
Abstract
Vacuole membrane protein 1 (VMP1), the endoplasmic reticulum (ER)-localized autophagy protein, plays a key role during the autophagy process in mammalian cells. To study the impact of VMP1-deficiency on midbrain dopaminergic (mDAergic) neurons, we selectively deleted VMP1 in the mDAergic neurons of VMP1fl/fl/DATCreERT2 bigenic mice using a tamoxifen-inducible CreERT2/loxp gene targeting system. The VMP1fl/fl/DATCreERT2 mice developed progressive motor deficits, concomitant with a profound loss of mDAergic neurons in the substantia nigra pars compacta (SNc) and a high presynaptic accumulation of α-synuclein (α-syn) in the enlarged terminals. Mechanistic studies showed that VMP1 deficiency in the mDAergic neurons led to the increased number of microtubule-associated protein 1 light chain 3-labeled (LC3) puncta and the accumulation of sequestosome 1/p62 aggregates in the SNc neurons, suggesting the impairment of autophagic flux in these neurons. Furthermore, VMP1 deficiency resulted in multiple cellular abnormalities, including large vacuolar-like structures (LVSs), damaged mitochondria, swollen ER, and the accumulation of ubiquitin+ aggregates. Together, our studies reveal a previously unknown role of VMP1 in modulating neuronal survival and maintaining axonal homeostasis, which suggests that VMP1 deficiency might contribute to mDAergic neurodegeneration via the autophagy pathway.
Similar content being viewed by others

Introduction
Vacuole membrane protein 1 (VMP1), an endoplasmic reticulum (ER)-resident protein, has attracted attention recently owing to its essential role on mediating autophagy. VMP1 gene encodes a 406-amino-acid transmembrane protein containing six hydrophobic regions1. Of them, an autophagy-related (ATG) domain is localized in the middle of the chain2. Once mutated at the ATG domain VMP1 fails to induce microtubule-associated protein 1 light chain 3 (LC3) recruitment and loses the interaction with Beclin-1, eventually blocking autophagy initiation2. However, recent work demonstrates that the VMP1 depletion reversely promotes the accumulation of LC3-labeled autophagic structures at ER in the VMP1 knockout (KO) mammalian cells upon aberrant nutrient conditions3, suggesting that VMP1 may play an important role in the autophagic process by regulating interactions between ER and autophagic-isolation membrane4. These studies indicate that VMP1 deficiency inhibits autophagosome maturation, disrupts the association with ER and blocks the fusion with lysosome4. Besides the animal studies, the research in Dictyostelium5, plants6, and Chlamydomonas7 also indicates that VMP1 is highly involved in the processes of protein secretion, phagocytosis, osmoregulation, and cytokinesis to mediate the diverse cellular process.
The autophagy-lysosome pathway is responsible for the clearance of intracellular protein aggregates, the dysfunction of which is associated with several neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease, amyotrophic lateral sclerosis, and Huntington’s disease8. So far, several mice models, such as Atg5fl/fl/Nestincre, Atg7fl/fl/Pcp2Cre, FIP200fl/fl/Nestincre, and ULK1/2fl/fl/Nestincre mice that specifically knock out autophagic genes in the nervous system have been generated, allowing the analysis of the roles of autophagy in neurodegeneration9. However, as a key autophagic component, the roles of VMP1 in central DAergic neurons have not been investigated in vivo. In the present study, we established VMP1fl/fl/DATCreERT2 mouse model (referred as VMP1cKO) that postnatally knocks out autophagic gene VMP1 in the cells expressing dopamine transporter (DAT) under the tamoxifen (TAM) treatment. We showed that the VMP1cKO mice developed severe motor deficits accompanied by a substantial loss of mDAergic neurons and striatal axon terminals. Additionally, enlarged mDAergic axonal terminals that contain α-syn+ inclusions in the striatum were characterized in VMP1cKO mice. Furthermore, the VMP1-deficient mDAergic neurons displayed damaged mitochondria, swollen ER, large vacuolar-like structures (LVSs), and accumulation of ubiquitin+ aggregates. Together, our studies provide strong evidence for the pathogenetic effects of VMP1 deficiency on autophagy-mediated mDAergic neurodegeneration.
Materials and methods
Chemicals and antibodies
TAM, corn oil, and routinely used chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). The information of antibodies used for WB and IFC staining was summarized in Table 1.
Generation of tissue-specific VMP1-deficient mice
The heterozygous mice VMP1Flox/wt were generated by ViewSolid Biotech Co. Ltd (Beijing, China). Briefly, CRISPR technology was used to cut the intron 3 of the VMP1 gene, and the donor vector having loxP-flanked exon 3 of VMP1 was provided to have the insertion of loxp sites at mouse genomic VMP1 DNA. According to the screen of cas9/gRNA activity and the target location, high-activity gRNAs (target DNA sequence: GAACAGAATTCTAGTCTCTGG; AAATATTGCTCTCCATTTGGG) were selected for microinjection into C57BL/6J fertilized eggs to construct conditional gene KO mice.
To achieve the mouse model of conditionally KO VMP1 in the mDAergic neuronal system, VMP1fl/fl/DATCreERT2 mice were produced by breeding mice carrying an inducible Cre recombinase under the DAT promoter with the heterozygous mice VMP1Flox/wt. The DATCreERT2 mice were kindly gifted by the Günther Schütz group, which were generated by recombining a construct containing an improved Cre recombinase fused to a modified ligand-binding domain of the estrogen receptor into a bacterial artificial chromosome containing the gene encoding DAT 10,11,12.
All experimental mice were maintained under SPF conditions (temperature, 22 ± 2 °C; air exchange per 20 min; 12 h/12 h light/dark cycle) with free access to food and water. Animal care and procedures were carried out in accordance with the Laboratory Animal Care Guidelines approved by the Institutional Animal Care Committee at Dalian Medical University. The protocol was approved by the Institutional Animal Care Committee at Dalian Medical University.
TAM treatment
TAM was dissolved in corn oil/ethanol (10:1) mixture at a final concentration of 10 mg/ml. Five to 6-week-old VMP1fl/fl (VMP1cWT) and VMP1fl/fl/DATCreERT2 (VMP1cKO) mice were both injected intraperitoneally with 1 mg TAM twice a day (total 2 mg/day) for 5 consecutive days, and then behavioral assessment was performed 2 weeks following the last injection of TAM. VMP1cKO mice showed significant body loss at the 4th week following TAM injection, but the weight of VMP1cWT mice was not altered. Both VMP1cWT and VMP1cKO mice were sacrificed at the 5th week following TAM injection. Some of the VMP1cWT and VMP1cKO mice were fasted for 6 h before sacrificed for IFC staining.
Genotyping
DATCreERT2 transgenic mice were identified by PCR screening (2 × EasyTaq PCR SuperMix, Transgen Biotech) of tail DNA using an antisense primer, CAG ACC AGG CCA GGT ATC TCT, and a sense primer, AGA ACC TGA TGG ACA TGT TCA GG, of which the transgene band size is 700 bp. The floxed VMP1 knock-in mice were identified using AGCCGTCTCCTACTCCCTG and TGGTGATGGTTTTGTGCTTG. The PCR product size of the wild-type allele is 170 bp and the knock-in flox allele 204 bp.
Locomotor activity
To measure the locomotor activity, VMP1cWT and VMP1cKO mice were placed into locomotor activity monitor instrument (25 × 25 × 30 cm, Med Associates Inc., St. Albans, USA) equipped with computer-controlled photocells. The activity was automatically recorded for 30 min, and total distance traveled, vertical time and counts, stereotypic time and counts calculated by the Med system. The behavioral assessment was performed between 13:00 and 16:00 on the 49th and 63rd postnatal day respectively.
Rotarod test
As described previously13, mice were trained on the IITC Rotarod (IITC Life Science, Woodland Hills, CA) at 10 r/min, three times per day (at 1-h intervals) for 2 days, and were tested on the rotating rod with speed auto accelerating from 4 to 40 r/min over a period of 5 min. The length of time the mouse stayed on the rotating rod was recorded across three trials at 1-h intervals. The behavioral assessment was performed on the 45th, 47th, 49th, 59th, 61st, 63rd, and 65th postnatal day, respectively.
Tail suspension test
Mice were suspended on a bar (50 cm above the floor) by the tail using a tape for 6 min. The cumulative immobility time during the last 4 min was recorded. The behavioral assessment was performed on the 60th postnatal day.
Y-maze test
The Y-maze test instrument (Beijing Zhongshidichuang Science and Technology Development Col., Ltd, Beijing, China) was implemented in a white background with three arms (labeled as a–c arm) that extended from a central platform at a 120° angle. Each mouse was placed in the center of the Y-maze and was allowed to explore freely through the maze for 6 min. The sequence and the total number of arms entered were recorded using the observer. An arm entry was considered to be complete when the whole body of the mouse was completely placed within the arm. The behavioral assessment was performed on the 60th postnatal day.
High-performance liquid chromatography (HPLC)
For the assessment of DA concentration, mice were sacrificed at the 5th week following TAM injection, and the striatum area was rapidly collected using a punch, weight and kept at −80 °C, which was performed for HPLC (EiCOM, HTEC-500, USA) as described in detail previously14.
Western blot (WB)
Mice were sacrificed at the 5th week following TAM injection, and the tissues were dissected rapidly on ice and homogenized in cold RIPA buffer (Beyotime Biotechnology, Shanghai, China) containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO, USA) and then lysed for 30 min on ice. The protein concentration in the supernatant was determined using protein assay kits (TaKaRa, Shiga, Japan). Forty micrograms of protein were loaded and separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). After blocking, membranes were incubated with appropriate primary antibodies (Table 1) at 4 °C overnight, followed by 1-h incubation at room temperature with a peroxidase-conjugated secondary antibody. Finally, the membrane was incubated with enhanced chemiluminescence (Millipore, Bedford, MA, USA), and the target protein bands were quantified using the FluorChem Q system (ProteinSimple, California, USA).
IFC staining
For histological analysis, mice were anesthetized with ketamine at the 5th week following TAM injection and perfused transcardially with 4% paraformaldehyde (PFA)15. After dehydrated in 30% sucrose, the brain tissues were cut into 40 μm coronal sections using Leica cryostat (CM-1950S, Leica, Germany). They were incubated with blocking buffer (10% normal goat serum, 1% bovine serum albumin, 0.3% Triton X-100, PBS solution) overnight at 4 °C and were then incubated with the primary antibodies overnight at 4 °C (Table 1). For Ub staining, the sections were performed antigen repair by using citrate buffer (pH 6.0). The stained sections were imaged using a laser scanning confocal microscope (A1 confocal, Nikon instruments (Shanghai) Co., Ltd). The paired images in the figures were collected at the same gain and offset settings.
Transmission electron microscope (TEM) analysis
Mice were sacrificed at the 5th week following TAM injection, and the midbrain area was collected rapidly on ice within 3 min into a fixative solution containing 2.5% glutaraldehyde (Servicebio, Wuhan, China) and then fixed at room temperature for 2 h followed by transferring to 4 degrees for storage. The tissues were washed three times in PBS (pH 7.4) before post-fixing in 1% osmium acid (diluted with 0.1 M PBS solution) at room temperature for 2 h and were successively dehydrated. After a series of embedding steps, the tissues were cut into 80 nm sections using the Leica ultrathin microtome (Leica UC7, Leica, Germany) and stained with 2% uranyl acetate saturated alcohol solution and lead citrate solution, respectively. The stained sections were imaged using a TEM (HITACHI, HT7700).
Image analysis
TH+ cells in SNc and VTA were calculated in every three sections from Bregma −2.70 to −3.88 mm at a magnification of ×10 by an observer who was blind to the genotype and grouping, and the data were collected from 8 to 10 slice per animal14. The outline of SNc and VTA was determined according to anatomical landmarks16. The analysis of IFC staining on the number of puncta, axon density, mean number of enlarged axon terminals were quantified using the Image J software, and the data were collected from 3 to 4 slices per animal.
Statistical analysis
Data were expressed as means ± SEM and were analyzed using GraphPad Prism software (Version 7.0). Protein bands were quantified using the FluorChem Q system (ProteinSimple, California, USA). Two-way ANOVA followed by Sidak’s multiple comparisons test was used for analyses across multiple groups, with Student’s t-test used to determine significant differences between two groups. Data were presented as means ± SEM, and p < 0.05 was considered significant. All experiments were repeated at least three times and sample sizes were estimated from pilot experiments. No statistical methods were used to predetermine sample size, but our sample sizes are similar to those reported in previous publications.
Results
VMP1cKO mice develop severe motor deficits
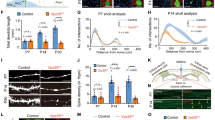
VMP1 is one of the core components of ATG machinery2,17. To determine its role in mammalian mDAergic neurons, we first established the mouse model with mDAergic neuron-specific deletion of VMP1 (the construction for TAM-inducible VMP1-deficient mice as shown in Supplementary Fig. 1A). At the 5 weeks old, VMP1cWTand VMP1cKO mice were both injected intraperitoneally with TAM (Fig. 1A). We recorded the body weight and physical condition of the mice and found VMP1cKO mice showing significant body loss at the 4th week following injection (Supplementary Fig. 1B, C). Mice were then sacrificed at the 70th postnatal day. The VMP1 expression profile in the SNc was firstly detected using immunofluorescence (IFC) staining. As expected, the expression level of VMP1 in the SNc mDAergic neurons (tyrosine hydroxy (TH)-labeled) of VMP1cKO mice was dramatically decreased (Fig. 1B, C), indicating the success of VMP1 depletion. Moreover, the biochemical analysis of midbrain and striatal homogenates from VMP1cKO and VMP1cWT mice by WB confirmed VMP1 depletion in the mDAergic neurons (Fig. 1D, E). To assess the impact of VMP1 depletion on motor activity, the open-field test was performed on the 49th and 63rd postnatal day, respectively. We found that the total distance traveled, vertical movement (vertical time and counts), and stereotypic movement (stereotypic time and counts) were significantly decreased in VMP1cKO mice at the 63rd postnatal day compared to the age-matched VMP1cWT mice (Fig. 2A–E), demonstrating that VMP1cKO mice developed progressive motor deficits. In addition, video recording at the 63rd postnatal day showed that VMP1cKO mice displayed an unbalanced, trembling walking pattern, which was not observed in VMP1cWT mice (Supplementary Videos 1 and 2). Furthermore, the rotarod task showed that VMP1cKO mice stayed less time on the rotating rod (Fig. 2F), suggesting a progressive pattern of impaired motor coordination in VMP1cKO mice. In addition, we performed a Y-maze test and tail suspension test at the 60th postnatal day and found no significant difference, suggesting no cognitive impairment in VMP1cKO mice (Fig. 2G). Moreover, the immobility time was comparable between VMP1cKO and VMP1cWT mice in the tail suspension test (Fig. 2H)18,19.

A A simplified scheme of the experimental timeline for the administration of TAM to both VMP1cWT and VMP1cKO mice. B IFC analysis for VMP1 expression in the SNc was performed by using an antibody against VMP1 (Green) together with TH (Red) in VMP1cWT and VMP1cKO mice. The nuclei were labeled with DAPI (Blue). Scale bar, 10 μm. C The percentage of TH+ and VMP1+ neurons in the SNc of VMP1cWT and VMP1cKO mice (N = 3 mice per genotype). D WB analysis for the VMP1 expression levels in the midbrain and striatum. E Quantification of WB analysis of (D) (N = 3 mice per genotype). C Was analyzed by using Student’s t-test. E Was analyzed by using two-way ANOVA followed by Sidak’s multiple comparisons test. Data were represented as mean ± SEM. ***p < 0.001, ****p < 0.0001.

Total traveled distance (A), vertical time (B), vertical counts (C), stereotypic time (D), and stereotypic counts (E) were presented, respectively (N = 24 mice per genotype). F The latency to fall from rotarod was recorded from VMP1cWT and VMP1cKO mice from the 45th to 65th postnatal day (N = 23–24 mice per genotype). G The mice entered a, b, or c arm were recorded, and the Y-maze alteration rate was calculated (N = 16–20 mice per genotype). H The immobile time was recorded in the tail suspension test (N = 9–12 mice per genotype). A–F were analyzed by using two-way ANOVA followed by Sidak’s multiple comparisons test. G, H were analyzed by using Student’s t-test. Data were represented as mean ± SEM. ****p < 0.0001.
VMP1cKO mice display a profound mDAergic neuronal loss and enlarged axonal terminals
To analyze the correlation between motor deficit and loss of mDAergic neurons, IFC staining of TH, a representative marker for DAergic neurons, was performed for the quantification of mDAergic neurons. The number of mDAergic neurons in the SNc and VTA from the 10-week-old VMP1cKO mice was markedly decreased by 59% and 20%, respectively, compared to the age-matched VMP1cWT mice (Fig. 3A–C). Furthermore, the fiber density of mDAergic axonal terminals sharply declined along with the presence of the massive enlargements (Fig. 3D–F), indicating that VMP1 deficiency in the mDAergic neurons leads to neuronal damage in both soma and axons. Dopamine (DA), the distinct catecholamine neurotransmitter synthesized by DAergic neurons, was measured by HPLC (Fig. 3G–I)20,21; TH, a key enzyme for DA synthesis, was examined by WB (Supplementary Fig. 2). The mean concentration of DA was notably reduced from 151.6 pg/μL in the midbrain of VMP1cWT mice to 36.85 pg/μL in that of VMP1cKO mice (Fig. 3H), and from 1287 pg/μL in the striatum of VMP1cWT mice to 340 pg/μL in that of VMP1cKO mice (Fig. 3I). Correspondingly, the TH protein levels in the midbrain and striatum were both decreased by 52% and 58% in VMP1cKO mice compared with VMP1cWT (Supplementary Fig. 2). Together, our data reveal the severe dysfunction of mDAergic nervous system in VMP1cKO mice.

A The IFC staining for the mDAergic neurons was performed by using the antibody against TH (red) in VMP1cWT and VMP1cKO mice. Scale bar, 100 μm. Scale bar of the high-magnification images, 15 μm. Quantitation of TH+ cells in the SNc (B) and VTA (C) from VMP1cWT and VMP1cKO mice, respectively (N = 3 mice per genotype). D IFC staining for the axonal density of the mDAergic neurons was performed by using the antibody against TH (green) in VMP1cWT and VMP1cKO mice. The nuclei were labeled with DAPI (blue). Scale bar, 500 μm. Scale bar of the high-magnification images, 25 μm. E Quantitation of TH+ fiber density in the striatum from VMP1cWT and VMP1cKO mice (N = 3 mice per genotype). F Mean number of the enlarged axon terminals (area >0.5 μm2) per 0.05 mm2 perspective (N = 3 mice per genotype). G The peak time of DA flux. DA concentration was assessed by HPLC in the midbrain (H) and striatum (I), respectively (N = 7 mice per genotype). Data were analyzed by using Student’s t-test and were represented as mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Necroptosis in the mDAergic neurons of VMP1cKO mice
Since VMP1cKO mice displayed a profound mDAergic neuronal loss, we tried to further elucidate the mechanism underlying cell death22. Necroptosis has been described as a highly regulated form of necrosis and considered to be a highly pro-inflammatory mode of cell death23,24,25. When necroptosis is induced, receptor-interacting protein kinase-3 (RIPK3) is activated by phosphorylation, and the activated RIP3 phosphorylates mixed lineage kinase like (MLKL) at the threonine 349, serine 345, and serine 347 residues26,27. Based on these previous studies, we thereby examined whether the mDAergic neuronal loss upon VMP1 deficiency is associated with necroptosis. Using the antibody against phospho-RIPK3 (Thr231/Ser232) and MLKL (phospho S345), we performed IFC staining and found that in the mDAergic neurons of VMP1cKO mice, p-RIPK3 was dramatically concentrated and formed discrete puncta at the cytoplasm (Fig. 4A, B). Correspondingly, p-MLKL was significantly accumulated and formed large puncta at the nucleus and cytoplasm of the mDAergic neurons (Fig. 4C, D), indicating necroptosis is induced in the mDAergic neurons due to VMP1 deficiency. Next, we examined the activation of caspase-3 in the midbrain region, showing that the mDAergic neurons in VMP1cKO mice displayed concentrated cleaved-caspase3 (Asp175) puncta, while the caspase-3 activation was barely observed in VMP1cWT mice (Fig. 4E, F). These results indicate that besides necroptosis, apoptosis might be also involved in the neuronal death caused by VMP1 deficiency.

Double-label immunofluorescence of p-RIP3 (Thr231/Ser232) (green) and TH (red) in the mDAergic neurons of VMP1cKO and VMP1cWT mice. Scale bar, 10 μm. B The proportion of TH+ neurons with p-RIPK3 puncta was quantified (N = 3 mice per genotype). C Double-label IFC staining of p-MLKL (phospho S345) (green) and TH (red) in the SNc of VMP1cKO and VMP1cWT mice. Scale bar, 10 μm. D The proportion of TH+ neurons with p-MLKL puncta was quantified (N = 3 mice per genotype). E Double-label immunofluorescence of cleaved-caspase3 (Asp175) (green) and TH (red) in the SNc of VMP1cKO and VMP1cWT mice. Scale bar, 10 μm. F The proportion of TH+ neurons with cleaved-caspase3 puncta was quantified (N = 3 mice per genotype). Data were analyzed by using Student’s t-test and were represented as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001.
Disrupted autophagic flux in the mDAergic neurons of VMP1cKO mice
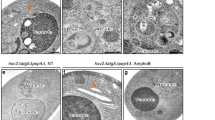
A constitutive level of autophagy is important for maintaining cellular homeostasis under normal conditions. Thus, we focused on whether VMP1 deficiency disrupts the autophagic flux in the mDAergic neurons. A large number of p62 puncta was accumulated in the soma of the mDAergic neurons of VMP1cKOmice (Fig. 5A, B). Similarly, large LC3+ puncta were concentrated in the cell body of the mDAergic neurons in VMP1cKO mice but not in VMP1cWT mice (Fig. 5C, D), suggesting that LC3-labeled autophagic luncta accumulate in the mDAergic neurons resulting from VMP1 deficiency. Double-staining results revealed that LC3 puncta were normally co-localized with lysosomal-associated membrane protein 1 (LAMP1)-labeled lysosomes in the mDAergic neurons of fasted VMP1cWT mice (Fig. 5E–H). However, such co-localization was disrupted in matched fasted VMP1cKO mice (Fig. 5E–H), showing most LC3 signals failed to merge with LAMP1 signals. Immunoblotting results also confirmed that p62 and LC3 levels were both increased in the midbrain of VMP1cKO mice compared with VMP1cWT mice (Fig. 5I–K). Neither LAMP1 nor Beclin1 protein expression level was altered when compared between the midbrain of VMP1cKO mice and VMP1cWT mice (Fig. 5L, M). Furthermore, TEM analysis showed an increase in the number of autophagosomes in VMP1cKO mDAergic neurons (Fig. 5N, O), indicating that autophagic flux at the fusion stage is impaired in the VMP1-deficient mDAergic neurons, in accordance with the previous report4. Although the fusion of the autophagosome with lysosome was blocked upon VMP1 depletion (Fig. 5E–H), the morphological alteration of VMP1 deficiency on the lysosome was not identified (Fig. 5P, Q). Degradative autophagic vacuoles (AVd) (including autolysosomes and amphisomes) typically have monolayer membrane, and usually contain electron-dense cytoplasmic material and/or organelles at various stages of degradation28. We found a substantial number of AVd in the mDAergic neurons from the VMP1cWT mice (Fig. 5R). However, in VMP1cKO mice, we hardly found AVd but saw abundant LVSs in irregular morphology with enlarged diameters (>500 nm) and clear content of lumenal material (Fig. 5S), indicating the disruption of degradation steps.

A IFC staining for p62 in the SNc was performed by using the antibody against p62 (green) together with TH (red). The nuclei were labeled with DAPI (Blue). Scale bar, 250 μm. Scale bar of the high-magnification images, 10 μm. B The proportion of TH+ cells with p62 puncta (>1 μm2) was quantified (N = 3 mice per genotype). C IFC staining for LC3 in the SNc was performed by using the antibody against LC3 (Green) together with TH (Red). The nuclei were labeled with DAPI (Blue). Scale bar, 250 μm. Scale bar of the high-magnification images, 10 μm. D The proportion of TH+ cells with LC3 puncta (>0.5 μm2) was quantified (N = 3 mice per genotype). E Double-label immunofluorescence of LC3 (green) and LAMP1 (red) in the mDAergic neurons. Scale bar, 10 μm. F The number of LAMP1+ puncta (>0.1 μm2) per TH+ neuron (N = 3 mice per genotype). G Quantification of the area of LC3+ puncta per TH+ neuron (N = 3 mice per genotype). H The proportion of LC3+ puncta co-localized with LAMP1 was quantified (N = 3 mice per genotype). I WB analysis for p62, LC3, LAMP1 and Beclin1 protein expression levels in the midbrain and striatum. GAPDH as an internal reference. J–M Quantification of p62, LC3II, LAMP1 and Beclin1 relative to GAPDH was shown, respectively (N = 3 mice per genotype). The representative TEM images of the AP (N), Ly (P), as well as AVd and LVSs (R) in cells. Scale bar, 0.5 μm. The quantification of the number of AP per cell section (O), the diameters of Ly (Q), and the number of LVSs per cell section (S) was shown. B, D, F, G, H, O, Q, S were analyzed by using Student’s t-test. J–M were analyzed by using two-way ANOVA followed by Sidak’s multiple comparisons test. Data were represented as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001. mid midbrain, stri striatum, Ly lysosome, AP autophagosome, AVd degradative autophagic vacuoles, LVSs large vacuolar-like structures.
Defects of mitochondria in the mDAergic neurons of VMP1cKO mice
Next, we determined the impact of VMP1 deficiency on the organelles of the mDAergic neurons. TEM analysis was also performed to assess the mitochondrial morphology. A lot of spherical mitochondria were found in the mDAergic neurons of VMP1cKO mice but not in VMP1cWT mice (Fig. 6A). Furthermore, compared to the VMP1cWT mice, the mean perimeter of the mitochondria was dramatically increased, an indication of swollen, in the mDAergic neurons of VMP1cKO mice (Fig. 6B). Additionally, more than 90% of the mitochondria cristae in the mDAergic neurons of VMP1cKO mice were reduced, broken or even disappeared, indicating that mitochondria are damaged upon VMP1 deficiency in the mDAergic neurons (Fig. 6C).

A The representative TEM images of the observed mitochondria. Scale bar, 0.5 μm. B Quantification of the mean perimeter of mitochondria (N = 3 mice per genotype). C The proportion of mitochondria with damaged cristae was quantified (N = 351 mitochondria collectively counted from 3 VMP1cWT mice, N = 291 mitochondria collectively counted from 3 VMP1cKO mice). Data were analyzed by using Student’s t-test and were represented as mean ± SEM. *p < 0.05, ****p < 0.0001. Mito mitochondria.
Altered rough ER structure in the mDAergic neurons of VMP1cKO mice
We next analyzed the rough ER (RER) structure and found a significant change in the morphology of RER in VMP1cKO mice compared with VMP1cWT mice (Fig. 7A). The mean width of RER tubules doubled in the mDAergic neurons of VMP1cKO mice (83.6 nm) compared to that in VMP1cWT mice (40.8 nm) (Fig. 7B), and the proportion of RER tubules (>100 nm) was increased from 0.05% in the mDAergic neurons of VMP1cWT mice to 16.65% in VMP1cKO mice (Fig. 7C). These results indicate that the mDAergic neurons suffer from severe RER damage because of VMP1 deficiency. It is known that SEC31A (SEC31 Homolog A, COPII coat complex component) is involved in vesicle budding from the ER and ER–Golgi transport29. We found that SEC31A was accumulated to form large, discrete puncta in the mDAergic neurons of VMP1cKO mice (Fig. 7D, E). Correspondingly, the mRNA levels of SEC16A (SEC16 Homolog A, ER export factor from ER to Golgi), SEC31A, and SEC31B (SEC31 Homolog B, COPII coat complex component) were significantly upregulated in VMP1cKO mice (Supplementary Fig. 3). These results suggest that vesicle budding of ER and ER–Golgi transport are abnormal upon VMP1 deficiency.

A The representative TEM images of the observed RER. Scale bar, 0.5 μm. B The mean width of RER tubules was shown (N = 487 RER collectively counted from 3 VMP1cWT mice, N = 500 RER collectively counted from 3 VMP1cKO mice). C The proportion of RER tubules (>100 nm) was quantified from (B). D Double-label immunofluorescence of SEC31A (green) and TH (red) in the mDAergic neurons. The nuclei were labeled with DAPI (blue). Scale bar, 10 μm. E Quantification of the number of SEC31A+ puncta (>0.1 μm2) per TH+ neuron (N = 3 mice per genotype). Data were analyzed by using Student’s t-test and were represented as mean ± SEM. **p < 0.01, ****p < 0.0001. RER rough endoplasmic reticulum, NM nuclear membrane, RER rough ER.
Accumulation of α-syn inclusion and abnormal protein aggregation in the nigrostriatal projection of VMP1cKO mice
To further clarify the potential role of VMP1 in the mDAergic neuron degeneration, we examined the aggregation of endogenous α-syn by IFC staining. We found that α-syn was diffusely present in the soma of the mDAergic neurons from the VMP1cWT mice (Fig. 8A), which is consistent with other reports that endogenous α-syn was presented in the SNc and VTA of C57Bl/6J mice30,31,32. However, in our age-matched VMP1cKO mice, the endogenous α-syn protein level in the soma was sharply reduced and no obvious presence of inclusions (Fig. 8A, B). Unexpectedly, we found that α-syn inclusions were highly accumulated at the enlarged axon terminals in the striatum of VMP1cKO mice (Fig. 8C, D), revealing the profound impact of VMP1 on the α-syn transportation at the axonal terminals of the mDAergic neurons. Our data thus provide evidence for VMP1’s role in regulating α-syn axonal transportation.

A IFC analysis for α-syn expression in the SNc was performed by using the antibody against α-syn (green) together with TH (red). The nuclei were labeled with DAPI (blue). Scale bar, 100 μm. Scale bar of the high-magnification images, 10 μm. B The proportion of α-syn+-TH+ cells in the SNc was quantified (N = 3 mice per genotype). C IFC analysis for α-syn expression in the striatum was performed by using the antibody against α-syn (green) together with TH (red). The nuclei were labeled with DAPI (blue). Scale bar, 25 μm. D Mean number of α-syn inclusions co-localized with TH in the striatum (N = 3 mice per genotype). E IFC analysis for Ub+ protein expression in the SNc by using the antibody against Ub (green) together with TH (red). The nuclei were labeled with DAPI (blue). Scale bar, 100 μm. Scale bar of the high-magnification images, 10 μm. F The proportion of TH+ neurons in the SNc with Ub+ puncta was quantified (N = 3 mice per genotype). G IFC staining for Ub in the striatum by co-staining Ub (green) together with TH (red). The nuclei were labeled with DAPI (blue). Scale bar, 25 μm. H Total area of Ub+ puncta (>0.5 μm2) per 0.05 mm2 in the striatum was quantified (N = 3 mice per genotype). Data were analyzed by using Student’s t-test and were represented as mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001.
We next examined protein aggregation in the midbrain and striatum using the antibody against ubiquitin, a classical marker of misfolded proteins. We found that large ubiquitin+ puncta were accumulated in the cytoplasm of mDAergic neurons of VMP1cKO mice, whereas such aggregations were not observed in VMP1cWT mice (Fig. 8E, F). Similarly, we also found large ubiquitin+ puncta concentrated in the striatum, but they were not significantly co-localized with axon terminal enlargements (Fig. 8G, H). These findings suggest the accumulation of misfolded proteins in both the midbrain and striatum, resulting from VMP1 deficiency.
Discussion
Increasing evidence indicates that VMP1 may act as a platform promoting the upstream autophagic events2,33,34. Extensive in vitro studies have demonstrated that the VMP1 deficiency blocks autophagic flux by suppressing the fusion of autophagosome with lysosome4. However, due to the absence of a physiological cellular context, it is unclear how these changes affect the specific neurons in vivo. Here, we thereby reported for the first time that conditional KO of VMP1 in the fully differentiated mDAergic neurons caused significant motor deficiency, such as imbalance, tremor, and ataxic walking. Meanwhile, these movement disorders were concomitant with the loss of mDAergic neurons and fibers, as well as the swollen terminals. Interestingly, the enlarged axonal puncta were highly colocalized with α-syn aggregates, suggesting the dysfunction of VMP1 may contribute significantly to α-syn-induced pathogenesis in the mDAergic neurons. At the molecular level, a proportion of the accumulated LC3II+ and p62+ aggregates was presented in the VMP1-deficient mDAergic neurons. LC3II, a standard marker for autophagosomes, is specifically associated with autophagosomes and autolysomes; p62, a classical receptor of autophagy participates in the cytoplasmic cargos’ transportation, and itself is autophagy substrate. Thus, the appearance of such LC3II+ and p62+ aggregates may indicate that VMP1 deficiency severely impairs the autophagy activity/flux in the mDAergic neurons35.
Autophagy is a key factor for keeping the homeostasis of cells by removing misfolded proteins or toxic components. VMP1 is an important component in the autophagic system in regulating interactions between the ER and the autophagic-isolation membrane. The conventional KO mice of VMP1 are embryonic lethal36. Therefore, for further mechanistic investigation, we have constructed conditionally KO mice that using TAM to induce postnatal deletion of the VMP1 gene specifically in the cells expressing DAT. Our VMP1cKO mice have about 5 weeks of survival after TAM treatment and they suffer from significant body loss at the 4th week and then die within one week with a profound mDAergic neuronal degeneration. Compared with Atg7DatCre mice that displayed a substantial loss of the mDAergic neurons at one year of age37, the VMP1cKO mice have a much more severe phenotype, nearly 60% of the mDAergic neuronal loss at 4th week following the TAM treatment, indicating that VMP1 is essential for the survival of the mDAergic neurons. As expected, we found massive Ub+ proteins accumulation and the LVSs formation in the cytoplasm of the mDAergic neurons of VMP1cKO mice, suggesting the severe impairment of protein degradation functions in the autophagic system because of VMP1 deficiency. It is likely the components of LVSs are autophagosome-related vacuoles. Thus, the accumulation of LVSs in VMP1cKO mDAergic neurons may indicate the dysfunction of local membrane trafficking and turnover. Furthermore, we have documented severely disrupted mitochondria in the mDAergic neurons of VMP1cKO mice. It is known that the removal of damaged mitochondria through autophagy, termed mitophagy, is indispensable for maintaining proper cellular functions38. Whereas, in our VMP1cKO mice autophagic flux was dramatically disrupted by the failure of the lysosome to fuse with autophagosome, resulting in the damaged mitochondria highly accumulated. These retained dysfunctional mitochondria are mainly characterized by the hallmarks of decreased ΔΨm and lower levels of OPA139. Mitochondria fusion is thought to be a less-selective process, meaning that the probability of mitochondria with lower ΔΨm to fuse is high40. Such aberrant fusion may largely affect the mitochondrial fission step and significantly contribute to the observed swelling phenotype. In addition, we have observed the extensive swollen ER in the mDAergic neurons of VMP1cKO mice, suggesting that the failure in handling of misfolded proteins is associated with the damage of several organelles. On the other hand, VMP1 is ubiquitously expressed in an extensive network of membranes, and ER establishes membrane contact sites with most organelles by signaling events and molecular trafficking. Previous studies suggest that VMP1 may be involved in a tight spatial and temporal regulations of PtdIns3P signaling and membrane source recruitment by restricting the ER–mitochondria contact sites4. Depletion of VMP1 in HeLa and Cos-7 cells causes an increase of ER–mitochondria contact sites and alters lipid and calcium exchanges between ER and mitochondria3. Such molecular re-establishment underlying cellular mechanism may consequently contribute to the ER and mitochondria morphologic defects as well.
Both the ER and mitochondria provide a membrane source for the formation of the phagophore, the initiation of autophagy41. Thus, the defects in those organelles may aggravate the autophagic-dependent pathology. Taken together, these data suggest that diverse cellular dysfunctions caused by VMP1 deficiency may collectively contribute to the mDAergic neuron loss. In particular, cell death may be processed by necroptosis via the RIPK3 phosphorylation pathway. RIPK1 and RIPK3 are both critical for forming the necrosome, a cytosolic complex required by necroptosis signaling. Dramatically concentrated p-RIPK3 was characterized in our VMP1cKO mice, indicating that VMP1-deficiency might promote the generation of necrosome. However, the linkage between autophagy induction and necroptosis still remains to be determined. Based on previous findings42, we speculate that the newly assembled necrosome may be associated with autophagosome membranes. Since a large amount of autophagosome was found to be aberrantly accumulated in VMP1cKO mice, which may trigger more necrosome forming and promote the necroptosis process. However, further detailed biochemical and IFC investigations on how autophagy interacts with necroptosis are needed in the future. Mechanistically, necroptosis is typically not associated with the activation of caspases24. However, in our VMP1cKO model higher activity of cleaved-caspase3 (Asp175) was characterized, suggesting apoptosis might cross talk with necroptosis upon the pathological process43. Therefore, underneath the cellular context, necroptosis and apoptosis may co-operate in the balanced interplay that involves autophagy.
In neurons, autophagy plays an essential role in the maintenance of axonal homeostasis, manifested by massive autophagosomes predominantly formed in the distal axons44. Whereas in our VMP1cKO mice the aberrant axonal swollen is so striking, suggesting that the impairment in the autophagic system may cause severe axonal transportation disturbance. Interestingly, a similar phenotype has been reported in the mice with Atg7 deficiency as well45. Moreover, these enlarged puncta extensively harbor α-syn inclusions, consistent with the previous report that in the brain of GBA−/− medaka α-syn inclusions were accumulated in the axonal swellings46. However, in contrast to the terminals α-syn expression, it is barely detectable in the soma of VMP1cKO mDAergic neurons. Comparably, in Atg7 conditional KO mice α-syn was aggregated in the swollen axons of TH+ neurons, but not in their cell bodies47. These data suggest α-syn may aggregate first in the axon rather than in the soma because of the axonal transportation disturbance48. It has been reported that α-syn fibrils are actively transported along microtubules both in the anterograde and retrograde directions49,50,51. We thereby speculate that the reduction of cytoplasmic α-syn in the soma may result from the defects of microtubule-based transport on terminals during the process of pathology.
In summary, VMP1 deficiency in the mDAergic neurons causes motor disorders, severe mDAergic neuronal loss, mitochondria abnormalities, and autophagy flux disruption. Strikingly, accumulation of α-syn inclusions were extensively identified in the enlarged striatal terminals of VMP1cKO mice, suggesting that VMP1 plays an essential role in the microtubule-based transport and ER–Golgi trafficking mediated by autophagy. It is speculated that the VMP1 deficiency in the nigro-striatal pathway may cause the misfolding protein aggregation in the terminals and eventually trigger the mDAergic neuron degeneration. Taken together, our findings reveal a novel role of VMP1 in modulating neuronal survival and maintaining axonal homeostasis, which suggests that our VMP1cKO mice may serve as a useful preclinical model to elucidate the mechanisms of mDAergic neurodegeneration induced by autophagy impairment.
References
Dusetti, N. J. et al. Cloning and expression of the rat vacuole membrane protein 1 (VMP1), a new gene activated in pancreas with acute pancreatitis, which promotes vacuole formation. Biochem. Biophys. Res. Commun. 290, 641–649 (2002).
Ropolo, A. et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 282, 37124–37133 (2007).
Tabara, L. C. & Escalante, R. VMP1 establishes ER-microdomains that regulate membrane contact sites and autophagy. PLoS ONE 11, e0166499 (2016).
Zhao, Y. G. et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol. Cell 67, 974–989 (2017). e976.
Calvo-Garrido, J., Carilla-Latorre, S., Lazaro-Dieguez, F., Egea, G. & Escalante, R. Vacuole membrane protein 1 is an endoplasmic reticulum protein required for organelle biogenesis, protein secretion, and development. Mol. Biol. Cell 19, 3442–3453 (2008).
Wang, P. et al. KMS1 and KMS2, two plant endoplasmic reticulum proteins involved in the early secretory pathway. Plant J. 66, 613–628 (2011).
Tenenboim, H., Smirnova, J., Willmitzer, L., Steup, M. & Brotman, Y. VMP1-deficient Chlamydomonas exhibits severely aberrant cell morphology and disrupted cytokinesis. BMC Plant Biol. 14, 121 (2014).
Ravikumar, B. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 (2004).
Komatsu, M. et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl Acad. Sci. USA 104, 14489–14494 (2007).
Parkitna, J. R., Engblom, D. & Schutz, G. Generation of Cre recombinase-expressing transgenic mice using bacterial artificial chromosomes. Methods Mol. Biol. 530, 325–342 (2009).
Chmielarz, P. et al. Dicer and microRNAs protect adult dopamine neurons. Cell Death Dis. 8, e2813 (2017).
Engblom, D. et al. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron 59, 497–508 (2008).
Cheng, J. et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 16, 2193–2205 (2020).
Xiao, Q., Yang, S. & Le, W. G2019S LRRK2 and aging confer susceptibility to proteasome inhibitor-induced neurotoxicity in nigrostriatal dopaminergic system. J. Neural Transm. 122, 1645–1657 (2015).
Tang, Y. et al. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 21, 369–380 (2014).
Baquet, Z. C., Williams, D., Brody, J. & Smeyne, R. J. A comparison of model-based (2D) and design-based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Neuroscience 161, 1082–1090 (2009).
Vaccaro, M. I., Ropolo, A., Grasso, D. & Iovanna, J. L. A novel mammalian trans-membrane protein reveals an alternative initiation pathway for autophagy. Autophagy 4, 388–390 (2008).
Cote, F., Collard, J. F. & Julien, J. P. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 73, 35–46 (1993).
Mangiarini, L. et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 (1996).
Warren, N., O’Gorman, C., Lehn, A. & Siskind, D. Dopamine dysregulation syndrome in Parkinson’s disease: a systematic review of published cases. J. Neurol. Neurosurg. Psychiatry 88, 1060–1064 (2017).
Birtwistle, J. & Baldwin, D. Role of dopamine in schizophrenia and Parkinson’s disease. Br. J. Nurs. 7, 832–834 (1998). 836, 838-841.
Galluzzi, L. et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 16, 1093–1107 (2009).
Kearney, C. J. & Martin, S. J. An inflammatory perspective on necroptosis. Mol. Cell 65, 965–973 (2017).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Welz, P. S. et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334 (2011).
Rodriguez, D. A. et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88 (2016).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016).
Tang, B. L. et al. Mammalian homologues of yeast sec31p. An ubiquitously expressed form is localized to endoplasmic reticulum (ER) exit sites and is essential for ER-Golgi transport. J. Biol. Chem. 275, 13597–13604 (2000).
Sumitomo, A. et al. A mouse model of 22q11.2 deletions: molecular and behavioral signatures of Parkinson’s disease and schizophrenia. Sci. Adv. 4, eaar6637 (2018).
Fares, M. B. et al. Induction of de novo alpha-synuclein fibrillization in a neuronal model for Parkinson’s disease. Proc. Natl Acad. Sci. USA 113, E912–E921 (2016).
Wegrzynowicz, M. et al. Depopulation of dense alpha-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol. 138, 575–595 (2019).
Wang, P., Kou, D. & Le, W. Roles of VMP1 in autophagy and ER-membrane contact: potential implications in neurodegenerative disorders. Front. Mol. Neurosci. 13, 42 (2020).
Tian, Y. et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141, 1042–1055 (2010).
Runwal, G. et al. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 9, 10147 (2019).
Jiang, P. H. et al. Expression of vacuole membrane protein 1 (VMP1) in spontaneous chronic pancreatitis in the WBN/Kob rat. Pancreas 29, 225–230 (2004).
Inoue, K. et al. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener. 7, 48 (2012).
Ding, W. X. & Yin, X. M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564 (2012).
Parone, P. A. et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 3, e3257 (2008).
Gomes, L. C. & Scorrano, L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta 1833, 205–212 (2013).
Bernard, A. & Klionsky, D. J. Autophagosome formation: tracing the source. Dev. Cell 25, 116–117 (2013).
Basit, F., Cristofanon, S. & Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 20, 1161–1173 (2013).
Nikoletopoulou, V., Markaki, M., Palikaras, K. & Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459 (2013).
Wang, Y., Song, M. & Song, F. Neuronal autophagy and axon degeneration. Cell. Mol. Life Sci. 75, 2389–2406 (2018).
Inoue, K. et al. Coordinate regulation of mature dopaminergic axon morphology by macroautophagy and the PTEN signaling pathway. PLoS Genet. 9, e1003845 (2013).
Uemura, N. et al. Viable neuronopathic Gaucher disease model in Medaka (Oryzias latipes) displays axonal accumulation of alpha-synuclein. PLoS Genet. 11, e1005065 (2015).
Friedman, L. G. et al. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J. Neurosci. 32, 7585–7593 (2012).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Volpicelli-Daley, L. A. et al. Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 25, 4010–4023 (2014).
Freundt, E. C. et al. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524 (2012).
Bieri, G., Gitler, A. D. & Brahic, M. Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis. 109, 219–225 (2018).
Acknowledgements
We thank Liaoning Provincial Center for Clinical Research on Neurological Diseases, the First Affiliated Hospital, Dalian Medical University for the research infrastructure and support. We thank Günther Schütz, David Engblom, Pierre Chambon, and Daniel Metzger for kindly gifting BAC-DATCreERT2 mouse strain. We would like to thank Professor Feng Du of Guangzhou Medical University for his professional guidance on the results of TEM, and Professor Jau-Shyong Hong, from Neurobiology Laboratory National Institute of Environmental Health Sciences, National Institutes of Health to give us a critical review and advice for this manuscript.
Funding
This work was supported by funding from the National Nature Science Foundation of China (NSFC 81771521), the National Key Research and Development Program of China (2016YFC1306600), and Key Realm R & D Program of Guangdong Province (2018B030337001). In addition, this manuscript was also contributed in part by the Intramural Research Program of National Institute on Aging, National Institutes of Health (Z01-AG000944, AG000928).
Author information
Authors and Affiliations
Contributions
W.P. and L.W. designed the experiments. W.P., C.X., W.Y.Y. contributed to imaging experiments and data analysis. W.P., L.X., W.Y., and W.H. contributed to behavior test and data analysis. C.X., W.Y.Y., and J.C. contributed to protein preparation and data analysis. W.P., C.X., W.Y.Y., C.H., S.H. and L.W. wrote and edited the manuscript. All authors agree to be accountable for the content of the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethics statement
Animal care and procedures were carried out in accordance with the Laboratory Animal Care Guidelines approved by the Institutional Animal Care Committee at Dalian Medical University. The protocol was approved by the Institutional Animal Care Committee at Dalian Medical University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Verkhratsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, P., Chen, X., Wang, Y. et al. Essential role for autophagy protein VMP1 in maintaining neuronal homeostasis and preventing axonal degeneration. Cell Death Dis 12, 116 (2021). https://doi.org/10.1038/s41419-021-03412-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03412-5
This article is cited by
-
Palmitoylation of vacuole membrane protein 1 promotes small extracellular vesicle secretion via interaction with ALIX and influences intercellular communication
Cell Communication and Signaling (2024)
-
α-Synuclein reduces acetylserotonin O-methyltransferase mediated melatonin biosynthesis by microtubule-associated protein 1 light chain 3 beta-related degradation pathway
Cellular and Molecular Life Sciences (2024)
-
Terahertz Irradiation Improves Cognitive Impairments and Attenuates Alzheimer’s Neuropathology in the APPSWE/PS1DE9 Mouse: A Novel Therapeutic Intervention for Alzheimer’s Disease
Neuroscience Bulletin (2023)
-
Protein clearance strategies for disease intervention
Journal of Neural Transmission (2022)
-
The essential role of transcription factor Pitx3 in preventing mesodiencephalic dopaminergic neurodegeneration and maintaining neuronal subtype identities during aging
Cell Death & Disease (2021)
