations of the MVP17 gene were responsible for rapidly progressive liver failure in infancy. Most of the human MDDS clinical cases were point mutant or deletion of the MVP17 [16, 17].
In 2013, the mpv17 defect in zebrafish was reported, and the phenotype of transparent (tra) zebrafish was characterized, whose mpv17 was a deletion resulting in a premature stop codon [18]. The tra mutant zebrafish showed a transparent phenotype with iridophore and melanophore defects. In 2017, D’Agati and colleagues provided genetic evidence to roy orbison (roy) and casper zebrafish [19, 20]. They indicated that a 19 bp deletion in the mpv17 mRNA caused the line roy [21]. They showed no other phenotype in the roy zebrafish, implying that there were no obvious defects in zebrafish liver, kidney, and muscle except the major defects of the skin color, compared to humans or mice mutant.
In this study, we developed a newly established zebrafish line with a truncate mpv17 to show a strong phenotype lacking iridophores and melanophores like tra and roy. Besides, our mpv17−/− zebrafish hardly survived more than 4 weeks; they had abnormal muscle and liver. The defects were consistent with human and mouse mutants but not the same as the zebrafish mutant line tra and roy. The mitochondria in the mpv17−/− possessed cristae defects, which is the key role for cellular respiration. Thus, the contents of TAG and ATP were decreased in the mpv17−/− zebrafish. Our work provided more details and evidence of the relationship between mpv17 and the development of muscles and the liver. The malfunction mitochondria played a key role in connecting the gene and these abnormal phenotypes. This newly established zebrafish mutant line could help us to elucidate the molecular mechanism of the function of the mpv17 gene on the MDDS.
Results
The generation of the mpv17 −/− zebrafish
We knocked out the zebrafish mpv17 using the CRISPR/Cas9 system to generate the mpv17−/− zebrafish. The target sites were designed at the second exon of the chromosome near the translation start code with an online tool ZIFIT Targeter (http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx) (Fig. 1A). The CRISPR/Cas9 system worked well for knocking out gene mpv17, which was confirmed by the T7E1 assay (Fig. 1B). The larvae were raised to adults for screening the homozygous mutant. Two mutants mpv17-2 and mpv17-5 were identified, which indicated that there were 2 and 5 bp deletions of the gene. For the details of mutants information, the sequences of these mutants were compared with those of wild-type and mutant transparent (tra) [18] (Fig. 1C). In the tra mutant, a deletion of 54–72 bp in mpv17 produced a frameshift following a stop code after 31 codons. The N-terminal 18 amino acid residues were identical with the wild-type. In our work, the deletion of 2 and 5 bp in the position 22 bp results in a frameshift and premature stop code. A 30 and 29 amino acid residues were left, and only 7 residues in the N-terminal were identical to the wild-type. Since the mpv17-5 showed only one residue less than mpv17-2, we performed all the following experiments using the mutant mpv17-5, which refers to mpv17−/−. The 1-month-old mpv17−/− zebrafish showed lack of iridophores and melanophores in the belly, dorsum, and eyes, which was similar to the tra mutant (Fig. 1D) [18]. The development stages of the wild-type and mpv17−/− larvae were recorded from zygotes to larvae at 9 days post-fertilization (dpf) (Supplementary Fig. S1). At 5 dpf, the eyes’ reflection of mpv17−/− larvae were significantly reduced due to the lack of iridophores (Supplementary Fig. S1E, F, G, H, Eʹ, Fʹ, Gʹ, Hʹ).

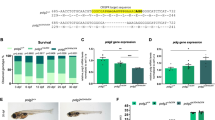
A The targeted loci were near the translation start code ATG of the gene mpv17. B The agarose gel electrophoresis results of the T7E1 assay for the screening of mpv17 knocked out zebrafish. The marker is DL1000 and the lane number shows different samples. The 397 bp band indicates the wild-type gene, and the lanes with 227 and 170 bp bands show the mutated genes. C The amino acid alignment of the mpv17 protein sequence in wild-type, tra mutant, and mpv17−/− zebrafish in this study. D One-month-old wild-type and mpv17−/− zebrafish larvae.
Poor growth and low survival beyond 4 weeks in the mpv17 −/− zebrafish
The heterozygotes mpv17+/− zebrafish were crossed, and the offspring eggs were used to check the survival rate assay by calculating the predictable proportion of mpv17−/−. We pooled mpv17+/+ and mpv17+/− for the survival calculation due to the mpv17+/− zebrafish was unable to distinguish from the mpv17+/+ zebrafish in the phenotype. The mpv17−/− zebrafish can be selected according to the reflection color of eyes at 5 days, but there was no difference between mpv17+/+ and mpv17+/−. The proportion was no significant in the larvae at 5 dpf (23.5%, 76.5%). At 10 days, the proportion was significantly different from the predicated, when 21.9% of mpv17−/−, 78.1% of the pooled samples were recovered. At 26 dpf, only 6.8% mpv17−/− zebrafish were still alive (Fig. 2A). The body length was also measured in the mpv17−/− and pooled mpv17+/+ and mpv17+/− groups. At the early stage of the development, the length of the mpv17−/− larvae was no different from the pooled group. After that, the mpv17−/− larvae were shorter than mpv17+/+ and mpv17+/− at 14 and 21 dpf. The mpv17−/− larvae almost stopped growing, compared to the 14 and 21 dpf samples (Fig. 2B). It indicated that the mpv17−/− zebrafish had poor growth and even stopped after 2 weeks. We did the rescue of the mpv17−/− larvae using the mpv17 mRNA injected into the eggs. The proportion of mpv17−/− larvae was significantly less in the rescue group, which indicated that the mpv17 mRNA could recover the phenotype of mpv17−/− mutant larvae (Fig. 2C).

A The mpv17−/− zebrafish exhibited a decreased survival ratio in the first month. n = 1160 for 5 dpf, 993 for 10 dpf, 227 for 15 dpf and 176 for 26 dpf. B The growth of the mpv17−/− zebrafish was decreased and even stopped after 14 days. The difference in the average length between mpv17−/− and the pooled (mpv17+/+ and mpv17+/−) larvae was increased. C The mpv17 mRNA can rescue the mpv17−/− zebrafish, and the proportion of mpv17−/− zebrafish in the rescue group was less than the control. Error bars were indicated with ±SEM, ***p < 0.005.
Aberrant skeletal muscle in the mpv17 −/− zebrafish
During the raising and screening of the homozygotes, we found an obvious phenomenon of the mpv17−/− zebrafish with abnormal movements (Supplementary Movie S1). According to the movie, we recorded the swimming behavior of the larvae with different color lines (Supplementary Fig. S2A). We counted the swing times of the caudal fin and the swimming distance (Supplementary Fig. S2B, C). Interestingly, the mpv17−/− larvae had long swimming distances for 15 s, and the swing times of caudal fin were significantly higher than those of the wild-type larvae. We considered that the muscle with defects was contributed to this phenotype. Thus, we performed in situ hybridization with the RNA probe for the muscle development marker gene myod. This gene is required for somatic and early cranial myogenesis, and some early pectoral fin development [22, 23]. The result showed that the signal of the myod was reduced in the mpv17−/− zebrafish (Fig. 3A). We confirmed this using qRT-PCR with more genes involved in the muscle development, and the expression levels of the myf5 [24], mstna, and mstnb were significantly decreased in the mpv17−/− zebrafish (Fig. 3D). For more details of the muscle development, we performed the whole-mount immunofluorescence with antibody F59 and F310, which predominantly recognizes slow muscle myosin fibers and fast muscle myosin fibers. The alterations in the arrangement and integrity of myofibers were observed apparently. The structure of myofiber was irregular, and the tissue displayed a disorganization pattern in the mpv17−/− zebrafish, compared to the wild-type (Fig. 3B). It’s indicated that the early development of skeletal muscle in the mpv17−/− zebrafish was influenced by the mutant mpv17 gene. The histological analysis was performed with hematoxylin-eosin (HE) staining of the section for the trunk skeletal muscle tissue (Fig. 3C). The mass of skeletal tissue was less than that of the control. The structure was loose and there were more interspaces between the myofibers in the mpv17−/− zebrafish.

A In situ hybridization analysis of myogenic marker gene myod at 24 hpf. Scale bar = 250 μm. B Confocal image of fast and slow muscle at 72 hpf. Antibodies F310 and F59 were used for staining the fast and slow muscle fiber, respectively. Scale bar = 50 μm. C Paraffin section and H&E staining analysis of the skeletal muscle structure in the 1-month-old fish. D The relative mRNA expression levels of the genes involved in the development of muscles.
Altered content of major energy depot and the change of related genes in the mpv17 −/− larvae
Due to the motility abnormality of the mpv17−/− zebrafish, we wonder whether the energy supply was reduced. Thus, we evaluated the contents of triglyceride (TAG), which is the major energy depot of all eukaryotic cells, and the metabolites diglyceride (DAG) and non-esterified fatty (NEFA) in the mpv17−/− larvae. The result showed that the TAG content was significantly reduced in the mpv17−/− larvae (0.007 mmol/g prot), compared to the control (0.039 mmol/g prot). For the content of NEFA, the mpv17−/− zebrafish was less than the wild-type control. There was no significant difference in the content of DAG between the mpv17−/− and control. Moreover, the glucose level was lower in the mpv17−/− zebrafish than that of the wild-type (Fig. 4A). These results suggested that the energy substrate of the mpv17−/− zebrafish was less than the wild-type. We tried to figure out whether the ATP contents of the mpv17−/− zebrafish were disturbed. As expected, the content of ATP in the mpv17−/− zebrafish was significantly reduced to half of the wild-type (Fig. 4B). Thus, we checked the expression levels of genes involved in TAG synthesis and degradation. The expression levels of all genes were changed in the mpv17−/− zebrafish except mogat3a, which can catalyze the first step in triacylglycerol synthesis. The genes atgl, hsla, and mgll that involved in the triglyceride breakdown were significantly increased in the mpv17−/− larvae. The increase of the gene expression enhanced the degradation of the triglycerides (Fig. 4C).

A The contents of the DAG, TAG, NEFA, and glucose were measured at 7 dpf stage. B The content of ATP was evaluated at 7 dpf. C The relative mRNA expression levels of genes involved in the TAG metabolism in mpv17−/− larvae with or without normal mitochondria injection. D The bodyweight of the mpv17−/− larvae with or without mitochondria injection. Error bars were indicated with ±SEM, *p < 0.05, **p < 0.01, ****p < 0.001.
Considering the mpv17 is a gene encoding a protein located in the mitochondrial inner membrane, we tried to figure out whether exogenous mitochondria can recover the gene expression of TAG metabolism or not. We performed the mitochondrial injection to the one-cell stage eggs of the mpv17−/−. After 7 days, the zebrafish were collected and detected the expression levels of genes involved in triglyceride metabolism. After injection, the expression levels of hsla and mogat3a were back to the wild-type level, indicating that the exogenous mitochondria may be a good solution to recover the mpv17 mutant (Fig. 4C). The bodyweight of the mpv17−/− larvae at 7 dpf was significantly reduced to approximately 16%, compared to control (Fig. 4D). Interestingly, injection with exogenous mitochondria, the bodyweight of the mpv17−/− larvae was also recovered closed to the wild-type level (Fig. 4D).
Delayed liver development and abnormal tissue
We performed the whole-mount in situ hybridization (WMISH) to evaluate the liver development of the mpv17−/− zebrafish with an RNA probe tfa (hepatogenic marker). The tfa gene signal was strong and presented on the left side of the body in the wild-type larvae from the dorsal view. However, the staining of the probe was absent in the mpv17−/− larvae at 3 dpf (Fig. 5A, upper and middle panel). We injected the mpv17−/− larvae with mpv17 mRNA to rescue the phenotype. After 3 days, the mpv17−/− larvae had a slight signal using the probe in the same position (Fig. 5A, lower panel). We also performed the experiment with larvae at 5 dpf, and the staining intensity was increased and spread from left to the right side in wild-type larvae from dorsal view. But, the mpv17−/− larvae had weak signals on the left side of the liver (Fig. 5B), implying that the development of liver in mpv17−/_ larvae was delayed, compared to the wild-type. We also performed the qRT-PCR to analyze the expression levels of the genes involved in liver development. The wnt2bb, bmp2b, and fgf play key roles as signaling molecules in the specification of hepatoblasts. The hhex and prox1 can promote liver budding and hepatoblasts migration, respectively [25]. The ppp1r12a mediates the liver primordia to promote liver bud growth [26]. The rbp4 plays a role in the formation of a liver bud, which requires the migration of liver progenitor cells [27]. All genes related to the initial development of the liver reduced significantly in the mpv17−/− larvae (Fig. 5C, D). These results confirmed that liver development was delayed in the mpv17−/− zebrafish. Further, the paraffin section and HE stain were used to check the structure of liver tissue with 1-month-old mpv17−/− zebrafish. As shown in Fig. 5E, the liver tissue was discrete and abnormal, compared to the control.

A In situ hybridization with RNA probe tfa at 3 dpf. Wild-type was in the upper, mpv17−/− in the middle, and the lower was the mpv17−/− larvae rescued with mpv17 mRNA. B In situ hybridization of larvae at 5 dpf using tfa probe. C The expression level of genes tfa. Error bars were indicated with ±SEM, *p < 0.05. D The expression levels of genes involved in liver development. Error bars were indicated with ±SEM, **p < 0.01, ***p < 0.005. E H&E staining paraffin-embedded sections of the liver in 1-month-old zebrafish. The black frames were enlarged as shown on the right.
Hollow mitochondrial cristae and reduced number of mitochondria in the mpv17 −/− zebrafish
The muscle tissue of mpv17−/− zebrafish at 7 dpf was collected for the mitochondria analysis by TEM. The mitochondria numbers in the mpv17−/− larvae muscles were significantly decreased, compared to the wild-type larvae (Fig. 6A, upper panel, Fig. 6B, upper panel). In each 100 μm2 of the tissue, the average number of mitochondria was about 6 and 3 in the wild-type and mpv17−/− larvae, respectively. The size of the mitochondria in mpv17−/− larvae was smaller than that of the wild-type (Fig. 6B, lower panel). The mitochondria were shrunk about 30% of the size in the mpv17−/− line. We also saw the hollow mitochondria with broken cristae in the mpv17−/− larvae muscle (Fig. 6A). These defects dramatically decreased the surface area of the inner-membrane system, which is the most important place for cellular respiration. Thus, the low content of ATP may be due to the defects of the mitochondrial cristae.

A TEM image of the skeletal muscle for the larvae at 7 dpf. The magnification was ×2500 in the upper panel and ×11,500 in the lower panel. Red arrowhead indicated the mitochondria in the skeletal muscles. B The number of the mitochondria per 100 μm2 (upper) and the diameter of the mitochondria in the skeletal muscle (lower). Error bars were indicated with ±SEM, *p < 0.05 and **p < 0.01.
Discussion
In this study, the mpv17 knockout zebrafish was generated using the CRISPR/Cas9 system. The mpv17−/− zebrafish showed developmental defects in muscles, liver, and energy supply via mitochondria dysfunction, which is similar to human MDDS. The clinical symptoms of human MDDS contain hepatopathy, peripheral neuropathy, failure to thrive, and rapidly progressive liver failure in infancy [14, 15]. In the previous study, a tra mutant was first reported in 1983 by Walker and Streisinger [28]. In 2013, this mutant line was well studied in phenotypic and molecular characterization by Krauss [18]. In this tra zebrafish, the iridophores and melanophores were significantly reduced due to the mutant of the mpv17 gene. In 2017, another work had figured out that the roy zebrafish has a 19 bp deletion of the mpv17 mRNA using the positional cloning method. They confirmed it by a rescue experiment with mpv17 mRNA [21]. However, In these works, the authors found that the mutant of mpv17 contributed to the zebrafish lacking iridophores and melanophores. Strikingly, they did not find any other defects in the liver, kidney, or muscle except the defects to the skin.
In this study, the homozygote hardly survived 1 month as indicated in the “Result” section, and the developments of liver, muscle, and mitochondria were all abnormal. Our finding was similar to the phenotype of mpv17 mutant mice that carried an insertion in the mpv17 gene and destroyed protein function. The mpv17 mutant mice developed mitochondrial DNA depletion, late-onset glomerulosclerosis, hair graying, and the neurodegeneration of the peripheral nervous system [1, 4, 5, 29]. In the humans, many mutations of this gene had been reported, and the mpv17 mutation was associated with mitochondrial DNA depletion syndrome and caused death during early childhood [17, 30, 31]. The different phenotypes between our mutant zebrafish and the tra or roy lines possibly because of the different truncated mpv17 proteins. In our work, the mpv17 protein had seven amino acid residues that were identical to the wild-type protein, while the tra had 18 amino acids. The 11 amino acids difference perhaps contributed to the diverse phenotype between these mutant lines. Another possible reason was that the deletion way of the gene was different. The mpv17−/− zebrafish in this work was established with the deletion of exon 2 using the CRISPR/Cas9 system, and this deletion was introduced by the non-homologous end joining (NHEJ). In the tra and roy zebrafish, the genotype was caused by a deletion of intron 2, following a cryptic splice donor site in exon 2 [21].
Compared to the wild-type, the movement was abnormal in the mpv17−/− zebrafish (Supplementary Movie S1, Supplementary Fig. S2). We considered that the abnormal movement was due to the defects of muscle tissues in the mpv17−/− zebrafish (Fig. 3B, C). Further, the number and size of muscle mitochondria were decreased, compared to the wild-type (Fig. 6). The phenomenon may come from mitochondrial remodeling, which is important in the early development of muscle in zebrafish [32]. In this study, we analyzed the mtDNA number of the whole zebrafish and did not find a significant difference between the mpv17−/− and wild-type larvae (data not shown). The potential reason was that the mtDNA number was decreased in a specific tissue, and the difference of the whole body was not obvious [33]. However, in the muscle tissue, the mitochondrial number was approximately 50% in the mpv17−/− zebrafish, compared to the control (Fig. 6B, upper panel). Besides, the disruption of the mitochondrial cristae was observed in the mpv17−/− zebrafish. Similarly, the loss of the cristae [13], dilated cristae [34], and unstructured cristae [35] were found in the liver of MVP17 mutant patients. Further, the mpv17 plays a vital role in maintaining mitochondrial structure and functionality [36]. The insufficient normal mitochondria with less energy ATP (Fig. 4B) fail to function well. Thus, the muscle fibers are developed irregularly, which may be due to the dysfunction of mitochondria.
Interestingly, the mpv17−/− larvae with weak muscles showed longer swimming distances and faster caudal fin swing (Supplementary Fig. S2B, C). Due to the defects of the muscles, the angle of caudal fin swing in the mpv17−/− is smaller than that of the wild-type. Thus, they need to move the caudal fin more times to keep balance in the water. The energy consumption for each caudal fin swing is lower. In other words, the mpv17−/− larvae have not enough energy for a longer distance of swing the fin each time. They swim in the water to a long distance by rapidly moving the caudal fin. A previous study showed that a 25-year-old male patient showed weakness and atrophy of both distal muscles and the intrinsic muscles due to the mutation of MPV17 [37]. Another clinical case of MPV17 mutation was reported in a 21-year-old male, who walked only with a slow unsteady gait due to the muscle wasting of hands and distal lower limbs [38]. In the mouse model, the mpv17−/− mice described by Viscomi indicated that a subclinical mitochondrial myopathy occurred in a 1-year-old mpv17−/− mouse [29], whose muscle tissue had only 25% mtDNA compared to the normal littermates. In the mitochondria, dysfunction of ATP synthase caused the deficiency of the flat lamellar cristae and the formation of separate vesicles with few or no cristae [39,40,41]. Thus, we hypothesized that the number of mitochondria and the defects in mitochondrial cristae contributed to the lower ATP content, which affected the muscle development and the movement of the mpv17−/− zebrafish.
Although the genetic difference exists between zebrafish and human, zebrafish is a good model for the studies of human diseases. The phenotype of mpv17−/− zebrafish is similar to human MDDS, because we knocked out mpv17 gene by deleting 5 bp coding sequences to destroy its function in zebrafish. Our mpv17−/− fish has defects in muscles, liver, and energy supply. We did not do the morpholino experiment for knocking down mpv17. D’Agati reported that ATG morpholino knockdown of mpv17 led to a strong roy phenocopy with loss of iridophores and visible pericardial edema [21].
In this study, the result of WMISH with probe tfa showed less signal intensity in the mpv17−/− larvae at 3- 5 dpf (Fig. 5A, B). Especially in the 3-day samples, the staining intensity almost disappeared. The low expression level of tfa gene, a hepatic function marker secreted by hepatocytes [42], implied that the secretory tfa proteins were suppressed in the hepatocytes. Moreover, the expression levels of the genes involved in liver development were all significantly reduced in the mpv17−/− zebrafish (Fig. 5D). These genes played important roles in the specification and migration of hepatoblasts [25, 43, 44], implying that they possibly reduced the development of hepatoblasts to a mature liver.
Taken together, our newly established mpv17−/− zebrafish can mimic the phenotypes of human MPV17 depletion diseases, which is firstly reported in the fish model. Due to the deficiency of mitochondria, abnormal defects of muscles, liver, and energy supply appeared in the mpv17−/− zebrafish, indicating that our mpv17−/− zebrafish can be applied for the therapy study of mitochondrial disease in the future.
Materials and methods
Ethical approval
The animal experiment is approved by the Animal Care and Use Committee of Chongqing, China and by the Institutional Animal Care and Use Committee of Chongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences (Approval ID: ZKCQY0106), which is performed according to the “Guide for the Care and Use of Laboratory Animals” (Eighth Edition, 2011).
Zebrafish culture
Adult zebrafish of the AB strain were raised and maintained in an automatic water cycle system at 28 °C with a 14-h light and 10-h dark cycling. The male and female were mated once a week, and the eggs were collected and cultured at 28 °C.
Generation of the mpv17 −/− zebrafish
The targeted sequence of zebrafish mpv17 was designed using an online tool ZIFIT Targeter (http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx). PCR was performed to amplify the target sequence for the transcript template. The mpv17 sgRNA was synthesized in vitro with a MAXIscript T7 Kit (Ambion, USA). A linearized plasmid pXT7-Cas9 as a template for producing the Cas9 capped mRNA transcription in vitro using Ambion mMESSAGE mMACHINE T7 Transcription Kit (Ambion, USA). The RNA products were purified according to the manual of the MicroElute RNA Clean-Up Kit (OMEGA, USA), and the quality and concentration were determined by agarose gel electrophoresis and NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, France). Then, the RNAs were stored at −80 °C before use.
Embryos at the one-cell stage were collected and distributed into a petri dish, and 1 nl mixture with Cas9 capped mRNA (300 ng/μL) and mpv17 sgRNA (30 ng/μl) was microinjected into embryos using a microinjector (Eppendorf, Germany). The injected eggs were incubated and raised in a petri dish at 28 °C. After 48 h, ten eggs were pooled to extract the total DNA, and the T7E1 assay was performed to detect the efficiency of the mpv17 targeted. The injected embryos were raised to adults for screening the mpv17−/− zebrafish.
Determination of mitochondrial DNA copy number
Absolute quantitative real-time PCR was performed for quantitating the mitochondrial DNA (mtDNA) copy numbers. A T-Vector pMD20 (Takara, Japan) was inserted with a PCR segment of zebrafish nd1. The vector molarity was calculated and diluted to 102, 103, 104, 105, 106, and 107 copies/μl. The genome DNA of 7 dpf wild-type and mpv17−/_ zebrafish were extracted and diluted to 3 ng/μl. The quantification used the qRT-PCR for targeting the mitochondrial gene nd1. The primers are listed in Table 1. Quantitative real-time PCR was performed using the TB Green™ Premix Ex Taq™ II (Tli RNaseH Plus) (Takara, Japan) on an ABI 7300 System (PerkinElmer Applied Biosystems, Foster City, CA, USA). Then, the standard curve was plotted as a logarithmic regression with Ct values against the pMD20 copy numbers. The mtDNA copy number was calculated according to the equation given by the standard curve with r2 > 0.99.
Analysis of mRNA expression level
Total RNA was extracted by RNAiso Plus (Takara, Japan) from 20 larvae at 5 or 7 dpf. Then the cDNA was synthesized using a PrimeScript RT reagent kit with gDNA Eraser (Takara, Japan). qRT-PCR was performed with a TB Green™ Premix Ex Taq™ II (Tli RNaseH Plus) (Takara, Japan) on an ABI 7300 System (PerkinElmer Applied Biosystems, Foster City, CA, USA). The expression levels of the genes were calculated with the 2−ΔΔCt method. All the primers used in this work are listed in Table 1.
Histological examination of the mpv17 −/− zebrafish
H&E staining of the muscle and liver tissue was performed. The 1-month-old mpv17−/− and wild-type zebrafish were selected and fixed in 4% paraformaldehyde (PFA) overnight. The samples were embedded in paraffin wax, dehydrated in ethanol, and sectioned into 5 mm slices. Then the paraffin sections of different samples were stained with H&E and photographed under a microscope (Nikon, Eclipse Ti, Japan).
Whole-mount in situ hybridization
For the WMISH, the embryos at different growth stages were fixed in 4% paraformaldehyde. We did the WMISH according to the protocol described by Thisse [45]. The antisense probe was designed to target gene transcript sequences. A T7 RNA polymerase promoter (T7: 5ʹ-TAATACGACTCACTATAGGG-3ʹ) was included in the appropriate primer for amplifying the probe template. The RNA probes used in this experiment were about 700 bp. The primers were synthesized by GENEWIZ (China) and the sequences are presented in Table 1. The probe templates were amplified and purified by TAINquick Midi Purification Kit (TIANGEN, China) and then transcribed with MAXIscript T7 Kit (Ambion, USA) in vitro. The products were purified by MicroElute RNA Clean-Up Kit (OMEGA, USA) and quantified with NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, France).
Whole-mount immunofluorescence
The whole-mount immunofluorescence was performed as follows. The 3 dpf zebrafish embryos were fixed in 4% PFA overnight, then washed three times with PDT (1× phosphate buffered saline (PBS), 0.1% Tween-20, 0.3% Triton-X, 1% dimethyl sulfoxide-DMSO). After blocking 1 h at room temperature with blocking buffer (1× PBS, 0.1% Tween-20, 10% fetal bovine serum (FBS), 2% bovine serum albumin (BSA)), the monoclonal antibody F59 (Developmental Studies Hybridoma Bank-DHSB, 1:20) and F310 (DHSB, 1:20) were used to anti-MHC and MLC, respectively. After incubation for 2 h at room temperature, the samples were washed and blocked again, then incubated with Alexa Fluor® 488 Goat Anti-Mouse (IgG) for 1.5 h at room temperature. Finally, the samples were submerged in 90% glycerol and imaged using an Olympus FluoView™ FV1000 Confocal Microscope (Japan).
Measurement of the contents of TAG, DAG, NEFA, and ATP
Wild-type and mpv17−/− larvae at 7 dpf were selected to measure the contents of triglyceride (TAG), diglyceride (DAG), non-esterified fatty acids (NEFA), and adenosine triphosphate (ATP) as described in the manufacturer’s protocol, respectively. For the tissue samples, the samples were washed with cold PBS, then resuspended and homogenized in extraction buffer. After centrifugation with 12,000 rpm at 4 °C, the supernatant was harvested for the following assay according to the manual instruction. The kits were used as follows: triglyceride assay kit and non-esterified free fatty acids assay kit (Nanjing Jiancheng, Nanjing, China), Zebrafish DAG ELISA kit (Mlbio, Shanghai, China), enhanced ATP assay kit (Beyotime, Shanghai, China).
Rescue of the mpv17 −/− larvae with mpv17 mRNA
The full open reading frame (ORF) of zebrafish mpv17 (NM_201165.2) was amplified using the PrimeSTAR HS Premix (Takara, Japan). Then, the amplicons were inserted into the pCS2+ vector for the in vitro transcription of mpv17 capped mRNA. We injected the mpv17 capped mRNA into the one-cell stage embryos from the mpv17+/− zebrafish parents at a concentration of 100 ng/μl. The samples were incubated at 28 °C before collection.
Analysis of the mitochondria structure of zebrafish muscle with TEM
TEM was used for further observation of the mitochondria. The zebrafish larvae at 7 dpf were selected and fixed in 2.5% glutaraldehyde solution, diced into 1 mm3, and immersed in 0.1 M phosphate buffer (pH 7.4) overnight. The treated samples were fixed in 1% OsO4 buffer for 1 h at 4 °C. Samples were dried in a graded series of ethanol and then further dehydrated using an epoxy resin. Finally, ultrathin sections (60−80 nm) of tissues were obtained using a Leica EM UC6 ultra-microtome and examined with a Hitachi (HT-7700) TEM at 80 kV. Three individual samples were randomly collected and used for TEM analyses. The number of the mitochondria was counted and calculated as number per μm2. The diameter of the mitochondria was measured by the long axis.
Mitochondria microinjection to the mpv17 −/− zebrafish embryos
The mitochondria were isolated from wild-type zebrafish liver using a mitochondrial extraction kit (Solarbio, Beijing, China). According to the manufacturer’s protocol, ten wild-type adult zebrafish livers were collected for the isolation procedure. The livers were washed three times with cold PBS, and then homogenized in the extraction buffer. The homogenate was centrifuged at 1000 × g for 5 min, and repeated once. Subsequently, the supernatant was centrifuged at 12,000 × g for 10 min, and the pellets were washed twice with wash buffer. The pellets were dissolved in PBS and the concentration was determined by BCA assay. Finally, the microinjection of mitochondria was done according to the previous description [46].
The survival ratio and the body length of mpv17 −/− zebrafish
The survival ratio of mpv17−/−, pooled mpv17+/−, and mpv17+/+ zebrafish was determined by comparing the observed phenotype with the expected ratio of 1:3. The eggs were harvested from individual crossbreeding of heterozygous parents and reared in beakers. Eggs were collected and the larvae number was counted and recorded. Pearson’s chi-square analyses were used to test whether expected genotype proportions were recovered at each time point. The body length of the zebrafish was measured in each group (mpv17−/−, pooled mpv17+/−, and mpv17+/+). The larvae at 4, 8, 14, and 21 dpf were anesthetized with MS222 (Aladdin, China) and taken pictures for recording the body length.
Movie record and analysis of the swimming behavior
A movie was made with five of each wild-type and mpv17−/− zebrafish larvae. We recorded the swimming trail with different color lines. The swing time of the caudal fin was counted for 15 s, and the swimming distance of each larva was calculated at the same time.
Statistics analysis
For statistical analysis, one-way analysis of variance (ANOVA) was applied to calculate the differences between the control and experimental groups by GraphPad Prism version 5.01 (GraphPad Software, USA). A value of p < 0.05 was considered statistically significant, and all values were expressed as the means ± standard error of the mean (SEM). Each experiment was performed three times independently.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
References
Weiher H, Noda T, Gray DA, Sharpe AH, Jaenisch R. Transgenic mouse model of kidney disease: insertional inactivation of ubiquitously expressed gene leads to nephrotic syndrome. Cell. 1990;62:425–34.
Zwacka RM, Reuter A, Pfaff E, Moll J, Gorgas K, Karasawa M, et al. The glomerulosclerosis gene Mpv17 encodes a peroxisomal protein producing reactive oxygen species. EMBO J. 1994;13:5129–34.
Meyer zum Gottesberge AM, Reuter A, Weiher H. Inner ear defect similar to Alport’s syndrome in the glomerulosclerosis mouse model Mpv17. Eur Arch Otorhinolaryngol. 1996;253:470–4.
Meyer zum Gottesberge AM, Massing T, Hansen S. Missing mitochondrial Mpv17 gene function induces tissue-specific cell-death pathway in the degenerating inner ear. Cell Tissue Res. 2012;347:343–56.
Müller M, Smolders JW, Meyer zum Gottesberge AM, Reuter A, Zwacka RM, Weiher H, et al. Loss of auditory function in transgenic Mpv17-deficient mice. Hear Res. 1997;114:259–63.
Meyer zum Gottesberge AM, Felix H, Reuter A, Weiher H. Ultrastructural and physiological defects in the cochlea of the Mpv17 mouse strain. A comparison between young and old adult animals. Hear Res. 2001;156:69–80.
Clozel M, Hess P, Fischli W, Löffler BM, Zwacka RM, Reuter A, et al. Age-dependent hypertension in Mpv17-deficient mice, a transgenic model of glomerulosclerosis and inner ear disease. Exp Gerontol. 1999;34:1007–15.
El-Hattab AW, Wang J, Dai H, Almannai M, Scaglia F, Craigen WJ. MPV17-related mitochondrial DNA maintenance defect. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. editors. GeneReviews((R)). Seattle, WA, University of Washington Press publisher; 1993.
Sarzi E, Bourdon A, Chrétien D, Zarhrate M, Corcos J, Slama A, et al. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. J Pediatr. 2007;150:531–4. 534 e531-536
Spinazzola A, Massa V, Hirano M, Zeviani M. Lack of founder effect for an identical mtDNA depletion syndrome (MDS)-associated MPV17 mutation shared by Navajos and Italians. Neuromuscul Disord. 2008;18:315–8.
AlSaman A, Tomoum H, Invernizzi F, Zeviani M. Hepatocerebral form of mitochondrial DNA depletion syndrome due to mutation in MPV17 gene. Saudi J Gastroenterol. 2012;18:285–9.
Mudd SH, Wagner C, Luka Z, Stabler SP, Allen RH, Schroer R, et al. Two patients with hepatic mtDNA depletion syndromes and marked elevations of S-adenosylmethionine and methionine. Mol Genet Metab. 2012;105:228–36.
Kim J, Kang E, Kim Y, Kim JM, Lee BH, Murayama K, et al. MPV17 mutations in patients with hepatocerebral mitochondrial DNA depletion syndrome. Mol Genet Metab Rep. 2016;8:74–76.
Karadimas CL, Vu TH, Holve SA, Chronopoulou P, Quinzii C, Johnsen SD, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544–8.
Wong LJ, Brunetti-Pierri N, Zhang Q, Yazigi N, Bove KE, Dahms BB, et al. Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology. 2007;46:1218–27.
Navarro-Sastre A, Martín-Hernández E, Campos Y, Quintana E, Medina E, de Las Heras RS, et al. Lethal hepatopathy and leukodystrophy caused by a novel mutation in MPV17 gene: description of an alternative MPV17 spliced form. Mol Genet Metab. 2008;94:234–9.
El-Hattab AW, Li FY, Schmitt E, Zhang S, Craigen WJ, Wong LJ. MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: new patients and novel mutations. Mol Genet Metab. 2010;99:300–8.
Krauss J, Astrinidis P, Astrinides P, Frohnhöfer HG, Walderich B, Nüsslein-Volhard C. transparent, a gene affecting stripe formation in Zebrafish, encodes the mitochondrial protein Mpv17 that is required for iridophore survival. Biol Open. 2013;2:703–10.
White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2008;2:183–9.
Ren JQ, McCarthy WR, Zhang H, Adolph AR, Li L. Behavioral visual responses of wild-type and hypopigmented zebrafish. Vis Res. 2002;42:293–9.
D'agati G, Beltre R, Sessa A, Burger A, Zhou Y, Mosimann C, et al. A defect in the mitochondrial protein Mpv17 underlies the transparent casper zebrafish. Dev Biol. 2017;430:11–17.
Weinberg ES, Allende ML, Kelly CS, Abdelhamid A, Murakami T, Andermann P, et al. Developmental regulation of zebrafish MyoD in wild-type, no tail and spadetail embryos. Development. 1996;122:271–80.
Shi J, Bi P, Pei J, Li H, Grishin NV, Bassel-Duby R, et al. Requirement of the fusogenic micropeptide myomixer for muscle formation in zebrafish. Proc Natl Acad Sci USA. 2017;114:11950–5.
Lin CY, Chen YH, Lee HC, Tsai HJ. Novel cis-element in intron 1 represses somite expression of zebrafish myf-5. Gene. 2004;334:63–72.
Tao T, Peng J. Liver development in zebrafish (Danio rerio). J Genet Genomics. 2009;36:325–34.
Huang H, Ruan H, Aw MY, Hussain A, Guo L, Gao C, et al. Mypt1-mediated spatial positioning of Bmp2-producing cells is essential for liver organogenesis. Development. 2008;135:3209–18.
Li Z, Korzh V, Gong Z. Localized rbp4 expression in the yolk syncytial layer plays a role in yolk cell extension and early liver development. BMC Dev Biol. 2007;7:117.
Walker C, Streisinger G. Induction of mutations by gamma-rays in pregonial germ cells of zebrafish embryos. Genetics. 1983;103:125–36.
Viscomi C, Spinazzola A, Maggioni M, Fernandez-Vizarra E, Massa V, Pagano C, et al. Early-onset liver mtDNA depletion and late-onset proteinuric nephropathy in Mpv17 knockout mice. Hum Mol Genet. 2009;18:12–26.
Meldau S, De Lacy RJ, Riordan G, Goddard EA, Pillay K, Fieggen KJ, et al. Identification of a single MPV17 nonsense-associated altered splice variant in 24 South African infants with mitochondrial neurohepatopathy. Clin Genet. 2018;93:1093–6.
Pyal A, Paramasivam A, Meena AK, Bhavana VB, Thangaraj K. MPV17 hepatocerebral mitochondrial DNA depletion syndrome presenting as acute flaccid paralysis—a case report. Mitochondrion. 2017;37:41–45.
Zhang M, Sun T, Jian C, Lei L, Han P, Lv Q, et al. Remodeling of mitochondrial flashes in muscular development and dystrophy in zebrafish. PLoS ONE. 2015;10:e0132567.
Moss CF, Dalla Rosa I, Hunt LE, Yasukawa T, Young R, Jones A, et al. Aberrant ribonucleotide incorporation and multiple deletions in mitochondrial DNA of the murine MPV17 disease model. Nucleic Acids Res. 2017;45:12808–15.
Vilarinho S, Choi M, Jain D, Malhotra A, Kulkarni S, Pashankar D, et al. Individual exome analysis in diagnosis and management of paediatric liver failure of indeterminate aetiology. J Hepatol. 2014;61:1056–63.
Piekutowska-Abramczuk D, Pronicki M, Strawa K, Karkucińska-Więckowska A, Szymańska-Dębińska T, Fidziańska A, et al. Novel c.191C>G (p.Pro64Arg) MPV17 mutation identified in two pairs of unrelated Polish siblings with mitochondrial hepatoencephalopathy. Clin Genet. 2014;85:573–7.
Martorano L, Peron M, Laquatra C, Lidron E, Facchinello N, Meneghetti G, et al. The zebrafish orthologue of the human hepatocerebral disease gene MPV17 plays pleiotropic roles in mitochondria. Dis Model Mech. 2019;12:dmm037226.
Baumann M, Schreiber H, Schlotter-Weigel B, Löscher WN, Stucka R, Karall D, et al. MPV17 mutations in juvenile- and adult-onset axonal sensorimotor polyneuropathy. Clin Genet. 2019;95:182–6.
Blakely EL, Butterworth A, Hadden RD, Bodi I, He L, McFarland R, et al. MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul Disord. 2012;22:587–91.
Davies KM, Anselmi C, Wittig I, Faraldo-Gomez JD, Kuhlbrandt W. Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci USA. 2012;109:13602–7.
Minauro-Sanmiguel F, Wilkens S, Garcia JJ. Structure of dimeric mitochondrial ATP synthase: novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc Natl Acad Sci USA. 2005;102:12356–8.
Blum TB, Hahn A, Meier T, Davies KM, Kuhlbrandt W. Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc Natl Acad Sci USA. 2019;116:4250–5.
Howarth DL, Yin C, Yeh K, Sadler KC. Defining hepatic dysfunction parameters in two models of fatty liver disease in zebrafish larvae. Zebrafish. 2013;10:199–210.
Zhao H, Han D, Dawid IB, Pieler T, Chen Y. Homeoprotein hhex-induced conversion of intestinal to ventral pancreatic precursors results in the formation of giant pancreata in Xenopus embryos. Proc Natl Acad Sci USA. 2012;109:8594–9.
Jung J, Zheng M, Goldfarb M, Zaret KS. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science. 1999;284:1998–2003.
Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69.
Bian W-P, Chen YL, Luo JJ, Wang C, Xie SL, Pei DS. Knock-In Strategy for Editing Human and Zebrafish Mitochondrial DNA Using Mito-CRISPR/Cas9 System. ACS Synth Biol. 2019;8:621–32.
Acknowledgements
This work was supported by the CAS Team Project of the Belt and Road (to D.S.P.), Research Program of Chongqing Science and Technology Commission (No. cstc2019jcyj-zdxmX0035, cstc2019jcyj-msxm0700, CSTCCXLJRC201714, and 2019-04 Beibei), and Program of China–Sri Lanka Joint Center for Water Technology Research and Demonstration by Chinese Academy of Sciences (CAS)/China–Sri Lanka Joint Center for Education and Research by CAS, and International Partnership Program of CAS (No. 121311kysb20190071).
Author information
Authors and Affiliations
Contributions
W.P.B. prepared the data and wrote the manuscript. S.Y.P., S.L.X., C.W., S.D., and P.R.S. analyzed the data. P.R.S edited the final version of the manuscript. D.S.P. conceived, designed and revised the manuscript. D.S.P. conceived, designed and revised the manuscript.
Corresponding author
Ethics declarations
COMPETING INTERESTS
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Maria Victoria Niklison Chirou
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bian, WP., Pu, SY., Xie, SL. et al. Loss of mpv17 affected early embryonic development via mitochondria dysfunction in zebrafish. Cell Death Discov. 7, 250 (2021). https://doi.org/10.1038/s41420-021-00630-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-021-00630-w
