
Abstract
To identify drivers of sarcoma cancer stem-like cells (CSCs), we compared gene expression using RNA sequencing between HT1080 fibrosarcoma and SK-LMS-1 leiomyosarcoma spheroids (which are enriched for CSCs) compared with the parent populations. The most overexpressed survival signaling-related gene in spheroids was phosphoinositide-3-kinase regulatory subunit 3 (PIK3R3), a regulatory subunit of PI3K, which functions in tumorigenesis and metastasis. In a human sarcoma microarray, PIK3R3 was also overexpressed by 4.1-fold compared with normal tissues. PIK3R3 inhibition using shRNA in the HT1080, SK-LMS-1, and DDLS8817 dedifferentiated liposarcoma in spheroids and in CD133+ cells (a CSC marker) reduced expression of CD133 and the stem cell factor Nanog and blocked spheroid formation by 61–71%. Mechanistic studies showed that in spheroid cells, PIK3R3 activated AKT and ERK signaling. Inhibition of PIK3R3, AKT, or ERK using shRNA or inhibitors decreased expression of Nanog, spheroid formation by 68–73%, and anchorage-independent growth by 76–91%. PIK3R3 or ERK1/2 inhibition similarly blocked sarcoma spheroid cell migration, invasion, secretion of MMP-2, xenograft invasion into adjacent normal tissue, and chemotherapy resistance. Together, these results show that signaling through the PIK3R3/ERK/Nanog axis promotes sarcoma CSC phenotypes such as migration, invasion, and chemotherapy resistance, and identify PIK3R3 as a potential therapeutic target in sarcoma.
Similar content being viewed by others


Introduction
Mesenchymal tissues are derived from the mesoderm (i.e., middle layer of the embryo) and include the musculoskeletal system, circulatory and lymphatic systems, and connective tissues. Sarcomas are malignant tumors of mesenchymal tissues, with an annual incidence of ~15,000 cases in the United States [1]. About 40% of sarcoma cases lead to death. Treatment of primary tumors often includes aggressive surgical resection and radiation therapy, but local recurrence remains a significant problem, especially for tumors in difficult anatomic locations such as the head and neck, paraspinal region, retroperitoneum, and pelvis [2]. Up to half of patients with large, high-grade sarcomas develop distant metastases, most frequently to the lung [3]. The efficacy of chemotherapy in treating local and distant recurrence is modest at best, and for the minority of patients who do respond, nearly all eventually develop chemotherapy resistance [4].
A likely driver of chemotherapy resistance as well as distant metastasis in sarcomas and other cancers are cancer stem-like cells (CSCs) [5, 6]. CSCs share characteristics with normal adult stem cells, specifically the capacity for self-renewal and pluripotent differentiation [7]. Methods to identify CSCs include tumor initiation in immunodeficient mice, spheroid colony formation in vitro, and expression of certain cell surface markers. The most commonly identified cell surface marker for CSCs in sarcomas is CD133 [5, 8]. There are over 80 distinct histologic subtypes of sarcoma, and some have suggested that sarcomas originate from multipotent cells such as mesenchymal stem cells [9].
To identify potential drivers of sarcomagenesis and sarcoma progression, we compared gene expression using RNA sequencing (RNAseq) between sarcoma cells grown as spheroids (to select for CSCs) and as monolayers. This analysis identified the phosphoinositide-3-kinase regulatory subunit 3 (PIK3R3), involved in PI3K/AKT signaling, to be the most upregulated survival signaling-related gene. Because PI3K has previously been implicated as essential to sarcoma survival [10,11,12], we sought to characterize the PIK3R3 signaling in spheroids and its role in the maintenance of CSC properties in sarcomas.
Results
PIK3R3 and Nanog are upregulated in sarcoma CSCs and human sarcomas
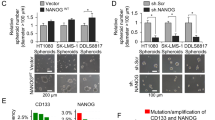
To uncover new genes important to the function of sarcoma CSCs, we compared gene expression in two sarcoma cell lines, HT1080 fibrosarcoma and SK-LMS-1 leiomyosarcoma, between monolayer and spheroid culture using RNAseq. Phosphoinositide-3-kinase regulatory subunit 3 (PIK3R3) was the most highly upregulated survival signaling-related gene in spheroid (CSC-enriched) vs. monolayer cells (Fig. 1A). Upregulation of PIK3R3 and the cancer stem cell marker CD133 in spheroid cells was verified using quantitative real-time PCR (qRT-PCR) in HT1080 and SK-LMS-1 cells as well as DDLS8817 dedifferentiated liposarcoma cells, which showed 3.9–6.2 and 2.1–3.8-fold higher expression, respectively, in spheroid vs. monolayer cells (Fig. 1B). Upregulation of PIK3R3 and CD133 in sarcoma cells grown as spheroids vs. monolayers was also confirmed at the protein level using Western blot analysis (Fig. 1C). To enrich for CSCs using an alternative approach, we sorted CD133+ cells from the three sarcoma cell lines, which composed 10–13% of the population when grown as spheroids as measured by flow cytometry (Fig. 1D, Supplementary Fig. S1A). Protein expression of PIK3R3 and the stem cell transcription factor Nanog were greater in CD133+ compared with CD133– cells (Fig. 1E), while levels of other stem cell transcription factors including Sox2, Oct-4, and c-Myc were not consistently increased. Finally, to examine whether PIK3R3 is generally overexpressed in human sarcomas, we immunostained commercially available tissue microarrays containing 79 human sarcomas (including diverse subtypes) and 4 human normal tissues (NBP2-30332, Novus Biologicals, and T242, US Biomax). Positive cells of CD133, Nanog, or PIK3R3 were 5.2–12.7-fold higher in tumor tissue compared with normal tissue as determined by immunostaining (Fig. 1F). Together, these results show that PIK3R3 and Nanog are overexpressed in a broad variety of sarcomas compared to normal tissues, and PIK3R3 and Nanog are also upregulated in sarcoma spheroid cells compared to unselected sarcoma cells.

A Heat map of expression of survival signaling-related genes in two sarcoma cell lines cultured as spheroids vs. monolayers as measured by RNA sequencing. Each sample was run in duplicate. Real-time RT-PCR (B) and (C) Western blot (C) for PIK3R3 and CD133 in three sarcoma cell lines grown as monolayers or as spheroids. D Proportion of CD133+ cells as measured by fluorescence-activated cell sorting (FACS) analysis in sarcoma cells grown as monolayers or as spheroids. E Western blot for PIK3R3, CD133, Nanog, Sox2, Oct-4 and c-Myc in CD133+ and CD133– populations of sarcoma cell lines cultured as spheroids. F Graph and immunostaining of commercially available tissue array slide containing 79 human sarcomas and 4 human normal tissues for CD133, Nanog, and PIK3R3. β-actin is the loading control. *p < 0.05 compared to control.
Inhibition of PIK3R3 inhibits stemness of sarcoma CSCs
We next examined whether PIK3R3 regulates stemness in sarcoma cells. In HT1080, SK-LMS-1, and DDLS8817 sarcoma cells, stable transduction of PIK3R3 shRNA did not alter proliferation in sarcoma cells grown as monolayers (Supplementary Fig. S1B) and had a small inhibitory effect in sarcoma cells grown as spheroids (Supplementary Fig. S1C). However, PIK3R3 shRNA decreased the size of spheroids grown from single cells by 68–73% compared to control shRNA (Fig. 2A) and reduced spheroid formation by 61–71% (Fig. 2B). PIK3R3 shRNA in sarcoma spheroids also resulted in decreased expression of CD133 and Nanog as determined by Western blot and immunostaining, but did not affect levels of Sox2, Oct-4, and c-Myc (Fig. 2C–D). Similarly, in CD133+ cells, PIK3R3 shRNA reduced expression of CD133 and Nanog (Fig. 2E). Conversely, overexpression of PIK3R3 in sarcoma spheroid cells increased expression of CD133 and Nanog as measured by Western blot (Fig. 2F) and immunofluorescence (Supplementary Fig. S1D) and increased spheroid size (Fig. 2G). Thus PIK3R3 promotes spheroid formation, expression of the sarcoma stem cell marker CD133, and expression of the stem cell transcription factor Nanog.

A–E Compare sh.PIK3R3- vs. sh.Scr-transduced sarcoma cells. All experiments except C were done in spheroid-cultured cells. Single-cell (A) and standard spheroid (>100 μm) formation (B), with quantitation of spheroid diameter at 7 days post-transduction. C Western blot for PIK3R3, CD133, Nanog, Sox2, Oct-4, and c-Myc. D Immunostaining for CD133 and Nanog (with DAPI counterstaining). E Western blot for PIK3R3, CD133 an Nanog in sorted CD133+ cells. F–G Compare WT PIK3R3 vs. carrier-transduced cells. F Western blot for PIK3R3, CD133 and Nanog. G Spheroid diameter. β-actin is the loading control. *p < 0.05 compared to control.
PIK3R3 promotes migration and invasion in sarcoma CSCs
We next sought to determine whether PIK3R3 promotes the migratory and invasive phenotypes of sarcoma CSCs, previously demonstrated in spheroid-cultured SK-LMS-1 and DDLS8817 cells [5]. We first confirmed these properties in HT1080 fibrosarcoma cells (Supplementary Fig. S2A). We also compared expression of matrix metalloproteinases (MMPs) 2 and 9, which are highly associated with tumor dissemination and invasiveness, and the pro-metastasis cell adhesion protein N-cadherin between spheroid- and monolayer-cultured cells and found that all were overexpressed in spheroid cells (Supplementary Fig. S2B). Gelatin zymography confirmed increased activity of MMP-2 and MMP-9 in spheroids (Supplementary Fig. S2B). CD133+ cells in all three sarcoma cell lines similarly overexpressed Nanog, N-cadherin, MMP-2, and MMP-9, and had greater MMP-2 and MMP-9 activity compared with CD133- cells (Supplementary Fig. S2C).
Having confirmed the migratory and invasive properties of sarcoma CSCs, we next examined the effects of PIK3R3 shRNA. In all three cell lines, PIK3R3 shRNA diminished the migratory and invasive capacities of spheroid cells by 72–85% and 79–81%, respectively, compared with control shRNA (Fig. 3A, Supplementary Fig. S2D). PIK3R3 knockdown in sarcoma spheroids decreased expression of N-cadherin and MMP-2 by Western blot and reduced the activity of MMP-2, but interestingly did not reduce expression or activity of MMP-9 (Fig. 3B). In CD133+ cells, we confirmed that PIK3R3 shRNA lowered expression of N-cadherin and MMP-2 and activity of MMP-2 by western blot and zymography (Fig. 3C). Following immunostaining of cells grown in spheroid formation conditions, cells treated with PIK3R3 shRNA showed downregulation of N-cadherin and Nanog (Fig. 3D).

All panels compare sh.PIK3R3 vs. sh.Scr-transduced sarcoma cells. Invasion (A) and migration (B) assay for sarcoma spheroid cells. B Western blot and zymography for PIK3R3, N-cadherin, MMP-2, and MMP-9 in sarcoma spheroid cells. C Western blot and zymography for PIK3R3, N-cadherin, MMP-2, and MMP-9 in CD133+ cells. D Immunostaining for N-cadherin and Nanog, with DAPI counterstaining in CD133+ cells. E Tumor volume graph and representative photos of HT1080 CD133+ cells grown in athymic nude mice (n = 5 mice in each group).
To determine the in vivo effects of PIK3R3 on tumor formation and invasiveness, HT1080 CD133+ cells with stable PIK3R3 knockdown were grown as flank xenografts in immunodeficient mice. PIK3R3 knockdown decreased tumor growth by 56% after 9 days (Fig. 3E).
PI3KR3 activates the AKT/ERK signaling pathway in sarcoma CSCs
We next searched for other signaling pathways upregulated in CD133+ compared to CD133− cells using a human phospho-kinase array. Phosphorylation of AKT1/2 (Ser308 and Thr473) and ERK1/2 were significantly greater in CD133+ vs. CD133–cells (Fig. 4A, Supplementary Fig. S3A), which we confirmed by Western blot (Fig. 4B). To investigate the relationship of PI3K/AKT signaling to stem cell properties, the PI3K inhibitor LY294002 was applied to spheroid-cultured HT1080, SK-LMS-1, and DDLS8817 cells. Pharmacologic PI3K inhibition decreased phosphorylation of AKT1/2 and protein levels of PIK3R3, CD133, Nanog, N-cadherin, and MMP-2 in CD133+ cells (Fig. 4C). This treatment also decreased spheroid formation, migration, and invasion of all three cell types (Fig. 4D, Supplementary Fig. S3B).

A–B Compare CD133+ vs. CD133–cells. A Quantitation of phosphorylation of indicated kinases in HT1080 cells as measured by human phospho-kinase array. B Western blot for CD133, total and phosphorylated AKT1/2, and total and phosphorylated ERK1/2. C–D Compare LY294002 (10 μM) vs. vehicle (DMSO)-treated CD133+ cells for 3 days. C Western blot and zymography for total and phosphorylated AKT1/2 and CD133, PIK3R3, Nanog, Sox2, Oct-4, N-cadherin, and MMP-2. D Spheroid formation. E Western blot and zymography for PIK3R3, total and phosphorylated ERK1/2, CD133, Nanog, N-cadherin, MMP-2, and MMP-2 in CD133+ spheroid cell lines transduced with sh.PIK3R3 or sh.Scr. β-actin is the loading control. *p < 0.05 compared to control.
To assess whether ERK signaling is downstream of PIK3R3 and AKT in CD133+ cells, we applied the ERK inhibitor U0126. ERK1/2 inhibition in CD133+ cells resulted in decreased phosphorylation of ERK1/2 and decreased expression of CD133, Nanog, N-cadherin, and MMP-2, but did not affect phosphorylation of AKT1/2 or PIK3R3 (Supplementary Fig. S3C). In contrast, knockdown of PIK3R3 in CD133+ cells resulted in decreased phosphorylation of AKT1/2 and ERK1/2 and decreased expression of CD133, Nanog, N-cadherin, and MMP-2 (Fig. 4E). Overexpression of PIK3R3 in sarcoma spheroid cells has the inverse effects (Fig. S3D). Thus, PI3KR3 activates AKT and ERK in sarcoma CSCs to promote spheroid formation, migration, and invasion.
Inhibition of the PIK3R3/ERK/Nanog pathway suppresses anchorage-independent growth
We next examined the effect of PIK3R3 signaling on anchorage-independent growth in sarcoma spheroid cells. Similar to prior results with CD133+ sarcoma cells [5], sarcoma cells initially grown as spheroids formed 2.5–4.6-fold more colonies than monolayer cells (Supplementary Fig. S4A). Overexpression of PIK3R3 further increased sarcoma spheroid cells’ anchorage-independent growth by 1.4– 1.6-fold (Supplementary Fig. S4B). Knockdown of PIK3R3, ERK1/2, or Nanog using shRNA, confirmed by western blot analysis (Supplementary Fig. S4C), reduced anchorage-independent colony formation by 77–82%, 76–81% and 85–901%, respectively (Supplementary Fig. S4D). Similarly, treatment of CD133+ cells with the PI3K inhibitor LY294002, the ERK1/2 inhibitor U0126, PIK3R3 shRNA, or ERK1/2 shRNA, significantly decreased anchorage-independent growth (Supplementary Fig. S4E). The efficacy of the inhibitors LY294002 and U0126 was confirmed by Western blot analysis (Supplementary Fig. S4F–G).
PIK3R3 inhibition reverses chemotherapy resistance
Numerous studies have demonstrated that a potential driver of chemotherapy resistance in tumors are CSCs [13,14,15]. We have previously shown that the same sarcoma cell lines grown as spheroids are less sensitive to doxorubicin, the most commonly used chemotherapy for sarcomas, compared with monolayer-grown cells [5]. In monolayer cells, the combination of PI3K inhibitor LY294002 and chemotherapy had little additive effect (Supplementary Fig. S5). However, combining the LY294002 with doxorubicin reduced spheroid cell proliferation by 77–84% compared with 11–13% with doxorubicin alone (Fig. 5A). Similarly, combining PIK3R3 shRNA with doxorubicin inhibited sarcoma spheroid cell proliferation by 74–78% vs. 24–33% for PIK3R3 shRNA alone (Fig. 5B). The combination of PIK3R3 shRNA and doxorubicin also led to significant induction of apoptosis, as measured by decreased levels of Bcl-2 and increased levels of cleaved caspase 3 (Fig. 5C). Similar results were obtained with ERK1/2 shRNA and doxorubicin, which decreased proliferation in HT1080, SK-LMS-1, and DDLS8817 sarcoma cells by 69–72% (Fig. 5D), and induced apoptosis to a greater degree than either alone (Fig. 5E). These data show that sarcoma CSCs’ chemotherapy resistance can be overcome with PIK3R3 or ERK1/2 inhibition.

All cells cultured as spheroids. Proliferation following treatment with combinations of (A) LY294002, doxorubicin, or DMSO (A), and doxorubicin, sh.PIK3R3, or sh.Scr (B). C Western blot for PIK3R3, Bcl-2, and cleaved caspase 3 following treatment with the same agents as in (B). D Proliferation following treatment with doxorubicin, sh.ERK1/2, or sh.Scr. E Western blot for PIK3R3, Bcl-2, and cleaved caspase 3 following treatment with same agents as in (D). β-actin is the loading control. *p < 0.05 compared to control.
Inhibiting PIK3R3 reduces stemness, malignancy, and chemoresistance in mouse xenograft models
To confirm in vivo relevance of the above results, the effects of combining PIK3R3 knockdown with doxorubicin were examined in sarcoma xenografts in mice. HT1080, SK-LMS-1 cells were stably transduced with control shRNA or PIK3R3 shRNA (Fig. 6A, Supplementary Fig. S6A), and growth rates of these stable lines in vitro were similar. In contrast, PIK3R3 knockdown reduced growth of xenografts in athymic nude mice by 38–42% (measured from the point at which tumors reached 100–200 mm3) similar to growth inhibition by doxorubicin (30-40%) (Fig. 6B, C, Supplementary Fig. S6B–S6C). The combination of PIK3R3 shRNA and doxorubicin dramatically inhibited tumor growth by 71–80.1%. The combination of PIK3R3 shRNA and doxorubicin did not dramatically reduce proliferation at 12–18 days of treatment as measured by Ki67 immunostaining (Fig. 6D, Supplementary Fig. S6D), but did cause more than additive increases in total apoptosis over doxorubicin or PIK3R3 shRNA alone (20 vs. 4.2 and 3.0 apoptotic cells per field). PIK3R3 shRNA and doxorubicin also reduced expression of CD133, Nanog, Bcl-2, and MMP-2 by 86–91% as measured by immunostaining (Fig. 6D, Supplementary Fig. S6D). To determine if these findings could be translated into the clinic, we performed the same HT1080 mouse xenograft experiment using the PI3K inhibitor LY294002 instead of PIK3R3 shRNA and fond similar results (Supplementary Fig. S7A-C). These results indicate that chemotherapy resistance in sarcoma CSCs can be overcome in vivo by blocking PIK3R3 or PI3K.

All experiments employ HT1080 fibrosarcoma cells. A Western blot confirming knockdown of PIK3R3. β-actin is the loading control. B Growth of xenografts in athymic nude mice following treatment with sh.Scr or sh.PIK3R3 and PBS or doxorubicin. C Photos of representative tumors. D Immunofluorescence of tumors for proliferation using Ki-67, apoptosis using cleaved caspase 3, Bcl-2, CD133, Nanog, and MMP-2, with quantitation at right. Scale bar, 50 µm. E A model of the function of PIK3R3 in sarcoma CSCs. *p < 0.01 compared to control.
Discussion
This study demonstrates the important role of PIK3R3, part of regulatory domain of PI3K, in maintaining sarcoma CSCs and promoting migration, invasion, and chemotherapy resistance. Sarcoma CSCs expressed higher levels of PIK3R3 than sarcoma cells generally, as did human sarcoma tumors relative to normal tissues. Eliminating PIK3R3 knockdown or inhibiting downstream signaling through AKT and ERK diminished expression of the self-renewal transcription factor Nanog and blocked migration, invasion, and chemotherapy resistance. Based on these findings, we developed a model of the function of PIK3R3 in sarcoma CSCs (Fig. 6E), in which PIK3R3 as part of the PI3K complex phosphorylates AKT, which activates the ERK signaling pathway for survival, cancer stemness, and epithelial-mesenchymal transition. Based on these data, inhibiting PI3KR3 or downstream signaling may represent a useful therapeutic strategy for sarcoma, which would be expected to prevent metastasis and reverse chemotherapy resistance.
Our finding that PIK3R3 is critical for sarcoma CSCs is in line with prior evidence on the role of PI3K/AKT signaling in tumorigenesis and metastasis generally [16,17,18,19], in CSCs [20, 21], and in sarcoma [10, 11, 22]. PIK3R3 is one of three members of the gene family encoding PI3K’s regulatory domain, P85 [23]. In addition, AKT expression has been shown to possess a significant prognostic value in sarcomas [24]. We also confirmed that PI3K/Akt signaling pathway plays an important role in promoting stemness, malignancy, and chemoresistance in a mouse xenograft models. PIK3R3 specifically is overexpressed in human hepatocellular carcinoma and other cancers [23, 25, 26] and is required for survival of multiple cancers [23, 26,27,28,29]. Most relevant to our study, variable expression of PIK3R3 in Ewing sarcoma has been shown to affect oncogenic phenotypes [30]. In this study, we also found that ERK was activated downstream of PIK3R3 and AKT. ERK has previously been implicated in the self-renewal and maintenance of CSCs [31]; targeting the ERK pathway has been shown to increase the likelihood of durable remission and prevent resistance to chemotherapy and radiotherapy [32, 33]. In this study, we showed that inhibition of ERK decreased anchorage-independent growth, cell migration, invasion, and chemotherapy resistance of sarcoma CSCs.
We focused on CSCs because they may represent the source of all cells within a tumor. Especially high expression of stemness-related genes, such as Nanog, Sox2, Oct-4 and c-Myc, is observed in histologically poorly differentiated tumors and is associated with worse outcomes [34, 35]. Thus, blocking CSCs has the potential to prevent tumor growth, metastasis, and chemotherapy resistance. In addition to PI3KR3 and ERK, our data implicate Nanog, a regulator of as a potential target within that strategy, as it is regulated by these proteins. Nanog is central to CSC maintenance, as it regulates the expression of Sox2, Oct-4, KLF4, and itself [36]. Specifically, we found that Nanog promotes CSC features, such as mesenchymal phenotype and chemotherapy resistance, and that its overexpression enhances anchorage-independent growth. In this study, we found that PIK3R3 plays a significant role in promoting sarcoma CSC migration and invasion, at least in part by promoting secretion of MMP-2.
Conclusion
In this study, we illustrated the importance of a potentially targetable signaling pathway in sarcoma CSCs, PIK3R3-ERK1/2-Nanog, to sarcoma CSC maintenance and malignant properties, including chemotherapy resistance. Our data show that inhibition of PIK3R3 expression attenuates oncogenic ERK1/2 signaling and Nanog in sarcoma CSCs. In a mouse xenograft model, combining PIK3R3 knockdown with doxorubicin had a synergistic effect in blocking tumor growth. Thus, targeting PIK3R3 in sarcoma CSCs may be an effective therapeutic strategy to prevent sarcoma metastasis and reverse chemotherapy resistance.
Materials and methods
Cell lines and reagents
HT1080 human fibrosarcoma cells and SK-LMS-1 human leiomyosarcoma cells were obtained from the American Type Culture Collection (Manassas, VA). The DDLS8817 dedifferentiated liposarcoma cell line was established from samples obtained from patients who signed informed consent forms and were confirmed by cytogenetic analysis and by DNA copy number arrays (Agilent 244 K) to harbor 12q amplification. HT1080 and SK-LMS-1 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), and DDLS8817 cells were maintained in DMEM-F12. All media were supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 μg/mL), and L-glutamine (2 mM) (“regular media”). Cancer cell lines were actively passaged for <6 months from the time that they were received, and United Kingdom Coordinating Committee on Cancer Research (UKCCCR) guidelines were followed [37]. Doxorubicin (RYG02) was purchased from TSZ Chem/BIOTANG Inc. (Lexington, MA). LY294002 (Cat. 440202), U0126 (Cat. 662005), and puromycin (Cat. 508838) were purchased from Calbiochem.
Spheroid culture
Cells were resuspended in “spheroid media” comprised of DMEM/F12 containing epidermal growth factor (EGF, E9644; Sigma-Aldrich, St. Louis, MO), basic fibroblast growth factor (bFGF, 341583; Sigma-Aldrich), N-2 supplement (A13707-01; Thermo Fisher Scientific, Waltham, MA), and B27 (0080085SA; Sigma-Aldrich), and then plated on Ultra-Low Attachment culture dishes (Corning Life Sciences, Tewksbury, MA). Spheroids were collected after 5–7 days except when noted otherwise. Proteins were extracted for analysis, or cells were dissociated with AccutaseTM (561527, BD Biosciences) and used for other experiments. Spheroid number was quantified by counting spheroids >50 μm in diameter in five fields after image processing using Imaris 7.6 (Bitplane, Concord, MA).
Gene expression analysis by RNA sequencing (RNAseq)
Monolayer cells were dissociated to single cell suspension, from which RNA was purified using TRIzol (Invitrogen) in accordance with manufacturer’s instructions. RNAseq was then performed at the sequencing core (Integrated Genomics Operation; IGO) of Memorial Sloan Kettering Cancer Center (MSKCC, New York) following previously reported procedures [38].
Real-time reverse transcription PCR (RT-PCR)
Total RNA was extracted from all cells using TRIzol® according to the manufacturer’s protocol. Using the RT [2] First Strand kit (Cat.330401, Qiagen), 500 ng RNA from each cell line/condition was converted to cDNA and mixed with SYBR green master mix (Cat. 201443, Qiagen) for qPCR using AIIA7 (Life Technologies). All primers were purchased from Integrated DNA Technologies, Inc. and the glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 5′-ACC ACA GTC CAT GCC ATC AC-3′ and 5′-TCC ACC ACC CTG TTG CTG TA-3′) gene was used to normalize expression levels in quantitative analyses. To amplify the genes encoding CD133 and PIK3R3, the following primers were used: CD133 (5′-GAT TAA GTC CAT GGC AAC AGC G − 3′ and 5′- GCT GGT CAG ACT GCT GCT AAG C − 3′), and PIK3R3 (5′-ATG TAC AAT ACG GTG TGG AGT ATG-3′ and 5′-GCT GGA GGA TCC ATT TCA AT-3′). The experiment was performed in triplicate.
Western blot
Proteins were extracted by collecting cells in RIPA buffer (Sigma-Aldrich) containing Complete Protease Inhibitor Cocktail (Roche Diagnostics USA, Indianapolis, IN), and protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Western blot analysis was performed using the following antibodies: PIK3R3 (sc-376615; Santa Cruz Biotechnology) CD133 (MBS462020; MyBioSource, San Diego, CA), Sox2 (#2748), Oct-4 (#2788), and Nanog (#4893) (all from Cell Signaling Technology), c-Myc (sc-40; Santa Cruz Biotechnology), N-cadherin (#14215; Cell Signaling Technology), MMP-2 (ab92536; Abcam), MMP-9 (ab119906; Abcam), p-AKT (Thr 308) (#9275; Cell Signaling Technology), p-AKT(Ser 473) (#9271; Cell Signaling Technology), AKT1/2 (sc-8312; Santa Cruz Biotechnology), p-ERK1/2 (#9101; Cell Signaling Technology), Erk2 (sc-154; Santa Cruz Biotechnology), Erk1/2 (sc-514302; Santa Cruz Biotechnology), Bcl2 (#2876; Cell Signaling Technology), cleaved caspase 3 (#9661; Cell Signaling Technology) and β-actin (A5441; Sigma).
Flow cytometry and flow-assisted cell sorting
Cells were dissociated using Accutase and resuspended in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA). The cells were stained with PE-conjugated CD133 (130-098-826; Miltenyi Biotec) or isotype control antibody (BD555742; BD Biosciences) on ice for 30 min. Cells were then washed with PBS and analyzed on a BD FACSCalibur™ (BD Biosciences) using BD CellQuest software (BD Biosciences).
Tumor microarray (TMA)
Commercially available paraffin-embedded tissue array slides containing 79 human sarcomas and 4 human normal tissues (NBP2-30332, Novus Biologicals, and T242, US Biomax, Inc., USA) were purchased. Sections were deparaffinized, then incubated with anti-human CD133, PIK3R3 (SR44-06; Novus Biologicals), and Nanog (ab80892; Abcam) in a solution of PBS with 1% BSA and 0.1% Triton X-100 at 4 °C overnight. Staining was visualized using anti-mouse Alexa Fluor 488 (A-11029; Thermo Fisher), and anti-rabbit Alexa Fluor 594 (A-11037; Thermo Fisher), with nuclear counterstaining using 4′,6-diamidino-2-phenylindole (DAPI, 28718-90-3; Sigma-Aldrich) and imaging on an inverted confocal microscope. Images were processed using Imaris 7.6.
Lentiviral transduction
Silencing of PIK3R3, Nanog, and ERK1/2 and was achieved via lentiviral transduction of and human PIK3R3 shRNA (sc-39124-V; Santa Cruz), human Nanog shRNA (sc-43958-V; Santa Cruz), human ERK1 shRNA (sc-29307-V; Santa Cruz), human ERK2 shRNA (sc-35335-V; Santa Cruz) per the manufacturer’s protocol. For the experiments involving spheroid cells transduced with shRNA, spheroids were grown from 5 to 7 days and then dissociated into single cells using Accutase. Cells were then transduced with shRNA and placed back into spheroid culture. Experiments were performed after 5 days expect where otherwise noted.
Overexpression of PIK3R3 was achieved via Nanog lentiviral activation particles (sc-402964-LAC; Santa Cruz) per the manufacturer’s protocol. Controls were scramble shRNA (sc-108080; Santa Cruz) or empty lentiviral activation particles (sc-437282, Santa Cruz). Maximal knockdown of genes occurred 72–96 h after transduction.
Single cell colony formation
Spheroid cells stably transduced with sh.PIK3R3 or carrier (sh.Scr) were dissociated to single cells and plated onto Ultra-Low Attachment 96-well plates at a density of one cell per well. After confirmation of clone establishment at 24 h, clone formation was monitored at sequential points of 4, 7, and 10 days. Clone size was measured using an inverted microscope (Leica Microsystems, Buffalo Grove, IL) and MetaMorph software, version 7.8.2 (Molecular Devices, Sunnyvale, CA).
Immunocytochemistry
Spheroid cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS, and incubated with antibodies against CD133 (MBS462020; Miltenyi Biotec), N-cadherin (#14215; Cell Signaling), Nanog (#4893, #8822; Cell Signaling Technology) and/or PIK3R3 (sc-376615; Santa Cruz Biotechnology) in a solution of PBS with 1% FBS and 0.1% Triton X-100 at 4 °C overnight. Cells were then stained with secondary antibodies tagged with anti-mouse Alexa Fluor 488 (A-11029; Thermo Fisher), and Alexa Fluor 568 (A-11061; Thermo Fisher). Nuclei were counterstained using DAPI. Stained cells were visualized on an inverted confocal microscope (Leica Microsystems) and images were processed using Imaris 7.6.
Proliferation, migration, and invasion assays
Spheroids were dissociated with AccutaseTM, and monolayer cells were collected with trypsin. For proliferation assays, 2 × 103 cells were plated onto 96-well flat-bottom plates and maintained in regular media overnight. A colorimetric MTT assay was used to assess cell number by optical density after the specified number of days, as previously described. [39] Data reflect the mean of six samples.
For migration and invasion assays, 2 × 103 cells were suspended in 0.2 mL of serum-free DMEM. The cells were loaded in the upper well of an 8 μm-pore Transwell chamber (Corning).
For invasion assays, the upper well was pre-coated with 10 mg/mL growth factor reduced BD Matrigel™ matrix (BD Biosciences). The lower well was filled with 0.8 mL of DMEM with serum. After incubation for 48 h at 37 °C, non-invading cells on the upper surface of the filter were removed with a cotton swab. The cells that migrated or invaded onto the lower surface of the filter were fixed and stained with Siemens Diff-Quick Stain Kit per the manufacturer’s protocol (B4132-1A; Siemens Healthcare Diagnostics, Newark, DE). The fixed cells were photographed at ×20 magnification on an inverted confocal microscope (Leica Microsystems) and images were processed using Imaris 7.6. Cells were counted in five microscopic fields per well, and the extent of migration or invasion was expressed as the average number of cells per microscopic field.
Gelatin zymography
Production of MMP-2 and MMP-9 was analyzed by gelatin zymography as previously described [40].
Mouse xenograft model
All mouse protocols were approved by the Memorial Sloan Kettering Institutional Animal Care and Use Committee. To generate subcutaneous flank tumors, 2 × 106 HT1080 and SK-LMS-1 cells (with shRNA knockdown of PIK3R3 where indicated) were resuspended in 100 µL of Hank’s balanced salt solution and injected subcutaneously into the right flank of athymic, nude, 6–8-week-old male BALB/c nu/nu mice (Taconic, Hudson, NY) following isoflurane anesthesia. Mice were assigned into treatment groups (five mice per group) when tumors reached 100–50 mm3 in volume, designated as day 0. Doxorubicin (4 mg/kg) or control DMSO carrier was administered two times per week by intraperitoneal injection. Tumor volume (TV) was calculated using the following formula: TV = length × (width)2 × 0.52.
Immunohistochemistry
At least four sections were analyzed from each tumor. Paraffin-embedded sections were deparaffinized [13] and incubated with primary antibodies against Ki67 (ab15580; Abcam), PIK3R3 (sc-376615; Santa Cruz Biotechnology), cleaved caspase 3 (#9661; Cell Signaling), MMP-2 (Abcam, ab92536), Bcl-2 (sc-65392, Santa Cruz), CD133 (MBS462020; Miltenyi Biotec), p-AKT (Thr 308) (ab8933; Abcam), p-AKT(Ser 473) (#4060; Cell Signaling Technology), AKT1/2 (sc-8312; Santa Cruz Biotechnology), or Nanog (ab54835; Abcam) in a solution of PBS with 1% FBS and 0.1% Triton X-100 at 4 °C overnight. Staining was visualized using anti-rabbit Alexa Fluor 488 (A-21206; Thermo Fisher) and Alexa Fluor 568 (A-11011; Thermo Fisher). Nuclei were counterstained using DAPI. Slides were digitally scanned with Panoramic Flash 250 (3DHistech, Budapest, Hungary) using a 20×/0.8NA objective and images were processed using MetaMorph version 7.8.2 (Molecular Devices). Staining was counted in five microscopic fields.
Human phospho-kinase array
Phospho-antibody array analysis was performed using the Proteome Profiler Kit ARY003B (R&D Systems) according to the manufacturer’s instructions [41].
Soft agar colony formation
To examine anchorage-independent growth, a cell suspension of 1 × 103 cells/mL was mixed in 0.4% agarose in either regular or spheroid media, as applicable, and seeded in triplicate onto previously set 0.9% soft agar in a 60 mm culture dish. Cells were incubated for 3–4 weeks during which growth was observed weekly under an inverted microscope (Leica). Colonies were then photographed and counted in 4–5 randomly chosen fields and expressed as means of the triplicate cultures.
Statistical analysis
Statistical analyses were performed using Microsoft Office Excel 2010 software. P values were calculated using the Student’s t test. For comparisons between more than two groups, treatment groups were compared to the control group using one-way ANOVA with the Bonferroni adjustment for multiple comparisons. All experiments were repeated independently at least twice and results shown were collected from a representative experiment. P values < 0.01 were considered significant.
Change history
19 March 2024
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41419-024-06609-6
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Brennan MF, Antonescu CR, Maki RG. Management of soft tissue sarcoma. New York:Springer;2013.
Billingsley KG, Burt ME, Jara E, Ginsberg RJ, Woodruff JM, Leung DH, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg. 1999, 229:602–10 discussion 610-602.
Singer S, Tap WD, Kirsch DG, Crago AM. Soft tissue sarcoma In: DeVita VT, Jr., Rosenberg SA, Lawrence TS, editors. Cancer: Principles and Practice of Oncology, 11 edn. Philadelphia: Lippincott Williams & Wilkins;2018. p. 1401–50.
Chang KK, Yoon C, Yi BC, Tap WD, Simon MC, Yoon SS. Platelet-derived growth factor receptor-alpha and -beta promote cancer stem cell phenotypes in sarcomas. Oncogenesis. 2018;7:47.
Yoon C, Chang KK, Lee JH, Tap WD, Hart CP, Simon MC, et al. Multimodal targeting of tumor vasculature and cancer stem-like cells in sarcomas with VEGF-A inhibition, HIF-1alpha inhibition, and hypoxia-activated chemotherapy. Oncotarget. 2016;7:42844–58.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8.
Glumac PM, LeBeau AM. The role of CD133 in cancer: a concise review. Clin Transl Med. 2018;7:18.
Yang J, Ren Z, Du X, Hao M, Zhou W. The role of mesenchymal stem/progenitor cells in sarcoma: update and dispute. Stem Cell Investig. 2014;1:18.
McKinnon T, Venier R, Yohe M, Sindiri S, Gryder BE, Shern JF, et al. Functional screening of FGFR4-driven tumorigenesis identifies PI3K/mTOR inhibition as a therapeutic strategy in rhabdomyosarcoma. Oncogene. 2018;37:2630–44.
Trautmann M, Cyra M, Isfort I, Jeiler B, Krüger A, Grünewald I, et al. Phosphatidylinositol-3-kinase (PI3K)/Akt Signaling is Functionally Essential in Myxoid Liposarcoma. Mol Cancer Ther. 2019;18:834–44.
Lim HJ, Wang X, Crowe P, Goldstein D, Yang JL. Targeting the PI3K/PTEN/AKT/mTOR Pathway in Treatment of Sarcoma Cell Lines. Anticancer Res. 2016;36:5765–71.
Yoon C, Park do J, Schmidt B, Thomas NJ, Lee HJ, Kim TS, et al. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin Cancer Res. 2014;20:3974–88.
Yoon C, Cho SJ, Aksoy BA, Park do J, Schultz N, Ryeom SW, et al. Chemotherapy Resistance in Diffuse-Type Gastric Adenocarcinoma Is Mediated by RhoA Activation in Cancer Stem-Like Cells. Clin Cancer Res. 2016;22:971–83.
Alison MR, Lin WR, Lim SM, Nicholson LJ. Cancer stem cells: in the line of fire. Cancer Treat Rev. 2012;38:589–98.
Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol: Off J Am Soc Clin Oncol. 2010;28:1075–83.
Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008,1784:159–85.
Jiang BH, Liu LZ. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res. 2009;102:19–65.
Elia U, Flescher E. PI3K/Akt pathway activation attenuates the cytotoxic effect of methyl jasmonate toward sarcoma cells. Neoplasia. 2008;10:1303–13.
Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci USA. 2009;106:268–73.
Martelli AM, Evangelisti C, Follo MY, Ramazzotti G, Fini M, Giardino R, et al. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in cancer stem cells. Curr Med Chem. 2011;18:2715–26.
Cen L, Hsieh FC, Lin HJ, Chen CS, Qualman SJ, Lin J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br J Cancer. 2007;97:785–91.
Wang G, Yang X, Li C, Cao X, Luo X, Hu J. PIK3R3 induces epithelial-to-mesenchymal transition and promotes metastasis in colorectal cancer. Mol Cancer Ther. 2014;13:1837–47.
Tomita Y, Morooka T, Hoshida Y, Zhang B, Qiu Y, Nakamichi I, et al. Prognostic significance of activated AKT expression in soft-tissue sarcoma. Clin Cancer Res. 2006;12:3070–7.
Cao G, Dong W, Meng X, Liu H, Liao H, Liu S. MiR-511 inhibits growth and metastasis of human hepatocellular carcinoma cells by targeting PIK3R3. Tumour Biol. 2015;36:4453–9.
Wang G, Yang X, Jin Y, Deng Y, Luo X, Hu J, et al. TGF-beta regulates the proliferation of lung adenocarcinoma cells by inhibiting PIK3R3 expression. Mol Carcinog. 2015;54:E162–71.
Zhang L, Huang J, Yang N, Greshock J, Liang S, Hasegawa K, et al. Integrative genomic analysis of phosphatidylinositol 3’-kinase family identifies PIK3R3 as a potential therapeutic target in epithelial ovarian cancer. Clin Cancer Res. 2007;13:5314–21.
Klahan S, Wu MS, Hsi E, Huang CC, Hou MF, Chang WC. Computational analysis of mRNA expression profiles identifies the ITG family and PIK3R3 as crucial genes for regulating triple negative breast cancer cell migration. Biomed Res Int. 2014;2014:536591.
Zhu Y, Zhao H, Rao M, Xu S. MicroRNA-365 inhibits proliferation, migration and invasion of glioma by targeting PIK3R3. Oncol Rep. 2017;37:2185–92.
Niemeyer BF, Parrish JK, Spoelstra NS, Joyal T, Richer JK, Jedlicka P. Variable expression of PIK3R3 and PTEN in Ewing Sarcoma impacts oncogenic phenotypes. PLoS ONE. 2015;10:e0116895.
Lee S, Wottrich S, Bonavida B. Crosstalks between Raf-kinase inhibitor protein and cancer stem cell transcription factors (Oct4, KLF4, Sox2, Nanog). Tumour Biol. 2017;39:1010428317692253.
Holck S, Klarskov LL, Larsson LI. Phospho-ERK levels as predictors for chemotherapy of rectal carcinoma. Oncotarget. 2019;10:1745–55.
Ciccarelli C, Vulcano F, Milazzo L, Gravina GL, Marampon F, Macioce G, et al. Key role of MEK/ERK pathway in sustaining tumorigenicity and in vitro radioresistance of embryonal rhabdomyosarcoma stem-like cell population. Mol Cancer. 2016;15:16.
Chang TS, Wu YC, Chi CC, Su WC, Chang PJ, Lee KF, et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res. 2015;21:201–10.
Li R, Huang J, Ma M, Lou Y, Zhang Y, Wu L, et al. Two-stage induced differentiation of OCT4+/Nanog+ stem-like cells in lung adenocarcinoma. Oncotarget. 2016;7:68360–70.
Torres-Padilla ME, Chambers I. Transcription factor heterogeneity in pluripotent stem cells: a stochastic advantage. Development. 2014;141:2173–81.
UKCCCR guidelines for the use of cell lines in cancer research. Br J Cancer. 2000,82:1495–509.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Yoon SS, Eto H, Lin CM, Nakamura H, Pawlik TM, Song SU, et al. Mouse endostatin inhibits the formation of lung and liver metastases. Cancer Res. 1999;59:6251–6.
Schmalfeldt B, Prechtel D, Harting K, Spathe K, Rutke S, Konik E, et al. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin Cancer Res. 2001;7:2396–404.
Lee HJ, Cao Y, Pham V, Blackwood E, Wilson C, Evangelista M, et al. Ras-MEK Signaling Mediates a Critical Chk1-Dependent DNA Damage Response in Cancer Cells. Mol Cancer Ther. 2017;16:694–704.
Acknowledgements
We thank MSKCC senior editor Jessica Moore for reviewing this paper.
Funding
This research was supported by the National Cancer Institute of the US National Institutes of Health through R01 CA158301 (MCS, SSY) and Cancer Center Support Grant P30 CA008748 (to MSK).
Author information
Authors and Affiliations
Contributions
SY designed research and approved the final paper; CY and JL analyzed the data, performed the research; SR and MC revised the paper; SY gave the financial support.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All mouse protocols were approved by the Memorial Sloan Kettering Institutional Animal Care and Use Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by R. Aqeilan
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41419-024-06609-6
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoon, C., Lu, J., Ryeom, S.W. et al. RETRACTED ARTICLE: PIK3R3, part of the regulatory domain of PI3K, is upregulated in sarcoma stem-like cells and promotes invasion, migration, and chemotherapy resistance. Cell Death Dis 12, 749 (2021). https://doi.org/10.1038/s41419-021-04036-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04036-5
This article is cited by
-
Circ_0021350 plays an oncogene role by regulating miR-1207-3p/PIK3R3 in glioblastoma
BMC Cancer (2023)
-
PIK3R3, a regulatory subunit of PI3K, modulates ovarian cancer stem cells and ovarian cancer development and progression by integrative analysis
BMC Cancer (2022)
-
Paired miRNA- and messenger RNA-sequencing identifies novel miRNA-mRNA interactions in multiple myeloma
Scientific Reports (2022)
