
Abstract
Hepatic stellate cells (HSC) and hydrogen sulfide (H2S) both play important roles in the development of hepatocellar carcinoma (HCC). Whereas, in the microenvironment of HCC, whether HSC participate in regulating the biological process of HCC cells by releasing H2S remains elusive. In vitro, Flow cytometry (FCM), CCK-8, RNA-sequencing, Western blotting, RT-qPCR, immunofluorescence and ChIP assays were carried out in the HCC cells to investigate the effect of H2S on biological functions and JNK/JunB-TNFSF14 signaling pathway. Specimens from HCC patients were analyzed by RT-qPCR and Western blotting assays for evaluating the expression of TNFSF14 and CSE. Statistical analysis was used to analyze the correlation between TNFSF14 expression and clinical data of HCC patients. Based on the FCM and CCK-8 results, we found the LX-2 cells were able to induce HCC cells apoptosis through releasing H2S. RNA-sequencing, RT-qPCR, and Western blotting results showed that TNFSF14 gene was upregulated in both LX-2 and NaHS group. NaHS treated in HCC cells led to JNK/JunB signaling pathway activating and greater binding of p-JunB to its responsive elements on TNFSF14 promoter. Impairment of TNFSF14 induction alleviated LX-2 and NaHS induced apoptosis of HepG2 and PLC/PRF/5 cells. Furthermore, TNFSF14 expression in HCC tissues was lower than the adjacent tissue. HCC patients with low expression of TNFSF14 had higher malignant degree and poor prognosis. In summary, demonstration of the involvement of HSC-derived H2S in JNK/JunB mediated expression of TNFSF14 gene strongly indicates H2S palys an important role in the regulation of HCC apoptosis.
Similar content being viewed by others
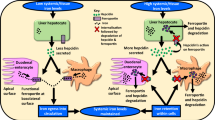


Introduction
Nowadays hepatocellular carcinoma (HCC) is the second most lethal cancer with persistently increasing mortality in the worldwide due to its highly complex and heterogeneous genetic aberrations [1, 2]. More and more researches suggest that tumor microenvironment (TME) plays an important role in the processes of hepatocarcinogenesis, epithelial-mesenchymal transition (EMT), invasion, and metastasis [3]. TME comprises various cells, including hepatic stellate cells (HSC), liver stromal endothelial cells (LSEC), immune cells and cytokines like extracellular matrix (ECM) [4].
HSC are usually considered to foster the tumor microenvironment formation and contribute to a malignant tumor phenotype. Activated HSC contribute to hepatocarcinogenesis through the secretion of growth factors, and producing ECM and matrix metalloproteinase (MMPs) [5,6,7]. But, there are more and more studies implicated that HSC could play an anti-tumor role in HCC progression [8, 9]. Hydrogen sulfide (H2S) is the most recently-discovered gas signal molecular, participated in regulating cell proliferation, apoptosis, oxidative stress, inflammation and angiogenesis [10]. H2S is mainly produced and cleared in the liver tissues by three enzymes: cystathionine-gamma-lyase (CSE), cystathionine-beta-synthase (CBS), and 3-mercapto-pyruvate sulfotransferase (MPST) [11]. Increasing evidence shows that H2S affect HCC through the regulation of cell proliferation [12], migration and invasion [13], angiogenesis and immune responses [14]. Intriguingly, our previous study found that exogenous H2S inhibit HCC progression through the PI3K/Akt/mTOR signaling pathway in vitro [15]. Our previous data found that activated HSC can produce H2S mainly regulated by CSE. Nowadays, the mechanisms involved in the capacity of HSC relased H2S to affect the HCC have not been worked out, all evidence up to this point suggests these mechanisms have great potential importance.
In this study, we investigate how activated HSC influence HCC fate. We found that activated HSC released H2S to induce HCC cell apoptosis. H2S induced cancer cell apoptosis through activating JNK/JunB signaling, which upregulated tumor necrosis factor superfamily member 14 (TNFSF14) expression.
Materials and methods
Patients
A total of 45 patients with HCC who received hepatectomy were enrolled from Beijing You’an Hospital, affiliated to Capital Medical University from July 2010 to January 2013. HCC was diagnosed and graded according to the criteria of the American Association for the Study of Liver Diseases [36]. Liver samples were collected from cancerous and adjacent non-cancerous liver tissues and stored in liquid nitrogen for further experimental use. Clinic pathologic characteristics of these patients are shown in Table S1. The study protocol was approved by the Ethics Committee of Beijing You’an Hospital affiliated to Capital Medical University. The informed consent was obtained from all enrolled patients.
Cells
PLC/PRF/5 (ATCC® Cat. No. CRL-8024TM), HepG2 (ATCC® Cat. No. HB-8065TM) and LX-2 cells were cultured in DMEM medium supplemented with 10% FBS in a 37 °C incubator with 5% CO2. The Transwell insert system was used for co-culture experiments to analyze the effect of LX-2 cells on HCC cells as previously described [37].
CCK-8
Cells proliferation was examined by a Cell Count KIT-8 assay (CCK8, AbMole, Houston, TX, America) according to the protocol. 2 × 103 cells with different treatments were pipetted into 96-well microplates for measurements. The absorbance values were measured under a wavelength of 450 nm on a Universal Microplate Reader (ELx800; Bio-Tek Instruments Inc, Winooski, VT, USA). The percentage of cell viability was calculated as: Cell proliferation % = [(A450sample-background)/ (A450control-backgroud)] × 100%.
Flow cytometry
Cell apoptosis was measured by FCM analysis using Annexin V-FITC/PI Apoptosis Detection Kit (40302ES20, Yeasen, Shanghai, China). Briefly, cells were collected and incubated with 5 µL Annexin V-FITC and 10 µL PI for 15 min at room temperature. Adjusted the volume to 500 µL with binding buffer. The apoptosis of HCC cells was analyzed with a FACScan-420 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
ELISA
The concentrations of H2S were examined by a Hydrogen sulfide Assay Kit (JEB-11780, Jin Yibai Biological Technology, Nanjing, China) according to the instruction.
RNA sequencing
RNA sequencing analysis was conducted by Novogene (Beijing, China). In brief, total RNA was isolated by TRIzol reagent (15596026, Thermo Fisher Scientific, Waltham, MA, USA). The RNA quantification and qualification were assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Sequencing of the libraries was carried out using the Illumina NovaSeq 6000 platform (Novogene, Beijing, China). All the raw data were uploaded on the Gene Expression Omnibus (GSE193660).
Quantitative real-time reverse transcription PCR (RT-qPCR)
Total RNA was isolated by TRIzol reagent and subsequently subjected to reverse transcription (4368814, High-Capacity cDNA Reverse Transcription Kits, Thermo Fisher Scientific, Waltham, MA, USA). RT-qPCR was performed using the TB Green (RR420A, Takara) on an ABI ViiA7 (GX-XVI R2, Applied Biosystems Life Tech, USA). The relative mRNA expression levels were calculated with 2−ΔΔCt method and presented as mean fold-change of samples to control. The primers used in the study were listed in the Table 1.
Western blotting
Cells or tissues were lysed in ice-cold lysis buffer. Total protein from cells or tissues was extracted using RIPA lysis buffer (R0010, Solarbio, Beijing, China). Nuclear and cytoplasmic fractions from cells were obtained by a Nuclear Protein Extraction Kit (R0050, Solarbio, Beijing, China). Forty μg of each protein were utilized for Western blotting. Membranes were blocked by TBST with 5% nonfat milk (w/v) for 1 h at room temperature. Incubated the membranes with primary antibodies at 4 °C for overnight. Next day, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h following washing with TBST. The immuno-complexes were incubated with the enhanced chemiluminescent system (34580 Thermo Scientific, Waltham, MA, USA) and were subsequently exposed by a Biorad Chemidoc XRS + imaging system (USA). Primary antibodies against JNK(#9252), phospho-JNK (#4668), JunB (#3753), phospho-JunB (#8053), GAPDH (#2118), β-actin (#4967), and HDAC1 (#34589) were purchased from Cell Signaling Technology (CST, USA). TNFSF14 antibody (#AF0329) was purchased from Affinity Biosciences (USA). Horseradish peroxidase-conjugated goat anti-mouse (#7076) and anti-rabbit (#7074) IgG secondary antibodies were purchased from CST. Full and uncropped western blots were uploaded as ‘Supplemental Material’.
Chromatin immunoprecipitation assay
The Chromatin immunoprecipitations (ChIP) assay was carried out according to a ChIP assay kit (#56383 CST). Cells were lysed with TNE buffer. Lysates were collected by centrifugation (15,000 g × 10 min) at 4 °C. Aliquots were incubated with Protein G–Sepharose beads (GE Healthcare) for 1 h. After centrifugation (1000 g × 5 min), the supernatants were incubated with phospho-JunB (#8053 CST) and IgG(#2729 CST) antibodies overnight at 4 °C. Next day, added Protein G–Sepharose beads for 8 h incubation. Then eluted the precipitate with the sample buffer. Finally, the immunoprecipitated chromatin was analyzed by PCR using primers targeting the human TNFSF14 promoter containing JunB binding sites. The primers used for ChIP-PCR are listed in Table 1.
Immunofluorescence staining
Cells were fixed in ice-cold 4% paraformaldehyde solution. Then incubated the cells with primary antibody (p-JunB) for overnight at 4 °C in the wet box. Next day, the cells were washed and subsequently incubated with the secondary antibody conjugated with Alexa Fluor 488 for 1 h at room temperature. Cell nuclei were counterstained with DAPI. Cells were viewed and captured with a fluorescence microscope (Nikon Eclipse 80i; Nikon, Tokyo, Japan).
Transient transfection
The small interfering RNAs (siRNAs) technique and TNFSF14-expressing plasmid were used to evaluate the effect of TNFSF14 on H2S-induced HCC cell apoptosis. The siRNAs sequences were listed in the Table 1. Briefly, HCC cells were seeded into the 6-well plates and cultured to the 80% confluence. SiRNAs and the plasmids were transfected to HCC cells according the instruction of LipofectamineTM 3000 transfection reagent (L3000-015, Thermo Fisher Scientific, Waltham, MA, USA). After 24 h of transfection, the expression of TNFSF14 was evaluated by RT-qPCR and Western blotting.
Data analysis of public databases
The GSE14520 gene expression dataset was downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo). According the annotation information in the platform, the probes were converted into the gene symbols. We divided the examined HCC patients into TNFSF14 high and low groups based on the median of the gene expression.
Statistical analysis
Each experiment was repeated at least three times independently. All data were presented as mean ± S.D. performed using the SPSS software (version 23.0). Differences between unpaired groups were compared by the Student’s t-test or Mann–Whitney U test. For paired groups, the Wilcoxon signed-rank test was used. Parametric or nonparametric multiple comparisons were performed using ANOVA-test or Kruskal–Wallis test. Pearson’s χ2 test or Fisher’s exact test was used to analyze categorical data. Correlations were assessed by the Spearman correlation. Survival analyses were performed by the Kaplan–Meier and the log rank test. P < 0.05 was considered significant.
Results
Activated hepatic stellate cells induce apoptosis of cancer cells
To examine how hepatic stellate cells (HSC) impact HCC cell behavior, we performed co-culture with activated HSC cell line LX-2 cells and HCC cell lines, HepG2 or PLC/PRF/5 cells. CCK-8 assays showed that cell viability of HCC cells was significantly reduced over time when the cells were co-cultured with LX-2 cells (Fig. 1A, C). Flow cytometry analyses further demonstrated that co-culture with LX-2 cells induced apoptosis of HepG2 and PLC/PRF/5 cells. The maximal apoptosis of these cancer cells was observed at 72 h following co-culture (Fig. 1B, D). These results suggest that activated HSC not only inhibit HCC cell proliferation, but also induce apoptosis of cancer cells.

A HepG2 and PLC/PRF/5 cells were collected after co-cultured with LX-2 for 24 h, 48 h, 72 h, and 96 h respectively. CCK-8 assay was used to determine the viability of HepG2 and C PLC/PRF/5 cells. B Flow cytometry was carried out to analyze the apoptotic rates of HepG2 and D PLC/PRF/5 cells. All Bars, ± SD; n = 3; *P < 0.05 compared with control.
LX-2 cells induce apoptosis of HCC cells through releasing H2S
How do activated HSC induce apoptosis of HCC cells? We hypothesized that H2S is a crucial mediator in the process given that activated HSC release H2S [16] and H2S is capable of inducing cancer cell apoptosis [15]. ELISA assay confirmed that LX-2 cells increased H2S release over time (Fig. 2A). LX-2 cells released the same amount of H2S at 72 h as 10–3 M NaHS did at 24 h (Fig. 2B). Subsequently, we observed the effects of NaHS, a donor of H2S, and DL-Propargylglycine (PPG), a CSE inhibitor, on LX-2 cell-dependent HCC cell apoptosis. As shown in Fig. S1, NaHS increased H2S release, whereas PPG dose-dependently inhibited mRNA and protein expression of CSE as well as H2S release in LX-2 cells. Flow cytometry analyses showed that NaHS resulted in HCC cell apoptosis (Fig. 2C, D). LX-2-dependent cancer cell apoptosis was significantly reduced by the administration of PPG (Fig. 2C, D). PPG alone did not affect HCC apoptosis (Fig. 2C, D). These results suggest that activated HSC result in HCC cell apoptosis through releasing H2S.

The amount of H2S released by cultured LX-2 cells (A) and 10–3 M NaHS (B) were measured by ELISA. C, D Flow cytometry was used to analyze the effects of exogenous H2S (NaHS) and endogenous H2S (LX-2 cells co-cultured or/and added PPG treated) on HCC cells apoptosis. All Bars, ± SD; n = 3; *P < 0.05 compared with D1 group in LX-2 cells, #P < 0.05 compared with D1 group in HepG2 cells.
H2S upregulates pro-apoptotic factor TNFSF14 expression in HepG2 cells
To clarify how H2S results in HCC cell apoptosis, we performed RNA-seq in HepG2 cells, which were co-cultured with LX-2 cells or treated with NaHS. A heat-map shows different transcriptome cluster in cells with different administrations (Fig. 3A). LX-2 co-culture and NaHS treatment respectively led to 1047 and 1183 gene upregulation as well 997 and 1144 gene downregulation in HepG2 cells. Among these altered genes, 659 up-regulated genes and 342 down-regulated genes were presented in HepG2 cells treated with either LX-2 co-culutre or NaHS (Fig. 3B). Based on the fold change, 30 genes with maximal fold changes were selected to further analyze (Fig. S2). Among these 30 genes, we identified a pro-apoptotic gene TNFSF14 (Fig. 3C). We examined TNFSF14 mRNA and protein in HepG2 and PLC/PRF/5 cells treated with different concentrations of NaHS. NaHS dose-dependently increased expression of TNFSF14 and reached a maximum at 10–3 M treated 24 h in both HCC cells (Fig. 3D–G). Furthermore, we observed that TNFSF14 expression was up-regulated when HCC cells were co-cultured with LX-2 cells (Fig. 3H, I). The effect was inhibited by PPG administration (Fig. 3H, I). These results imply that TNFSF14 might be the key factor mediating H2S-induced HCC apoptosis.

A The heatmap. B Venn diagram of gene predictions based on the control group and HepG2 cells treated with NaHS and co-cultured with LX-2 cells. C The expression levels of TNFSF14 mRNA changed in HepG2 cells treated with NaHS and co-cultured with LX-2 cells. D RT-qPCR and (E) Western blotting were performed to examine the effects of various concentrations of NaHS on expression of TNFSF14 gene in HepG2 and PLC/PRF/5 cells. F RT-qPCR and (G) Western blotting analysis of the expression of TNFSF14 under 10−3 M NaHS treatment with different time in HepG2 and PLC/PRF/5 cells. H RT-qPCR and (I) Western blotting were used to detect the effects of PPG treated on TNFSF14 expression in HCC cells. All Bars, ± SD; n = 3; *P < 0.05 compared with control in HepG2 cells, #P < 0.05 compared with control in PLC/PRF/5 cells, &P < 0.05 compared with the co-culture group in HCC cells.
TNFSF14 is indispensable for H2S-induced HCC cell apoptosis
To confirm the role of TNFSF14 in H2S-induced HCC cell apoptosis, we knocked down TNFSF14 expression in both HepG2 and PLC/PRF/5. RT-qPCR and Western blotting analyses confirmed that NaHS and LX-2 co-culture-induced TNFSF14 mRNA and protein expression was inhibited by siRNA-TNFSF14 in both HepG2 (Fig. 4A, B) and PLC/PRF/5 cells (Fig. 4D, E). Flow cytometric analyses further showed that NaHS and LX-2 co-culture-induced apoptosis of cancer cells was significantly inhibited when TNFSF14 expression was knocked down (Fig. 4C, F). On the other hand, the effect of TNFSF14 was also confirmed through transfection of the tnfsf14 gene into HCC cells, which showed that transfection with pcDNA3.1-tnfsf14 results in apparent increase of apoptosis in HepG2 and PLC/PRF/5 cells with different treatments (Fig. S3). These results suggest that TNFSF14 expression is required for H2S-induced HCC cell apoptosis.

TNFSF14 mRNA and protein expressions levels were evaluated by (A, D) RT-qPCR and (B, E) Western blotting assays in HepG2 and PLC/PRF/5 cells under different treatments. Flow cytometry was carried out to analyze the effects of different TNFSF14 expression levels on HepG2 (C) and PLC/PRF/5 (F) cells apoptosis. All Bars, ± SD; n = 3; *P < 0.05 compared with NC group, #P < 0.05 compared with NC + NaHS group, &P < 0.05 compared with NC + LX-2 group.
H2S regulates TNFSF14 transcription through JNK/JunB signal
To clarify how H2S regulates the expression of TNFSF14, we analyzed PROMO dataset and adopted JASPAR software to predict potential transcript factors binding to the TNFSF14 promoter. JunB got the highest scored among all the analyzed factors (Fig. 5A upper panel) and possessed multiple binding sites on the TNFSF14 promoter (Fig.5A lower panel). ChIP assays confirmed the binding of p-JunB to the TNFSF14 promoter in both HepG2 and PLC/PRF/5 cells (Fig. 5B). The binding was increased by NaHS treatment (Fig. 5B). Immunofluorescence staining and Western blotting showed that administration of NaHS remarkably induced p-JunB expression and nuclear translocation in both HCC cell lines (Fig. 5C–D). Besides NaHS, LX-2 cell co-culture also increased p-JunB expression and nuclear translocation (Fig. 5E). JunB phosphorylation and nuclear translocation required upstream p-JNK. Administration of SP600125, a p-JNK inhibitor, completely inhibited NaHS- or LX-2 cell co-culture-induced p-JunB expression and nuclear translocation in both cancer cells (Fig. 5F). The effects of NaHS- or LX-2 cell co-culture-induced p-JunB expression and nuclear translocation was also inhibited by PPG (Fig. 5F). These results suggest that JNK/JunB signaling mediates H2S to regulate TNFSF14 transcription.

A Schematic overview of the predicted transcription factors of TNFSF14 gene by PROMO dataset (upper) and JunB binding sites on TNFSF14 promoter predicted with JASPAR website (lower). B ChIP assays for JunB binding onto TNFSF14 promoter were carried out in HepG2 (upper) and PLC/PRF/5 (lower) cells. The right panel showed the statistical analysis for the ChIP assay. C NaHS was used to treat HepG2 (left) and PLC/PRF/5 (right) cells for 0, 1, 2, 8, and 24 h. Expression of p-JunB in nuclear of HCC cells was detecting using immunofluorescence staining, and Nuclei and p-JunB were stained with DAPI (blue) and FITC (green), respectively. The image is representative of three independent experiments. D The effects of different time of NaHS treatment on the expression of p-JunB in the nuclear and cytoplasm were analyzed with Western blotting in the HepG2 cells and PLC/PRF/5 cells. E The expression levels of p-JunB protein were detected by Western blotting in the HCC cells under exogenous and endogenous H2S treatment. F Western blotting for the effects of PPG (CSE inhibitor) and SP600125 (p-JNK inhibitor) in the expressions of JNK/p-JNK, JunB/p-JunB, and TNFSF14 proteins in HepG2 and PLC/PRF/5 cells. All Bars, ± SD; n = 3; *P < 0.05 compared with control group in HepG2 cells, #P < 0.05 compared with control group in PLC/PRF/5 cells.
TNFSF14 expression is associated with the clinical outcome of HCC
Subsequently, we observed the association between TNFSF14 expression and clinical outcome of 45 HCC patients. TNFSF14 and CSE mRNA expression were significantly downregulated in tumor tissues compared with surrounding non-tumor tissues (Fig. 6A). Western blotting analyses performed in 6 patients confirmed the results (Fig. 6B). CSE mRNA expressions in tumor tissues were positively correlated with that of TNFSF14 (r = 0.438, P < 0.05, Fig. 6C). We further investigated the association between TFNSF14 expression and tumor characteristics. Patients with high TNFSF14 expression in HCC showed less portal invasion, lower Barcelona Clinic Liver Cancer (BCLC) HCC stages and longer survival and disease-free time than those with low TNFSF14 expression (Table 2 and Fig. 6D, E). These results were similar as the data in GSE 14520: TNFSF14 and CSE gene expression were decreased in tumor tissues compared with non-tumor tissues (Fig. S4). CSE gene expressions in HCC tissues were positively associated with TNFSF14 expression (r = 0.168, P <0.05, Fig. S4). In addition, patients with high TNFSF14 expression showed less alpha-fetoprotein (AFP) levels, lower ALT, lower BCLC HCC stages, and lower metastasis risk than those with low TNFSF14 expression (P < 0.05, Fig. S4). The patients with high TNFSF14 expression had longer survival and disease-free time (Fig. S4).

A TNFSF14 mRNA and CSE mRNA expression levels in tumorous tissues (HCC; N = 45) and nontumorous tissues (NT; N = 45). B Western blotting analysis for the expression of CSE and TNFSF14 in the tumorous and nontumorous tissues in 6 patients with HCC. C Correlation between CSE mRNA expression levels and TNFSF14 mRNA expression levels in tumorous tissues (N = 45). D, E Overall survival and disease-free survival curves showed that patients with high expression levels of TNFSF14 have better prognosis. All Bars, ± SD; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
HSC, as the central modulators of the tumor microenvironment, have long been considered as a tumor promoter during hepatocarcinogenesis by the secretion of growth factors such as hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF) and connective tissue growth factor (CTGF), altering components of the ECM, regulating angiogenesis and immune surveillance [17,18,19,20,21]. In stark contrast, HSC are a source of transforming growth factor beta (TGF-β), which exert tumor-suppressing activity by inducing cytostasis and apoptosis of hepatocytes in early tumor phases [8]. Recently, HSC were reported to be negative regulators of HCC progression through upregulating endosialin which is capable of inhibiting tumor-promoting cytokines, including IGF2, RBP4, DKK1, and CCL5 [9]. The findings of our study shed further insight into the increasing appreciation that HSC do not just act as pro-tumorigenic role, but may inhibit tumor progression. In our study, we found that HSC cells co-cultured with HepG2 and PLC/PRF/5 cells could promote cell apoptosis which was time-dependently manner. Further research found that HSC cells through releasing H2S induced HCC cells apoptosis, which was relieved by adding PPG (CSE inhibitor) to reduce the amount of H2S.
As a gas signaling molecule, the role of H2S in HCC is still unclear. The previous reports shown that in the HepG2 and PLC/PRF/5 cells, endogenous H2S/CSE pathway regulated cell growth mainly through EGFR pathway [12]. Another similar study found that in the HepG2 cells, H2S/CSE pathway was activated after irradiated, which led to the long-term cell invasion and tumor metastasis [22]. The function of exogenous H2S has also been studied in HCC cells. In the PLC/PRF/5 cells, 500 μM NaHS could induce cell proliferation, migration, and anti-apoptotic via activating NF-κB pathway [13]. Other researchers found that exogenous H2S effectively restricted the tumor development in H22 HCC-bearing mice and HCC cell lines proliferation by blocking STAT3 and NF-κB pathways [14, 23]. Similar with our pervious study, 10−3 M NaHS treated HepG2 and HLE cells could inhibit cells proliferation, migration and pro-apoptotic via mTOR signaling pathway [15]. In this study, our results showed that both endogenous and exogenous H2S promoted HCC cells apoptosis, and the apoptosis was reduced when endogenous H2S was inhibited by adding PPG.
How do H2S regulate HCC cells apoptosis? Based on the RNA-sequencing results, we found that both endogenous and exogenous H2S could up-regulate TNFSF14 in HepG2 cell. TNFSF14, mainly expressed on activated T cells, is activated in Natural Killer (NK) cells, and immature dendritic cells (DC) [24]. Previous studies reported that TNFSF14 act as an immunomodulatory factor to induce tumor regressions and apoptosis through enhancing T cells proliferation, differentiation, and cytokines secretion [25,26,27]. In human epithelial cancer cell lines, TNFSF14 promotes apoptosis and inhibits proliferation, which effect is independent of T cells [28,29,30,31]. However, there are few reports about the effect of TNFSF14 in HCC. In our study, we found that H2S induced HCC apoptosis partly due to the TNFSF14 gene. TNFSF14 mRNA and protein expression levels was upregulated in HepG2 and PLC/PRF/5 cells under H2S treatment. Knockdown of TNFSF14 reduced H2S-induced HCC apoptosis.
To figure out the precise molecular mechanism of H2S regulating the transcription of TNFSF14 gene, the transcription regulatory region of TNFSF14 gene was analyzed on the JASPAR website. We found transcription factor JunB binds to the TNFSF14 gene. The transcriptional activity of JunB is enhanced through phosphorylation-JNK [32]. In HCC cells, JNK/JunB exerts anti-tumor effects by promoting tumor cell apoptosis and inhibiting cell growth [33]. In this study, we found that H2S regulated TNFSF14 transcription mainly through activating JNK/JunB singal in both HepG2 and PLC/PRF/5 cells.
All these in vitro results demonstrated that H2S, released by LX-2 cells, participated in the JNK/JunB-TNFSF14 pathway to promote HCC cell apoptosis. Further validation was performed in the GEO dataset and our cohort, the results showed that patients with high CSE and TNFSF14 expression in HCC showed less portal invasion, lower BCLC stages and longer survival and disease-free time compared with lower TNFSF14 expression patients. Similar with our results was found in the primary colorectal cancer patients, TNFSF14 expression was decreased on tumor infiltrating lymphocytes in resected colorectal cancer liver metastases patients and significantly associated with overall survival and recurrence-free survival [34]. But in the nonalcoholic steatohepatitis (NASH)-related HCC patients, NKT cells secreted TNFSF14 to promote NASH-to-HCC transition [35]. These discrepant researches suggested that TNFSF14 may play different roles in different liver diseases.
It is presently unclear how significant the impact of H2S mediated TNFSF14 expression through JNK/JunB pathway on the occurrence and high mortality of HCC. However, taking into consideration the effects of H2S in the HCC, it is reasonable to speculate that H2S has great potential significance and importance in HCC apoptosis. Taken our results together, HSC released H2S could upregulate the TNFSF14 expression through JNK/JunB axis to induce HCC cells apoptosis, and TNFSF14 was significantly associated with prognosis of HCC patients. Thus, enhance the circulating H2S to promote cells apoptosis may be a potential strategy for HCC. Nonetheless, further clinical study of this approach will be required.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Change history
30 November 2022
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41419-022-05464-7
References
Kocarnik JM, Compton K, Dean FE, Fu W, Gaw BL, Harvey JD, et al. Global Burden of Disease 2019 Cancer Collaboration. Cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life years for 29 cancer groups from 2010 to 2019: A systematic analysis for the global burden of disease study 2019. JAMA Oncol. 2021;e216987. https://doi.org/10.1001/jamaoncol.2021.6987.E1-E24.
Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol. 2018;68:526–49.
Sevic I, Spinelli FM, Cantero MJ, Reszegi A, Kovalszky I, García MG, et al. The role of the tumor microenvironment in the development and progression of hepatocellular carcinoma. In: Tirnitz-Parker JEE, editor. Hepatocellular Carcinoma [Internet]. Codon Publications: Brisbane, AU, 2019, Chapter 2.
Tahmasebi Birgani M, Carloni V. Tumor microenvironment, a paradigm in hepatocellular carcinoma progression and therapy. Int J Mol Sci. 2017;18:405.
Rhee H, Kim HY, Choi JH, Woo HG, Yoo JE, Nahm JH, et al. Keratin 19 expression in hepatocellular carcinoma is regulated by fibroblast-derived HGF via a MET-ERK1/2-AP1 and SP1 axis. Cancer Res. 2018;78:1619–31.
Yan C, Yang Q, Gong Z. Activation of hepatic stellate cells during liver carcinogenesis requires Fibrinogen/Integrin αvβ5 in zebrafish. Neoplasia. 2018;20:533–42.
Scheau C, Badarau IA, Costache R, Caruntu C, Mihai GL, Didilescu AC, et al. The role of matrix metalloproteinases in the epithelial-mesenchymal transition of hepatocellular carcinoma. Anal Cell Pathol. 2019;2019:9423907.
Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-β in hepatic stellate cell activation and liver fibrogenesis-Updated 2019. Cells. 2019;8:1419.
Mogler C, König C, Wieland M, Runge A, Besemfelder E, Komljenovic D, et al. Hepatic stellate cells limit hepatocellular carcinoma progression through the orphan receptor endosialin. Embo Mol Med. 2017;9:741–9.
Szabo C, Papapetropoulos A. International union of basic and clinical pharmacology. CII: pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharm Rev. 2017;69:497–564.
Mani S, Cao W, Wu L, Wang R. Hydrogen sulfide and the liver. Nitric Oxide. 2014;41:62–71.
Pan Y, Ye S, Yuan D, Zhang J, Bai Y, Shao C. Hydrogen sulfide(H2S)/cystathionine γ-lyase (CSE) pathway contributes to the proliferation of hepatoma cells. Mutat Res. 2014;763–764:10–8.
Zhen Y, Pan W, Hu F, Wu H, Feng J, Zhang Y, et al. Exogenous hydrogensulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-κB pathway in PLC/PRF/5 hepatoma cells. Int J Oncol. 2015;46:2194–204.
Yang D, Li T, Li Y, Zhang S, Li W, Liang H, et al. H2S suppresses indoleamine 2, 3- dioxygenase 1 and exhibits immunotherapeutic efficacy in murine hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38:88.
Wang SS, Chen YH, Chen N, Wang LJ, Chen DX, Weng HL, et al. Hydrogen sulfide promotes autophagy of hepatocellular carcinoma cells through the PI3K/Akt/mTOR signaling pathway. Cell Death Dis. 2017;8:e2688.
Damba T, Zhang M, Buist-Homan M, van Goor H, Faber KN, Moshage H. Hydrogen sulfide stimulates activation of hepatic stellate cells through increased cellular bio-energetics. Nitric Oxide. 2019;92:26–33.
Cho Y, Park MJ, Kim K, Park JY, Kim J, Kim W, et al. Tumor-stroma crosstalk enhances REG3A expressions that drive the progression of hepatocellular carcinoma. Int J Mol Sci. 2020;21:472.
Makino Y, Hikita H, Kodama T, Shigekawa M, Yamada R, Sakamori R, et al. CTGF mediates tumor-stroma interactions between hepatoma cells and hepatic stellate cells to accelerate HCC progression. Cancer Res. 2018;78:4902–14.
Li Q, Wang C, Wang Y, Sun L, Liu Z, Wang L, et al. HSCs-derived COMP drives hepatocellular carcinoma progression by activating MEK/ERK and PI3K/AKT signaling pathways. J Exp Clin Cancer Res. 2018;37:231.
Morse MA, Sun W, Kim R, He AR, Abada PB, Mynderse M, et al. The role of angiogenesis in hepatocellular carcinoma. Clin Cancer Res. 2019;25:912–20.
Hsieh CC, Hung CH, Chiang M, Tsai YC, He JT. Hepatic stellate cells enhance liver cancer progression by inducing myeloid-derived suppressor cells through interleukin-6 signaling. Int J Mol Sci. 2019;20:5079.
Zhang H, Song Y, Zhou C, Bai Y, Yuan D, Pan Y, et al. Blocking endogenous H2S signaling attenuated radiation-induced long-term metastasis of residual HepG2 cells through inhibition of EMT. Radiat Res. 2018;190:374–84.
Lu S, Gao Y, Huang X, Wang X. GYY4137, a hydrogen sulfide (H2S) donor, shows potent anti-hepatocellular carcinoma activity through blocking the STAT3 pathway. Int J Oncol. 2014;44:1259–67.
Skeate JG, Otsmaa ME, Prins R, Fernandez DJ, Da Silva DM, Kast WM. TNFSF14:Lighting the way for effective cancer immunotherapy. Front Immunol. 2020;11:922.
Harrop JA, McDonnell PC, Brigham-Burke M, Lyn SD, Minton J, Tan KB, et al. Herpesvirus entry mediator ligand (HVEM-L), a novel ligand for HVEM/TR2, stimulates proliferation of T cells and inhibits HT29 cell growth. J Biol Chem. 1998;273:27548–56.
Zou W, Zheng H, He TC, Chang J, Fu YX, Fan W. LIGHT delivery to tumors by mesenchymal stem cells mobilizes an effective antitumor immune response. Cancer Res. 2012;72:2980–9.
Qiao G, Qin J, Kunda N, Calata JF, Mahmud DL, Gann P, et al. LIGHT elevation enhances immune eradication of colon cancer metastases. Cancer Res. 2017;77:1880–91.
Han B, Wu LQ, Ma X, Wang ZH, Li JP, Bi CY, et al. Synergistic effect of IFN-γ gene on LIGHT-induced apoptosis in HepG2 cells via down regulation of Bcl-2. Artif Cells Blood Substit Immobil Biotechnol. 2011;39:228–38.
Zhu X, Su D, Xuan S, Ma G, Dai Z, Liu T, et al. Gene therapy of gastric cancer using LIGHT-secreting human umbilical cord blood-derived mesenchymal stem cells. Gastric Cancer. 2013;16:155–66.
Hu X, Zimmerman MA, Bardhan K, Yang D, Waller JL, Liles GB, et al. Lymphotoxin beta receptor mediates caspase-dependent tumor cell apoptosis in vitro and tumor suppression in vivo despite induction of NF-kappaB activation. Carcinogenesis. 2013;34:1105–14.
Kim YS, Nedospasov SA, Liu ZG. TRAF2 plays a key, nonredundant role in LIGHT-lymphotoxin beta receptor signaling. Mol Cell Biol. 2005;25:2130–7.
Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83.
Guo C, Liu Q, Zhang L, Yang X, Song T, Yao Y. Double lethal effects of fusion gene of wild-type p53 and JunB on hepatocellular carcinoma cells. J Huazhong Univ Sci Technol Med Sci. 2012;32:663–8.
Maker AV, Ito H, Mo Q, Weisenberg E, Qin LX, Turcotte S, et al. Genetic evidence that intratumoral T-cell proliferation and activation are associated with recurrence and survival in patients with resected colorectal liver metastases. Cancer Immunol Res. 2015;3:380–8.
Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–64.
Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American association for the study of liver diseases. Hepatology. 2018;68:723–50.
Li ZQ, Wu WR, Zhao C, Zhao C, Zhang XL, Yang Z, et al. CCN1/Cyr61 enhances the function of hepatic stellate cells in promoting the progression of hepatocellular carcinoma. Int J Mol Med. 2018;41:1518–28.
Acknowledgements
This work was supported by the National Natural Science Foundation (81672725, 81970525), Beijing Natural Science Foundation(7212052), Beijing Hospitals Authority Youth Programme (QML20211701), Sino-German Cooperation Group (GZ1517), and Sino-German Mobility program (M-0200). The authors express their gratitude to the National Natural Science Foundation (81672725, 81970525), Beijing Natural Science Foundation(7212052), Beijing Hospitals Authority Youth Programme (QML20211701), Sino-German Cooperation Group (GZ1517), and Sino-German Mobility program(M-0200) for financial support.
Author information
Authors and Affiliations
Contributions
HD designed and supervised the study; YM, SW, YW, BL, LL, and WW conducted experiments or interpreted the data; HW provided technical support; YM and SW wrote the paper. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
The study protocol was approved by the Ethics Committee of Beijing You’an Hospital affiliated to Capital Medical University. The informed consent was obtained from all enrolled patients.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Boris Zhivotovsky
This article has been retracted. Please see the retraction notice for more detail:https://doi.org/10.1038/s41419-022-05464-7
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, Y., Wang, S., Wu, Y. et al. RETRACTED ARTICLE: Hepatic stellate cell mediates transcription of TNFSF14 in hepatocellular carcinoma cells via H2S/CSE-JNK/JunB signaling pathway. Cell Death Dis 13, 238 (2022). https://doi.org/10.1038/s41419-022-04678-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-04678-z
