
Abstract
Vital osteocytes have been well known to function as an important orchestrator in the preservation of robustness and fidelity of the bone remodeling process. Nevertheless, some key pathological factors, such as sex steroid deficiency and excess glucocorticoids, and so on, are implicated in inducing a bulk of apoptotic osteocytes, subsequently resulting in resorption-related bone loss. As much, osteocyte apoptosis, under homeostatic conditions, is in an optimal state of balance tightly controlled by pro- and anti-apoptotic mechanism pathways. Importantly, there exist many essential signaling proteins in the process of osteocyte apoptosis, which has a crucial role in maintaining a homeostatic environment. While increasing in vitro and in vivo studies have established, in part, key signaling pathways and cross-talk mechanism on osteocyte apoptosis, intrinsic and complex mechanism underlying osteocyte apoptosis occurs in various states of pathologies remains ill-defined. In this review, we discuss not only essential pro- and anti-apoptotic signaling pathways and key biomarkers involved in these key mechanisms under different pathological agents, but also the pivotal role of apoptotic osteocytes in osteoclastogenesis-triggered bone loss, hopefully shedding new light on the attractive and proper actions of pharmacotherapeutics of targeting apoptosis and ensuing resorption-related bone diseases such as osteoporosis and fragility fractures.
Similar content being viewed by others

Facts
• Osteocyte apoptosis under various pathological agents has a link to resorption-associated bone fragility and bone loss.
• The morphology and vitality of osteocytes is likely an indicator of bone quality.
• A crucial cross-talk mechanism between autophagy and osteocyte apoptosis exists in maintaining osteocytic vitality.
• Key pathological agents and intrinsic signaling pathways have important roles in initiating and mediating osteocytic pro- and anti-apoptotic mechanisms.
• Apoptotic osteocytes function as a direct and indirect role in developing resorption-related bone loss.
Open questions
• Are underlying molecular mechanisms the same whereby osteocyte cell death occurs upon various pathological agents?
• How do autophagy and osteocyte apoptosis interplay in maintaining the osteocytic vitality?
• What roles does osteocyte apoptosis have in developing the resorption-related bone loss?
Introduction
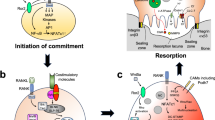
The normal rate of osteocyte apoptosis thought as the default fate for osteocytes could result in the removal of the “dead” bone through autophagic programmed cell death, which permits the self-renewal of the bone cells and preserving of bone strength1,2. Nevertheless, a bulk of apoptotic osteocytes are implicated in several key pathological conditions, including aging3, fatigue/microdamage4, unloading/disuse5, excess glucocorticoids (GCs)6, estrogens (Es) or androgens (As) deficiency7, and inflammation8, which are correlated with decreased bone mineral density (BMD) and increased bone loss. Indeed, targeted ablation of osteocytes was shown to induce osteoporosis with the deterioration of bone microstructure and mechanotransduction in the diphtheria toxin receptor (DTR)-9.6 kb transgenic mice, which mimicked the aging skeleton9. As well known, bone has a fascinating but complex hierarchical structure, whereas, osteocytes function as a pivotal role in orchestrating the preservation of bone mass and efficient load bearing9,10. Currently, it is widely accepted that the pulsatile fluid flow (PFF) around osteocytes may cause fluid shear stress (FSS) which affects osteocytic metabolism and vitality. Accordingly, the osteocytic gap junctions via connexin43 (Cx43) hemichannel proteins control both intercellular communication and mechanotransduction in the bone through affecting the PFF within the lacunar-canalicular network (LCN)11. Notably, nutrient and oxygen are transported by PFF within LCN to preserve the vitality of osteocytes, which is of paramount importance in maintaining bone mechanosensation and mechanotransduction10,11. Intriguingly, it was found that PFF-induced [Ca(2+)]i oscillations could be attenuated due to estrogen deficiency in MLO-Y4 osteocytes, which ultimately alter osteocyte function and differentiation12. Nevertheless, how osteocytes orchestrate the maintenance of bone mass and efficient load-bearing is not yet completely understood. Also, accumulating evidence indicates that differences in osteocyte mechanosensitivity are strictly controlled by osteocytic morphology and the lacunae size13. This is supported by the fact that the difference in osteocytic morphology exists in various states of pathologies. For instance, osteocytes were relatively elongated and small in the skeleton with osteoarthritis, whereas relatively round and large in osteopenic skeleton13. Apparently, the difference in osteocytic morphology and lacunae size would impact the cell to cell communication, subsequently reducing the structural integrity and vitality of osteocytes14,15. Accordingly, it is no surprise that changes to the size and density of osteocytes could be correlated with the deterioration of biomechanical property of bone tissue, consequently contributing to bone regeneration failure, as well as bone loss16. Specifically, irrevocable and cumulative effects, due to osteocyte apoptosis, could occur through disruption of osteocytic LCN and suppression of the repair of bone microfractures, thereby resulting in the increase of bone fragility, or maybe osteoporosis14,15. Based on the above reasons, the morphology of osteocytes is likely an important structural marker of osseointegration in adapting mechanoresponsivity of bone to mechanical loading, as well as an indicator of bone quality. Yet, the intrinsic mechanism underlying how osteocytic morphology affects bone architecture and function is complex and needs to be further investigated.
It is intriguing that increasing experimental evidence has suggested that there exists an intrinsic cross-talk mechanism between cell death and autophagy, in which pro-apoptotic biomarkers could participate in autophagy or vice versa17,18,19,20. Also, it was suggested that cellular stress causes the displacement of B-cell lymphoma/leukemia 2 (BCL2) from Beclin-1 and BCL2-associated X protein (BAX), thereby triggering autophagy and apoptosis, respectively18 (Table 1). Based on this reason, the imbalance between apoptosis and autophagy might give rise to undesirable pathophysiological consequences. As well known, autophagy is a programmed cell survival mechanism, whereby autophagy might protect the amount of osteocyte cellular projections and retain endoplasmic reticulum and mitochondria in osteocytes21. Meanwhile, it is referred as a “double-edge sword” implicated in both protecting cells and enhancing the cell death21,22,23. It should be kept in mind that autophagy can be triggered by such stress factors as hypoxia, starvation, and increased oxidative stress24,25. Specifically, the osteocytes responded with autophagy in an attempt to “save themselves”. However, higher, or more prolonged stress could result in extensive recycling of damaged organelles and the accumulation of autophagosomes, eventually evoking cell death or apoptosis26. In fact, the communication of autophagy with apoptosis could be undertaken by the interplay between apoptotic proteins (e.g., cleaved-Caspase-3) and autophagic proteins (e.g., Beclin-1) (Table 1). This is manifested by the fact that the reduction of beclin-1-immunolabeled osteocytes was in parallel with the increase of cleaved-Caspase-3-immunolabeled osteocytes, thus weakening its anti-apoptotic activity27. In agreement with this notion, ovariectomized (OVX)-induced osteocyte apoptotic has been observed to be accompanied by a reduced Beclin-1 and microtubule-associated protein light chain 3A (MAP LC3A)-immunolabeled osteocytes and increased p62-positive osteocytes, which is abolished by the Es replacement. All these findings indicate that the intrinsic cross-talk mechanism between osteocyte autophagy and apoptosis has a crucial role in mediating cell metabolism and preserving osteocytic homeostasis28 (Fig. 1).

Pro-apoptotic stimuli that stimulate death receptors and mitochondria-mediated apoptotic pathways give rise to the triggering of pro-survival autophagy-related biomarkers including Beclin-1 and BCL-2. In the process of continued exposure to apoptotic stimuli, phosphorylation of BCL-2, which separates from Beclin-1, translocates to mitochondria and sensitizes cells to apoptotic signals and blocks BCL-2 from suppressing pro-apoptotic biomarkers, thereby enhancing cells apoptosis. This demonstrates a mechanism of a positive-feedback loop for amplifying the death of cells.
All in all, despite the progress made in our understanding of the mechanism of osteocyte apoptosis and its pivotal role in resorption-related bone diseases, numerous key aspects, such as therapeutic strategies that prevent osteocyte apoptosis under pathological conditions in humans, remain enigmatic. Therefore, it might be necessary to utilize novel approaches to elucidate the findings in humans, such as the application of human-derived bone cells and immortalized bone cell lines with CRISPR-Cas9 knockout. Until recently, it has been reported that the establishment of two novel osteogenic cell lines (OmGFP66 and OmGFP10) forms 3D bone-like structures with an extensive interconnected LCN structure that mimics bone in vivo29. In addition, a modified method to isolate primary human osteocytes from bone also has been reported to significantly improve the yield of osteocytes30. The elucidation of mechanisms by more selective cells and models may have tremendous implications to inform clinical studies in humans to alleviate osteocyte apoptosis under pathologies and resorption-related bone diseases. In the present review, we summarize osteocytic pro- and anti-apoptotic mechanisms under key different pathological agents and roles of apoptotic osteocytes for a better understanding of the regulatory landscape on osteocytic life and death.
The roles of pathological agents in osteocyte apoptosis
Aging and senescence
Bone aging is characterized by increased “osteocyte loss”, which is associated with changes in osteocytic LCN with respect to the shape and number density31. Also, it was concluded that the capability to respond and repair is diminished in aged bone cells32,33. Morphologically, the disruption of osteocytic LCN with age decreases the provision of nutrients for osteocytes, attenuates their mechanoresponsiveness, and ultimately lead to increased bone loss and bone fragility in the elderly14. Indeed, the shape of osteocytes, with aging, will be smaller and more spherical. Moreover, LCN density will have a majority of the reduction in the human cancellous bone16, human iliac crest cortical bone34, and murine femoral cortical bone35. At large, these adverse outcomes could originate from osteocyte cell death and subsequent mineralization of osteocytic lacuna referred to as micropetrosis16. Recent accumulating data suggest that osteocyte death by apoptosis and the prevalence of empty lacunae are implicated age-related bone abnormalities3,36,37 (Table 2). It seems that the reduction of physical activity could result in, with aging, skeletal unloading to a certain extent, which gives rise to apoptosis of osteocytes38,39,40. Considering aging is commonly concomitant with increased bone loss and bone fragility, it is proposed that changes in osteocytes shape and LCN density could be correlated with an altered capability to respond to mechanical loads, which is of great importance in the preserving of bone mass and architecture31. Accordingly, due to aging-induced osteocyte apoptosis, the decrease in the number of osteocytes and the accumulation of mineralization in bone lacuna disturb the bone remodeling process, eventually making bone more susceptible to fragile fracture36,37 (Table 2). Of note, the osteocyte, one of the “permanent” cells of bone, regulates cell-to-cell communication of signals via Cx43 gap junctions to maintain cell survival, whereas, production of phosphorylated Cx43, prostaglandin E2 (PGE2), and nitric oxide (NO) would decrease with aging41,42,43,44 (Table 1). It should be emphasized that miR21 deletion in miR21fl/fl bones increased apoptosis-related gene expression, such as phosphatase and tensin homolog (PTEN), in contrast, a miR21 analog prevented apoptosis in Cx43def osteocytes, indicating that disconnection of the Cx43/miR21 pathway leads to aging-induced osteocyte apoptosis41 (Fig. 2). In addition, it was shown that age-related changes that occur in mitochondrial senescence or apoptosis of osteocytes comprise the reduction of mitophagy and increased uncoupling of the mitochondrion, as well as nuclear fission and fusion and increased superoxide production45,46. Importantly, oxidative stress and resultant DNA damage (e.g., p53 and p66Shc), due to excessive production of reactive oxygen species (ROS), is regarded as a crucial pathway in the aging process of many cell lines including bone cells47,48 (Fig. 1). This is in line with the findings that AP20187, an anti-senescent drug, led to the significant reduction of both p16Ink4a mRNA expression (by −59%) and enhanced green fluorescent protein (EGFP) mRNA expression (by -48%) in bone, compared with that in vehicle-treated mice, respectively, which is consistent with the clearance of both senescent cells and ensuing age-related bone loss3. Taken together, it is conceivable to assume that aging-induced senescence and apoptosis of osteocytes could result from the initiation of excessive oxidative stress, ensuing activation of p53, mitochondrial membrane rupture and DNA damage, as well as unleashing of cytochrome C49.

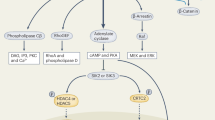
These signaling pathways, including pro-apoptotic pathways (red arrows) and anti-apoptotic pathways (green arrows), are closely related to increased bone resorption and decreased bone formation.
Unloading, nutrition deprivation, and hypoxia
As well known, osteocyte apoptosis could be decreased and bone accrual could be enhanced upon mechanical stimulation. Instead, skeletal weightlessness or unloading could result in the reverse consequence on both sides. On one hand, the data analysis suggested that apoptotic osteocytes were reduced by 40% upon short periods of mechanical stimulation, which led to peak compressive strains of 3000–4000 microstrain, compared with the unloading control38. It should be emphasized that FSS and muscle mass have been shown to be of great importance in the mechanical control of osteocyte apoptosis in vitro38,50. Similarly, the in vivo application of strains in human bone resulted in the reduction of apoptotic osteocytes, whereas unloading led to high levels of apoptosis51. On the other hand, due to the loss of stimuli to preserve the viability of osteocytes, unloading/disuse might contribute to more senescence and apoptosis in osteocytes. This has been manifested in spaceflight studies with rodents that help identify the pivotal role of mechanical force in maintaining osteocytic vitality. Since the effect of the decreased muscle activity during unloading or disuse, the reduction or disruption of nutrient supply could eventually result in the lipid accumulation in both osteoblasts and osteocytes5,38 (Table 2). Importantly, it has been assumed that the loss of mechanosensitivity, the resulting uncoupling of bone remodeling, and the generation of the senescence-associated secretory phenotype (SASP) may mainly result from osteocytic senescence and apoptosis, whether it is induced directly or indirectly during unloading38,51. However, it needs further research to identify the role of unloading or disuse on osteocytic senescence and apoptosis. Increasing evidence shows that osteocyte apoptosis and ensuing bone loss are both increased in both trabecular and cortical skeleton upon unloading in mice models5,38, which indicates that osteocyte apoptosis might have a pivotal role in regulating the adaptive response of the skeleton to changes in loading. Intriguingly, osteocyte apoptosis, during hindlimb unloading, does not occur nearer to the periosteal surface, the reason for which is likely that the fluid transport system still operates more effectively in disuse due to muscle contractions50. Furthermore, more apoptotic osteocytes, in disuse, occur near the endocortical surface than in the deep bone matrix, which may suggest that osteocytes near the endocortical surface are more in demand for growth factors and survival cytokines52. It is noteworthy that the SOST- mice (the ablation of SOST gene) were resistant to disuse-induced bone loss, suggesting that sclerostin (SOST) mediates skeleton response to mechanical unloading through antagonizing Wnt/β-catenin signaling53. Alternatively, PFF could ameliorate disuse-induced apoptosis in osteocytic MLO-Y4 cultures, dependent on, at least partially, stimulation of nitric oxide synthase (NOS) via alterations in Caspase-3 and B-cell lymphoma-2 (BCL-2) gene expression54, and the involvement of mitogen-activated protein kinases (MAPKs) and initiation of the integrin/Src/extracellular-regulated protein kinases (ERK) pathway to maintain osteocytic viability55 (Fig. 2). Strikingly, PGE2, produced by osteocytes upon mechanical loading, may protect these osteocytes against apoptosis56. Nevertheless, The precise mechanism underlying how unloading/disuse induce osteocyte apoptosis is not yet completely understood. In addition, it was reported that integrins, tethering osteocytes to the canalicular wall, function as a mediator in the interaction of osteocytic processes with extracellular matrix in vivo. Hence, it has been assumed that PFF in osteocytic LCN could give rise to tension on the integrins, which in turn stimulate integrin signaling to prevent osteocyte apoptosis55,57. Also, it was found that the level of irisin, a peptide cleaved from Fndc5, is increased in the circulation during and following exercise in mice and humans, and its receptor, an integrin alphaV/beta 5 (αV/β5), is characterized in osteocytes58. Interestingly, irisin was shown to inhibit apoptosis through regulating the expression of Caspase-9 and -3 in MLO-Y4 cells and in cortical bone of osteoporotic murine models57. Admittedly, Cx43, as a docking platform for signal transduction, is referred to as “quality control checkpoint” in preserving the mechanosensation and mechanotransduction of osteocytic LCN41,42,43. Notably, It has been shown that fibroblast growth factor 7 (FGF7) can increase Cx43 expression and promote gap junction elongate through inducing the accumulation of cytoplasmic β-catenin and partial nuclear translocation59 (Table 1). As described above, the intact LCN is necessary for nutrient and oxygen exchange for osteocytes. Thus, the decrease of PFF during disuse may lead to disruption of LCN, as well as localized hypoxia in the bone. Of note, growth differentiation factor 15 (GDF15), secreted from adjacent osteocytes upon disuse and/or ischemia could function as an essential role in the promotion of osteoclastogenesis-triggered bone loss60. Accordingly, it is assumed that oxygen deprivation might be one main cause of the osteocyte apoptosis during unloading/disuse. This is in line with the notion that loss of mitochondrial ATP, production of reactive oxygen species (ROS), and decrease of intracellular PH (PHi) could result in cellular damage or even cell death once hypoxia and ischemia61. Thus, it is not surprising that mechanical stimuli are likely to promote oxygen diffusion through the movement of PFF around osteocytes, which would prevent hypoxia-induced osteocyte apoptosis. Intriguingly, a large amount of cell death may be by apoptosis, significantly precede the expression of oxygen-regulated protein 150 (ORP150) mRNA62, while expression of the pro-apoptotic BCL2 and adenovirus E1B19 kDa interacting protein 3 (BNIP3) is also dramatically upregulated during hypoxia60. It has been shown that activation of hypoxia-inducible factor-1 (HIF-1) is likely to directly stimulate the expression of BNIP3 upon hypoxia, similar to the mechanism of hypoxic induction of glycolytic enzymes (GE), vascular endothelial growth factor A (VEGF-A), and erythropoietin (EPO)62. It should be emphasized that ORP150 (150 kDa) deeply embedded osteocytes, a novel endoplasmic reticulum-associated chaperone62, has been identified to be highly expressed upon hypoxia for the formation of dendritic processes, and protection within a hypoxic environment63, which could have a pivotal role on protecting osteocytes from oxygen deprivation. Yet, to date, the normal scope of concentration of oxygen maintaining osteocytic vitality in LCN is still unclear. Also, the role of BNIP3 in osteocyte apoptosis needs further investigation.
Microdamage and plasma membrane disruption
Apoptosis of osteocytes can also be induced by fatigue loading, as in the case of unloading4,64. The fatigue may give rise to the microdamage in the skeleton mineralized matrix, and ensuing larger bone lesions, ultimately leading to obvious fracture4,64. Data analysis has shown that strains of 8000 microstrains enhanced a remarkably increased osteocyte apoptosis in ulnae of rodents65. Similarly, it has been indicated that osteocytes will undergo apoptosis once the fatigue induces bone structural microdamage upon overloading4,64. All these results strongly suggest that a healthy osteocytic LCN might be of great importance in stimulating skeleton remodeling responses and/or protecting against damage, which aims to preserve bone mass and bone strength. Nevertheless, the intrinsic mechanism underlying how osteocytes die in response to microdamage or how microdamage occurs after osteocyte cell death, or both, is still ill-defined. Of interest, osteocytes are prone to give rise to apoptosis in the site close to microdamage, which is correlated with elevated expression of the pro-apoptotic biomarkers (i.e., Caspase-3, BAX). Unlike this, the expression of the anti-apoptotic protein (e.g., BCL2) is increased farther away from the damage focus66 (Table 1), implying that the cross talk mechanism between pro- and anti-apoptotic signals pathways has a central role in avoiding widespread cell death and maintaining bone homeostasis. In fact, the integrity of osteocytic LCN will be disrupted once microcracks occur, thus leading to the rupture of dendritic processes, which indicates that some detrimental agents including the disruption of PFF, loss of nutrients, hypoxia-induced oxidative stress, and destroy of the Cx43 gap could cooperatively give rise to osteocyte apoptosis64 (Table 2). Until recently, the interesting evidence indicates that tiny tears of the osteocytic membrane (from nanometers to microns), referred to as plasma membrane disruptions (PMD), are generated upon mechanical loading, which activates the osteocytic calcium signaling pathway and stimulates the upregulation of c-fos associated with the mechanoresponsivity. As a result, the ability of bone to translate mechanical stimulus into bone formation might be impaired due to loss of slower-repairing osteocytes and elevated mineralized osteocyte lacunae with aging67. Importantly, vitamin E, an antioxidant of interest in the musculoskeletal system, functions as an essential mediator in control of PMD-driven osteocytic adaptation and mechanoresponsivity. It has been corroborated in vitamin E-deficient diet (VEDD) mice model that the formation of PMD induced by oxidative stress was increased due to vitamin E deficiency, therefore impairing osteocyte survival68. In this line, the upregulation of cellular communication network factor 2 (CCN2) in osteocytes due to excessive mechanical stress is frequently addicted to osteocyte apoptosis through extracellular signal-regulated kinase 1/2 (ERK1/2) pathway via binding to integrin αVβ369 (Fig. 2). Indeed, CCN2, as an essential growth factor, is well known to remarkably mediate the development of osteogenesis including endochondral ossification69. Based on these findings, it is conceivable that CCN2 might be upregulated in osteocytes, but not produced due to the aging-related reduction of osteocytic processes, as mechanical loading was exerted on mature osteocytes originated from the elderly mice. Consequently, apoptosis signalings might be activated since CCN2 is accumulated into mature osteocytes69.
Sex steroids deficiency and oxidative stress
Es/As, as a systemic hormone, has a crucial role in the maintenance of bone remodeling homeostasis7,70. Indeed, osteocyte apoptosis and bone loss are frequently linked with women at menopause70 and animal models with Es deficiency7,71. In support of this notion, osteocyte apoptosis could, due to depletion of estrogen, be significantly induced in the alveolar process of OVX rodent model28. Likewise, because of Es deficiency, osteocyte apoptosis has been demonstrated to be dramatically increased in the long bone of OVX rodent model48,71. Intriguingly, apoptotic osteocytes are not located in sites of the cortex with younger osteocytes, but in sites of the cortex with oldest osteocytes. The difference of location is likely due to more sensitivity of the oldest osteocytes to the elevation of reactive oxygen species (ROS) resulted from Es deficiency48,72 (Table 1). In this line, in the OVX C57BL/6J rat model, it was found that increases in Casp+ (Caspase-positive) osteocytes were mostly positioned in the posterior cortex of diaphysis of a long bone, indicating estrogen deficiency-induced osteocyte apoptosis might be positioned locally, rather than wholely, in sites of the cortex, and apoptosis is necessary to activate endocortical remodeling following Es loss71. All results strongly suggest that estrogen deficiency could give rise to osteocyte apoptosis, followed by resorption-related bone loss48,73. It should be noted that removal of sex steroids could trigger the activation of oxidative stress in bone, including decreased antioxidant enzymes (AE) and increased phosphorylation of p53 and p66shc, eventually leading to accumulation of apoptotic osteocytes72. Consistent with this finding, oxidative stress has been shown to function as an essential pathway in regulating osteocytic viability upon Es or As deficiency via gonadectomy. Instead, oxidative stress induced by Es or As deficiency is rescued by sex steroids replacement48. Moreover, oxidative stress reaction and accumulation of ROS may be nullified by the administration of the antioxidant N-acetyl cysteine (NAC), consequently alleviating osteocyte apoptosis in gonadectomized mice48. Besides the antioxidant role, Es also has a pro-survival effect on osteocytes, as evidenced by the result that miR-199a-3p is involved in estrogen-mediated autophagy through the insulin-like growth factor-1 (IGF-1)/mamalian target of rapamycin (mTOR) pathway in osteocyte-like MLO-Y4 cells73. Indeed, autophagy could prevent estrogen deficiency-induced osteoporosis74. Of note, estrogen receptors (ERs), namely ERα, ERβ, and G-protein-coupled ER (GPER) have a crucial role in mediating autophagy via key signaling molecules, such as BCL2-associated athanogene 3 (BAG3)75. This is in line with the finding that oxidative stress induced by Es or As deficiency has been shown to be a stimuli for activation of the autophagy pathway24. In agreement with this notion, it is demonstrated that Es may not only activate MAPK-dependent antioxidant signaling72 but also the ERK1/2 pathway via binding to its receptors (e.g., ERs and ARs) to rescue osteocytes apoptosis induced by estrogen deficiency in vitro72,74. Importantly, the ERK1/2 pathway is one of the important pro-survival pathways that activate autophagy76 (Fig. 2). In addition, it was suggested that low-intensity electrical stimulation (LIES) could dramatically suppress the increased apoptotic osteocytes, and ensuing elevated empty lacunae, as well as the decreased expression of inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) in OVX rats7. Nevertheless, it has been shown that estrogen deficiency is responded differently by the jaw, vertebral, and long bones in animal models76, the underlying mechanism is still not completely understood. Until recently, it was reported that osteocyte cell death, compared with littermate controls, was more significantly increased in aged Dmp1-Cre+ Sema3Aflox/∆ and Dmp1-Cre+ Nrp1flox/Sema- mice77. Moreover, it was found that semaphorin3A (Sema3A) or soluble guanylate cyclase (sGC) could initiate the activation of biomarkers in the cyclic guanosine monophosphate (cGMP) signaling pathway, including protein kinase G (PKG), ERKs, AKT, and vasodilator-stimulated phosphoprotein (VASP), which were suppressed by a PKG antagonist77. Based on these findings, it was suggested that the Sema3A-Nrp1-sGC-PKG pathway functions as an essential anti-apoptotic pathway in protecting osteocytes against estrogen deficiency-induced apoptosis. Of note, Sema3A deficiency in osteocytes may result in severe osteopenia in aged mice, which could be repressed by the Sema3A-targeting miRNAs, subsequently enhancing Sema3A expression77 (Table 2). Here, it should be emphasized that the serum level of Sema3A could decrease with age or after menopause in humans77.
GCs and autophagy
GCs are known as potent immunosuppressive and anti-inflammatory agents with the potential to inhibit the expression of several cytokines involved in inflammatory responses6. Thus, it is often used for the therapy of inflammatory non-infectious diseases, including rheumatoid arthritis, systemic lupus erythematosus, organ transplantation, asthma, and malignancies, etc78,79,80. Nevertheless, excessive GCs treatment could be associated with the development of over 30% of osteoporotic fracture and over 10% of osteonecrosis6. Importantly, it is suggested that GCs-induced bone disease is commonly characterized by the accumulation of apoptotic osteocytes in mice and humans6,78. In addition, the collapse of the femoral head, following GC-induced osteonecrosis, is also associated with apoptotic osteocytes localized adjacent to the subchondral region17. Accumulating evidence in vitro and in vivo indicates that GCs could induce osteocyte apoptosis6,78,79,80,81,82. In this line, GCs also inhibit several proteases expression, required for perilacunar skeleton remodeling, which could eventually lead to the disruption of osteocytic LCN, disorganization of collagen, and hypermineralization of the matrix, which are all found in necrotic bone lesions of human79. This also is supported by the fact that Wnt inhibitor genes (e.g., SOST) were expressed by osteocytes with GCs exposure, whereas concomitant autophagy mechanism also was activated, which has an anti-apoptotic role in GCs-induced osteocyte apoptosis80,81,82. Following a cumulative and unrepairable defect, bone strength could be reduced, eventually leading to the deformity of the femoral head. This is likely due to the interfering of GCs-induced apoptosis with the production of VEGF and VEGF-induced endothelial angiogenesis (Table 2). Also, tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) are highly expressed in the GCs-induced apoptotic osteocytes, which in turn affects the vitality of neighboring healthy osteocytes78. As previously reported, it seems to exist an interplay between BCL-2 (an anti-apoptotic biomarker) and Beclin-1 (an autophagy-related biomarker), which has a crucial role in controlling the switch of autophagy and apoptosis80 (Fig. 1). Hence, GCs treatment could affect the expression level of the antioxidant gene in a dose-dependent manner. Specifically, GCs might activate the autophagy pathway at a lower dose level, whereas apoptosis pathways could be triggered by GCs at a higher dose level81. It is intriguing that the mechanism underlying GC-induced osteocyte apoptosis is also addicted to factor associated suicide (FAS)/CD95 signaling pathway, except it is mediated through the mitochondrion6 (Fig. 2). In addition, GCs were shown to induce apoptosis in osteocytes overexpressing wild-type proline-rich tyrosine kinase 2 (Pyk2), which was rescued in osteocytes that lacks kinase activity (K−; Pyk2 mutant). It was suggested that activation of Pyk2 is implicated in the process of GCs-induced osteocyte apoptosis82. On the contrary, mechanical stimulus-induced chemokine (C-C motif) ligand 7 (CCL7) could, in an autocrine manner, protect osteocytes from GCs-induced apoptosis selectively. Nevertheless, it was demonstrated that CCL2 expressed by MLO-Y4 cells could not only protect against osteocytic apoptosis induced by GCs, but also by TNF-α83. Until recently, GCs-induced apoptosis of osteocytes is shown to produce higher expression of sclerostin (SOST), receptor activator of nuclear factor-kB ligand (RANKL), and dental matrix protein 1 (DMP-1) mRNA, which could be rescued by recombinant human parathyroid hormone (rhPTH) (1–34)84. Importantly, DMP-1, as a crucial biomarker in mediating osteocyte homeostasis, may be required for osteocyte apoptosis84. Of interest, it has been found that the protective effect of rhPTH (1–34) for GCs-induced femoral head necrosis, at least in part, from enhanced autophagy due to the upregulation of LC3II and Beclin-184, implying there might exist an interplay between PTH pathway and autophagy pathway. Furthermore, it is also indicated by a recent study in vivo that vitamin E, as antioxidant medication, has a crucial role in counteracting skeletal oxidative stress after excess GCs treatment, and consequently promotes osteocytic survival85. Nonetheless, the intrinsic cross-talk mechanism among oxidative stress, autophagy, and PTH signaling pathways in the anti-apoptotic process of osteocytes needs to be further investigated.
TNF-α and inflammation
Currently, the burgeoning field of osteoimmunology mainly focuses on the investigation of the interaction of the immune system in the skeleton with bone physiology, of which there exist many chronic inflammatory diseases concomitant with resorption-associated bone loss, including rheumatoid arthritis (RA), spinal cord injury (SCI), and aging-related osteoporosis (OP), etc85,86,87,88,89. Though the underlying mechanism whereby some pro-inflammatory factors induce resorption-associated bone loss remains fully unknown, it is hypothesized that osteocyte apoptosis might have a profound role in regulating bone homeostasis under these chronic inflammatory diseases. It is noteworthy that osteocyte density was shown to decrease accompanied by the increased osteocyte apoptosis when the expression of TNF-α and RANKL was elevated in rats with inflammatory bowel disease90,91. Likewise, apoptotic osteocytes were shown to be increased in a mouse infectious osteomyelitis model, while the expression level of pro-inflammatory biomarkers was was also elevated including TNF-α, interleukin-1β (IL-1β), interleukin-17 (IL-17), and IL-6, and so on. Conversely, in the context of TNF-α deficiency mice, the inflammatory responses to osteomyelitis were significantly weakened, concomitant with fewer apoptotic osteocytes8 (Table 1). Accumulating evidence suggests that pro-inflammatory cytokines such as TNF-α and IL-6 could induce directly osteocyte apoptosis in cell culture models, and therefore trigger the unleashing of the bulk of pro-inflammatory cytokines by a positive-feedback loop, which is abolished by binding of CD40 ligand (CD40L) independent of signaling or transcriptional mechanisms92. In the line with this notion, it has been shown that apoptotic osteocytes themselves could produce pro-inflammatory biomarkers including IL-1β and interleukin-18 (IL-18)93 (Table 2). Intriguingly, due to the inefficient clearance of apoptotic cells, many chronic inflammatory diseases are propagated such as rheumatoid arthritis, systemic lupus erythematosus, and cystic fibrosis89. Based on these findings, it is tempting to speculate that apoptosis of osteocytes functions as a gatekeeper of the regulatory inflammatory responses in pathological bone diseases. Until recently, it was found that receptor for advanced glycation end products (RAGE) has an essential role in upregulating the expression level of IL-6 and VEGF-A in MLO-Y4 cells, as well as in advanced glycation end products (AGEs)-induced osteocyte apoptosis93,94. Specifically, it is indicated that AGEs activate the downstream signals pathways (e.g., ERK1/2, p38, and STAT3) via RAGE, thus allowing both upregulating the production of IL-6 and VEGF-A in osteocytes in a dose- and time-dependent way and inducing osteocytic apoptosis93 (Fig. 2). Typically, RAGE is upregulated in states of chronic inflammation, such as rheumatoid arthritis, aging- and diabetes-related osteoporosis94. Once AGEs bind to RAGE, the upregulation of nuclear factor kappa-B (NF-κB) is exacerbated, which in turn increases the production of AGEs, IL-6, and VEGF-A from apoptotic osteocytes in an amplifying loop mechanism, consequently promoting osteocyte apoptosis93. In addition, it also was suggested that not only osteocytic apoptosis but also its dysfunction both could be induced by AGEs. Of note, FOXO1, associated intensely with apoptosis, functions as a pivotal transcription biomarker in regulating the expression of Caspase-3, SOST, and RANKL, and enhancing AGEs-induced osteocytic apoptosis and dysfunction. Furthermore, FOXO1 might induce apoptosis by regulating TNF-related apoptosis-inducing ligand (TRAIL)94.
The key molecular mechanisms in promoting the osteocytic apoptosis
Oxidative stress pathway
It has been suggested that the oxidative stress pathway could be activated in a wide variety of pathophysiological agents in humans, including aging, cancer, chronic inflammatory processes95. Nevertheless, the precise mechanisms whereby oxidation stress signaling pathways induce cellular damage or even death remain elusive. Growing experimental evidence indicates that while oxidative stress is mediated by many oxidant and antioxidant enzymes, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (e.g., Nox1 and Nox2) are dominant oxidative stress-inductive enzymes, which could be nullified by supplementation of superoxide dismutase (SOD)96,97. Mitochondria is well known to be the switchboard controlling the apoptosis machinery. Once initiated, it will, as an important source of ROS, primarily targeted by the damaging effect of ROS95 (Fig. 1). It should be emphasized that once mitochondria are deficient in cytochrome C, ROS will be amplified to generate the initial insult, thus leading to the induction of either apoptosis or senescence since ROS accumulation95. Conversely, glutathione peroxidative 4 (Gpx4) has been shown to serve as a crucial phospholipid hydroperoxide glutathione peroxidase (PHGP) in defending oxidative damage of mitochondria and protecting cells from apoptosis97. It was reported that the levels of ROS and p16Ink4a, a principal effector of senescence, were upregulated in aging wild-type (WT) female and male mice. Meanwhile, it was concomitant with a reduction of glutathione reductase (GR), which is well known to protect cells from excessive oxidative stress48,98. Here, it should be kept in mind that p53 is implicated in cellular redox regulation as a crucial orchestrator, as well as known as a kind of SASP98 (Table 1). Once the activation of p53, the expression of the BCL2-associated X protein (BAX) and p53 upregulated modulator of apoptosis (PUMA) may be triggered, which targets the mitochondria to induce the rapid release of cytochrome C and subsequent acceleration of the caspase cascade98. It was found that increased phosphorylation of p53 and p66shc activate oxidative stress pathway, therefore leading to ROS-induced osteocyte apoptosis, which indicates that oxidative stress pathway could have an important role in triggering osteocytic senescence and apoptosis upon aging48 (Fig. 2). In this line, a previous study by Kobayashi et al.47 showed that the level of ROS was significantly increased in 24 months aged mice, compared with 2 months aged mice. Intriguingly, it also be found that when osteocyte is targeted to delete the gene of mitochondrial SOD2, the levels of ROS are elevated concomitant with a low bone mass phenotype, resembling accelerated aging. Based on this finding, it is suggested that loss of SOD2 could result in degeneration of the osteocytic LCN, decreased osteocytic numbers, as well as reduced osteocytic connectivity, thereby inducing the process of osteocyte apoptosis. In addition, Piemontese et al.99 reported that increased bone loss and bone porosity, in the cortical skeleton of elderly C57BL/6J mice, was associated with the increased DNA damage, cellular senescence, and SASP (e.g., p53, p21), as well as the elevated expression level of RANKL in osteocytes. Thereby, it is assumed that oxidative stress evokes DNA damage, subsequently activates cellular senescence pathways, consequently leading to the development of aging-induced osteocyte apoptosis and bone loss. In support of the notion, it was shown that the age-related bone loss in mice model was rescued by suppressing the senescence of cells through novel transgenic approaches or “senolytic” drugs3. Besides, the oxidation stress pathway also is regarded as an essential pathway in the osteocyte apoptosis resulted from Es or As deficiency48. For instance, it is suggested that the production of ROS might be correlated with suppression of the NO-sGC-cGMP pathway in aging or sex steroid deficiency skeleton (Fig. 2) since excessive oxidative stress impairs the transduction of the sGC signal pathway, which is known as an essential pathway in enhancing the osteogenic effect100. Instead, increasing evidence has shown that 5′-AMP-activated protein kinase (AMPK)-peroxisomeproliferator-activated receptor-C coactivator-1α (PGC-1α)-ROS axis has an essential role in triggering anti-oxidative mechanisms to decrease the damage because of the unleashing of ROS. Specifically, PGC-1α has a pivotal role in regulating mitochondrial homeostasis and cell differentiation due to its ability to prevent mitochondria from ROS-controlled damage101,102. It should be noteworthy that osteocyte apoptosis could be induced by homocysteine (Hcy) in MLO-Y4 cells via the increased expression of Nox97, which was ameliorated by activation of AMPK-PGC-1α-ROS axis96. Until recently, it has been found that the p16−/−-OVX mice, compared with the WT-OVX mice, could significantly decrease the expression levels of p21 marker, indicating that p16del can prevent estrogen deficiency-induced osteoporosis through suppressing the oxidative stress, osteocytic senescence, and ensuing osteoclastogenesis-triggered bone resorption. As a result, osteoblastic bone formation and osteogenesis could be enhanced95. All in all, the continuing investigation of how the oxidation stress pathway mediates the stimulation of osteocyte apoptosis might discover essential signaling pathways, which eventually could be applied for therapeutic intervention in resorption-associated bone diseases induced by osteocyte apoptosis.
AGEs-RAGE/HMGB1-RAGE pathways
As described above, RAGE is expressed in many cell types including osteoblasts, osteoclasts, and osteocytes. Normally, it is, under physiological conditions, expressed in the human body at a low level, and has a crucial role in regulating bone metabolism93,94. Importantly, RAGE signaling is suggested to be mainly correlated with age- and inflammation-related bone diseases (Fig. 1). Once the activation of RAGE, downstream signaling proteins (e.g., NF-κB, AP-1, CREB, STAT3, NFAT) might be stimulated to induce cytokines/chemokines transcription, to control cellular processes, as well as to influence cell viability by regulating autophagy and apoptosis. However, AGEs accumulation patterns, intensities of binding RAGE/AGEs, or pro-inflammatory biomarkers components may change with aging, dependent on different organs103. It is well known that AGEs are irreversible Amadori products derived from protein glycation (PG), and might be present in many tissues including bone104,105. While AGEs can be bound to several receptors to enhance the oxidative stress reaction and effects of inflammation, RAGE is of great importance among these receptors. Once AGEs are bound to RAGE, production of ROS and pro-inflammatory cascade will be stimulated, which in turn promote AGEs formation in a positive-feedback loop104,106 (Fig. 2). It should be emphasized that the crosslink of proteins is implicated in the formation of AGEs. Specifically, more crosslink of matrix proteins is required for elevating the number of AGEs, which, at least in part, gives rise to deterioration of biomechanical properties in skeleton105. As noted, accelerated AGEs generation and accumulation are implicated in osteopenia in people with diabetes and the elderly. Instead, the diabetes-related bone loss may be rescued by irbesartan (an angiotensin II receptor antagonist) via blocking the AGEs/RAGE-mediated oxidative stress pathway105. Increasing evidence indicates that AGEs are crucial regulators of the pathogenesis and development of resorption-related bone diseases93,94,106. This is in line with the notion that Tanaka et al.107 reported that AGEs could enhance the production of osteocytic DNA fragments correlated with apoptosis. Also, AGEs and high glucose (HG) could give rise to deterioration of the cortical skeleton by propelling osteocyte cell death. Instead, PTH could have a positive protective role in AGEs-induced osteocyte apoptosis107. Similarly, a study by Notsu et al.108 in MLO-Y4-A2 cells has also demonstrated that osteocytic apoptosis, as well as the expression of SOST, was dramatically induced by AGE3 via the increased expression of TGF-β. However, this study did not investigate the underlying mechanisms whereby AGEs induce osteocyte apoptosis. Until recently, it is reported that the AGEs-RAGE signaling pathway is an essential pathway to induce cellular apoptosis via activating downstream proteins (e.g., ERK1/2, p38, and STAT3) in MLO-Y4 cells culture. It was found that the number of apoptotic osteocytes was more significantly elevated in the AGEs-incubated MLO-Y4 cells group (approximate 11.27%), compared with the control group (only 2.88%)93. Intriguingly, consistent with a previous study, AGEs were shown to promote the expression of RAGE and the unleashing of pro-inflammatory biomarkers (e.g., TNF-α, IL-6, RANKL, VEGF-A, etc) from osteocytes via a positive-feedback loop94,107, which might enhance the effect of AGEs-induced osteocyte apoptosis. It also is manifested by the result that when the FPS-ZM1R (an AGEs antagonist) was applied to pre-conditional osteocytes, the number of AGEs-induced apoptotic osteocytes was decreased significantly93. It should be noteworthy that other factors, such as ROS levels, intra- and extracellular redox conditions, and metabolic status, can also alter RAGE ligand properties and modify downstream signaling109, implying there might exist a cross-talk mechanism between oxidative stress pathway and RAGE pathway. In addition, high mobility group box 1 (HMGB1) has been demonstrated, as a damage-associated molecular pattern protein, to regulate essential cellular processes by interacting with two receptors including RAGE and the toll-like receptor 4 (TLR4), subsequently mediating bone homeostasis, turnover, and repair109,110,111,112. Importantly, HMGB1 functions as an alarmin in a warning and notifying cells about the hazardous extracellular environment in distress111 (Table 1). Similar to HMGB1, S100A4, bound to RAGE, also promotes the secretion of pro-inflammatory biomarkers in osteoblasts and osteocytes, and then affects the vitality of osteocytes, ultimately resulting in osteoclastogenesis-triggered bone resorption113. Some preliminary evidence suggests that HMGB1, thought as a pro-inflammatory cytokine, might be released by senescent or apoptotic osteocytes, thereby stimulating the generation of pro-inflammatory cytokines (e.g., TNF-α, IL-6, RANKL, etc) by activating RAGE from osteoclasts and osteocytes111,112,114. Accumulating evidence suggests that pro-inflammatory cytokines (e.g., TNF-α, IL-1β, etc) directly induce osteocyte apoptosis in cell culture models, therefore triggering the release of cytokines that influence bone turnover92,111,115. Also, TNF-α binds to its receptor, TNF receptor 1(TNFR1), triggering Caspase-8 activation, and ensuing Caspase-3 stimulation, which results in the induction of apoptosis116 (Fig. 1). In addition, the formation of a multinuclear actin ring induced by HMGB1 was suppressed in RAGE−/− pre-osteoclasts, implying there might exist an interplay between HMGB1 and RAGE112. Therefore, it is assumed that HMGB1-RAGE signaling pathways might be a crucial pathway in enhancing osteocyte apoptosis in the inflammation-related bone diseases, which, nevertheless, needs to be further corroborated by more studies (Table 2).
FAS/FASL pathway
The FAS/FAS ligand (FASL) pathway is well thought as an essential mechanism in removing misplaced and/or displaced cells in immune tissues or organs6. It should be kept in mind that FASL is one of the TNF family and is mainly synthesized as a membrane protein (type II). Specifically, the cytoplasm contains N terminus of FASL, whereas, the region of C terminus, in common, is stretched out to the extracellular space117. When the FAS receptor (FASR) is bound to FASL, it will oligomerize and recruit the FAS-associated protein with a novel death domain (DD), then interact with Caspase-8, and, in consequence, trigger a caspase cascade leading to apoptosis6 (Fig. 1). It has been suggested that FAS/FASL apoptotic pathway is implicated in GCs-induced apoptosis in several different cell types6,117 (Fig. 2). Nevertheless, whether bone homeostasis is controlled by FAS-mediated apoptosis is still poorly understood. It is noteworthy that the FASL deficiency mice (FASLCKO) showed an osteopenic phenotype more remarkably, compared with the littermate control (FASLfl/fl). Meanwhile, the number of apoptotic osteoclasts (TUNEL+; TRAP+) was significantly decreased, indicating that the enhanced osteoclastic activity is most likely due to the reduction of apoptotic osteoclasts118. Importantly, because of the detection of apoptosis-associated biomarkers (e.g., BCL-2, FAS, BAX, and FASL, etc) and cells apoptosis, it is assumed that part of osteoblasts and osteocytes could give rise to apoptosis when osteoblasts differentiate to form osteocytes early, which is tightly controlled by the appropriate number of mature osteocytes in the bone to maintain the optimal condition118. Similarly, the receptor of FAS/CD95 mRNA was found to be expressed in MLO-Y4 cells whether these osteocytes were pre-treated by GCs or not117. In addition, it was shown that GCs could, in a Caspase-8-dependent pattern, induce osteocyte apoptosis via the FAS/CD95 death receptor6, which is inhibited by all variants of the bisphosphonates (BPs) molecules including non-N-BPs and N-BPs6. It is intriguing that the upregulation of FAS was found to not be accompanied by high expression of FASL, pointing to the possible existence of FAS-mediated osteocyte apoptosis independent of FASL117.
The key molecular mechanisms in preserving the osteocytic vitality
Autophagy pathway
Autophagy is a well-conserved mechanism among species and is involved in various biological events20. Specifically, autophagy, as a “self-devouring” autophagosome-lysosomal process, is triggered to adapt to the stressful environment to preserve the vitality and function of long-lived cells by degrading and recycling damaged organelles and macromolecules21. Importantly, starvation, referred to as a crucial stimulus of autophagy, could promote the generation of metabolites through degrading the intracellular constituents to preserve cell vitality, which is required for fueling the respiration of mitochondria and production of ATP119. Not surprisingly, this pathway should also be implicated in osteocyte metabolism. Currently, several specific autophagy-related receptors and key biomarkers, known as crucial mediators in the selective degradation of pathogens, dysfunctional organelles, and aggregated proteins, have been screened and characterized partly. However, how selective autophagy has a physiological role is not yet completely understood120. Of note in this regard, the punctuate distribution of MAP1LC3A was both shown in osteocytes of the murine and human cortical skeleton, indicative of autophagy. In addition, autophagy flux, in an in vivo study, was upregulated when osteocytes encounter stress conditions such as deprivation of nutrition and hypoxia21. Therefore, it is suggested that the elevation of autophagic flux is required for the survival of differentiated osteocytes in stressful conditions. Accumulated evidence indicates that the autophagy pathway has a protective role in preserving the osteocytic vitality under the state of low oxygen pressure (PO2) or low-dose GCs therapy80,81,121. In this line, it was demonstrated that autophagy, as an anti-apoptotic factor, could be negatively mediated via the mTOR pathway with aging, thereby leading to cell senescence and apoptosis122. Likewise, bone turnover was reduced and p66shc phosphorylation was increased in L6 vertebrae of Dmp1-Cre; Atg7f/f mice, suggesting inhibition of osteocytic autophagy could generate the evidence of bone aging in many ways123. Consistent with this findings, it was suggested that the secretion of LC3II/I, Unc-51 like kinase 1 (Ulk-1), and Beclin-1 was significantly reduced with aging, while the expression of SQSTM1/p62 (sequestosome 1) and osteocytic apoptosis were dramatically elevated, implying senile osteoporosis concomitant with bone loss might be attributed to weakening osteocytic autophagy, which is independent of apoptosis124 (Table 1). This also is in line with the result that autophagy might protect the skeleton against age-related bone loss, in part at least, through decreasing the activity of apoptosis, whereas, suppression of autophagy could result in the elevation of damaged mitochondria and oxidative stress product (e.g., ROS)121,122,123,125 (Table 1). It is intriguing that Beclin-1, an anti-apoptotic biomarker in autophagy, could be cleaved by caspases, thereby acquiring a new function and amplifying mitochondrion-mediated apoptosis due to the C-terminal fragment of Beclin-1126,127, implying a cross-talk mechanism between autophagy and apoptosis (Fig. 1). Indeed, osteocyte apoptosis, under physiological conditions, is in an optimal state of balance tightly controlled by pro- and anti-apoptotic mechanism pathways. Once this balance is upset, masses of osteocytes may undergo apoptosis (Fig. 3). In this line, it was reported that GCs-induced apoptosis was increased in MLO-Y4 osteocytes in a dose- and time-dependent pattern. Meanwhile, autophagy markers (LC3II and Beclin-1) were increased at the low dose of dexamethasone (10−7 or 10−6 M), and yet decreased at the high dose (10−5 M), indicating that the self-activation of autophagy may be a protective mechanism against GCs-induced apoptosis84.

Under physiological conditions, it is in an optimal state of balance tightly controlled by pro- and anti-apoptotic mechanism pathways. Once this balance is upset, masses of osteocytes may undergo apoptosis, thereby resulting in bone metabolism disorder.
Nevertheless, there are controversial observations, including (1) no significant relationship between autophagy and apoptosis in osteocytes during aging124 and (2) no any changes in the number of osteocytes or osteocytic apoptosis in the transgenetic mice (i.e., Atg7del by the Dmp1-Cre transgene)123. It should be emphasized that if autophagy can never become inactive until osteoblasts differentiate into the stage of matrix-synthesizing, thus it is not impossible to allow autophagy to continue until osteocytes were fully formed123. Accordingly, it is no surprise that the formation of osteocytes and maintenance of vitality may be influenced if autophagy becomes inactive at earlier stages of osteoblast differentiation. Based on above reasons, the controversial problems in these studies could be, at least in part, explained. Nonetheless, the molecular mechanisms about potential effect of autophagy on osteocytic apoptosis need to be further established.
PTHrP/PTH1R and VEGF/VEGFR2 pathways
As well known, osteocytes could express parathyroid hormone-related protein (PTHrP) type 1 receptors (PTH1R), which are fully activated by PTH fragments and coupling to multiple signal transducers, including adenylyl cyclase(AC) and phospholipase C128,129. It has been found that osteocyte-selective PTH/PTHrP receptors (PPR) knockout (Ocy-PPRcKO mice) revealed a reduction in trabecular bone and slight osteopenia correlated with the elevated expression of SOST, which, yet, were unable to be suppressed by PTH in the Ocy-PPRcKO animals128. Accordingly, it is suggested that PTH functions as an important protective role in mediating the skeleton remodeling and calcium homeostasis. Of note, intermittent parathyroid hormone (iPTH) is highly correlated with its anti-apoptotic effect on both osteoblasts and osteocytes, however, production of excessive PTH following primary hyperparathyroidism or calcium deficiency could give rise to bone loss, which is attributed to the upregulation of fibroblast growth factor 23 (FGF23), and suppression of osteoblast differentiation and mineralization78,130,131. It should be emphasized that SOST-positive osteocytes of the cortical skeleton in Dmp1Cre.Pthlhf/f mice models were more significantly than that in Dmp1Cre.Pthlhw/w controls, and decreased bone mass and increased bone resorption were shown in Dmp1Cre.Pthlhf/f mice, indicating the full length of PTHrP derived from osteocytes stimulate skeleton formation and enhances skeleton strength in an autocrine and paracrine manner, in which actions might either be mediated by PTH1R or independent of PTH1R129. Pharmacologically, it was shown that application of iPTH (1–34) could remodel and strengthen skeleton structure in a time- and dose-dependent pattern, which was especially obvious in the trabecular region of the long bone in mice model131, as evidenced by the findings that rhPTH (1–34) could reverse GCs-induced osteocyte apoptosis not only in cell numbers but also in the function84. In this line, it was found that deletion of PTH1R in transgenic mice displayed slight osteopenia and disruption of calcium homeostasis, indicating that PTH/PTH1R pathway has a crucial role in preserving bone mass128 (Fig. 2). Importantly, PTHrP, much like PTH, displays anti-apoptotic features in osteoblasts and osteocytes132,133,134. Similarly, it also was corroborated that many of the osteocyte-related genes, such as SOST, DMP-1, were downregulated by PTH in an expected manner in OmGFP66 cells (immortalized osteogenic cell lines) derived from primary osteocytes of transgenic mice (DMP1-promoter)29. Based on these findings, it is demonstrated that the PTHrP/PTH1R pathway functions as an essential pathway in preserving the osteocytic vitality upon mechanical stimulus (Fig. 3). Nevertheless, the mechanism whereby PTH prevents osteocyte apoptosis remains ill-defined. As noted, it was reported that activation of PTH1R in osteocytes could promote gap junction-mediated intercellular coupling, increase expression of MMP-9, and potentiate calcium influx via stretch-activated Cai2+ channels, as well as amply the osteogenic response to mechanical loading in vivo, therefore leading to protect osteocytes from apoptosis135. Consistent with this notion, it is suggested that G-protein coupled receptors (GPCR) may participate in the cell transduction of mechanical signals, PTH1R and stretch-activated Cai2+ channels might both be part of the same mechanosensor system136. Of note, both Cai2+ and cAMP-dependent pathways activated by upregulation of PTHrP, as exhibited in an in vitro study, could have a cooperative role in maintaining MLO-Y4 cell viability133. Different from these results, it has been demonstrated that the expression level of SOST is decreased and Wnt/β-catenin signalings are activated in the transgenic mice (DMP1-caPTH1R), whereas, while these mice were crossed with mice lacking the low-density lipoprotein (LDL) related receptor 5 (LRP5) (Wnt co-receptor, LRP5−/−) or with mice overexpressing SOST (the Wnt antagonist) (DMP1-SOST), the skeleton phenotype is, in part, reversed in the DMP1-caPTHR1; LRP5−/− mice and abolished in DMP1- caPTHR1; DMP1-SOST mice, indicating that PTH1R from osteocytes has a pivotal role in promoting bone formation via Wnt/β-catenin signaling137. Strikingly, the inactivation of the pro-apoptotic protein, such as BCL2-associated death promoter (BAD), and increased synthesis of the anti- apoptotic protein (e.g., BCL2) are required for the PTH-induced cell survival (Table 1). It should be emphasized that Cx43 mutants (Cx43∆245 or Cx43S368A), thought as disruption of Cx43 communication and function, was shown to fail to recover PTH-induced osteoblast survival in Cx43 deficiency bone cells. Likewise, over-expression or phosphorylation of Cx43 in wild-type mice, but not Cx43S368A or Cx43∆245, also decreased the interplay between PTH1R and β-arrestin138, implying interaction Cx43 with β-arrestin is a prerequisite for osteoblast survival induced by PTH. It is intriguing that whether PTH produces an anti-apoptotic or pro-apoptotic effect is dependent on cAMP activation in the cAMP signaling pathway, which has a critical role in controlling autophagy via ERK-mediated induction of cyclin E and recruitment of Beclin-184,139 (Fig. 4). Besides the PTHrP/PTH1R system, VEGF/VEGF receptor2(VEGFR2) system is well known as an important mediator in proliferation, differentiation, and survival of bone cells, including osteoblasts, osteoclasts, and osteocytes140. Indeed, VEGF, as a major angiogenic factor, has a crucial role in coupling angiogenesis and osteogenesis via binding to VEGFR2140,141. It is noteworthy that VEGF, secreted by neighboring healthy osteocytes, was shown to be decreased in apoptotic osteocytes, and to be increased in sites far away from microdamage, suggesting that VEGF from osteocytes could have a pro-survival effect on osteocytes141. In agreement with this notion, it has been demonstrated that mechanical stimulus and VEGF both stimulated the secretion of anti-apoptotic biomarker BCL2, whereas, pro-apoptotic biomarker BAX was suppressed via ERK signalings140 (Table 1). Strikingly, different from non-transfected or Scr siRNA-transfected MLO-Y4 cells, PFF-induced VEGFR2 phosphorylation, via both Tyr-1059 and Tyr-1175, was dramatically impaired in caveolin-1 siRNA-transfected MLO-Y4 cells, which is correlated with re-localization of β-catenin into the osteocytic membrane140 (Fig. 4). Hence, it is supposed that VEGFR2/caveolin-1 might be an essential pathway, as parts of the signalosomes, in the anti-apoptotic osteocyte protection associated with mechanical loading140.

Initiation of the mitogen-activated protein kinase (MAPK) cascade is triggered by key pathological agents including Es, FSS, and so on, thereby promoting cell survival. Besides, the cAMP/PKA pathway, Sema3A-Nrp1-sGC-cGMP pathway, PTHrP/PTH1R system, and VEGF/VEGFR2 system activation also functions as a cooperator during this process. Intriguingly, there exists a cross-talk mechanism among caveolin-1/ERKs signalings and Wnt/β-catenin signalings and PI3k/AKT signalings.
Src/MEK/ERK pathway
It has been demonstrated in an in vitro study that the stretching of MLO-Y4 cells stimulated an anti-apoptotic effect via a complex signaling pathway including such signalosomes as integrins, Src kinases, and extracellular signal-regulated kinases (ERKs)55. It has been suggested that mechanical stimuli promote osteocyte viability via activation of Src/MEK/ERK signal pathway (Fig. 2). Importantly, these signalsomes are involved in the activation of Src/MEK/ERK signal pathway including integrins, actins, microtubular cytoskeletons, FAK, Src kinase, and Shc, which cooperatively transduct mechanical signals into ERK activation69. Specifically, Src is required not only for phosphorylation of substrates (e.g., Shc), but also for assembling of the signalosomes, which makes it possible to transduct the stretching stimulus signals via the interplay of protein SH2 domain, indicating the SH2 domain of Src and the kinases are both necessary for stretching stimulus to have an anti-apoptotic role74. Consistent with the fact that there exists the high expression level of β1-integrin in MLO-Y4 osteocytic cells, but not β2-integrin, it was demonstrated that β1-integrin, ɑ2-integrin, and ɑ5-integrin stimulated the initiation of ERK pathway induced by stretching through activating intracellular anti-apoptotic biomarkers93. It should be emphasized that activation of ERK and mechanical stimulus-induced osteocytic survival were reversed by β-cyclodextrin, which disrupted the microdomains of the cell membranes as a cholesterol chelator74. Of note, caveolin-1, known as an important structural constituent of caveolae, could interact with β1-integrin and ERKs in MLO-Y4 cells55, indicating caveolin-1 is involved in transduction of mechanical signals in osteocytes. Indeed, sex steroids (e.g., Es, As) also eliminate the osteocyte cell death through promptly triggering the Src/ERK pathway142 via ERs α/β and ARs70 (Fig. 4). Whereas, BPs or alendronates (ALNs) functions as anti-apoptotic drugs by activating ERKs signaling pathway and opening Cx43 hemichannels138. Nonetheless, how ERK pathway is exactly activated and how ERKs stimulate the osteocytic survival both remain ill-defined. As noted, the function of Cx43 hemichannels are correlated with the activation of kinase Src in the Src/MEK/ERK signal pathway, in which the binding sites of SH2 and SH3, domains of Src, are involved138. Specifically, the pore formation of C-terminal domain of Cx43, activation of Src kinase, stimulation of kinase ERK, and the binding actions of SH2 and SH3 are required for the transduction of mechanical signals via Src/MEK/ERK signal pathway41,42. It is intriguing that once Cx43 hemichannels are phosphorylated by Src or ERKs, its closure could be induced by a feedback loop mechanism41,138. In this situation, activation of Src could not only stimulate signals of cell survival via activation of ERKs, but also turn off Cx43 hemichannels to maintain the balance of cell homeostasis42. As described above, it is shown that Cx43 hemichannels are necessary for BPs or ALNs to have the anti-apoptotic effects on osteocytes41,138. Earlier in vitro studies has corroborated that the opening of Cx43 hemichannels is involved in the mechanism whereby BPs or ALNs generate the anti-apoptotic effect on osteocytes via triggering the Src/MEK/ERK pathway42,138. Of interest, unlike most agents that induce ERKs activation, BPs could activate ERKs, independent of the mevalonate pathway, to promote the osteocytic survival, which is mimicked by IG9402 (a bisphosphonate analog) independent of osteoclasts143.
cAMP/PKA and PI3K/AKT/β-catenin pathways
As described above, FSS might share the same pathways with GCs in the process of osteocyte apoptosis. It has been demonstrated that FSS could suppress GCs-induced apoptosis in MLO-Y4 osteocyte-like cells via the cross-talk mechanism between β-catenin pathway and the AKT/phosphatidylinositol 3-kinase (PI3K) pathway, as well as the classic protein kinase A (PKA)/cAMP pathway, in which PGE2 has an essential role of mediator56 (Fig. 4). Indeed, PGE2, as a factor maintaining osteocytic vitality, is shown to be produced by osteocytes in response to mechanical loading. Notably, the anti-apoptotic role of PGE2 is controlled by binding to EP2 and EP4 receptors, and ensuing activating cAMP/PKA pathway in the protection of osteocytic vitality upon FSS56. Intriguingly, not only the cAMP/PKA pathway but also β-catenin signaling pathways is implicated in protecting osteocyte against apoptosis by FSS. It has been suggested that there exists a cross-talk mechanism between these two pathways56. In agreement with the notion, it also has been indicated that sex steroids-activated PI3K might trigger the phosphorylation of the pro-apoptotic biomarker BAD and ensuing its inactivation, which is most likely due to an action of ribosomal S6 kinase 2 (Rsk2), apart from the anti-apoptotic effect via ERKs/JNK-triggered signalings pathway142. In addition, it was found that mechanical stimulus resulting in osteocytic survival is tightly mediated by bidirectional cross-talk mechanism between Wnt/β-catenin signalings pathway and ERKs pathway (Fig. 3), as evidenced by the result that blockade of the caveolin-1/ERK pathway obliterates phosphorylation of glucogen synthase kinase 3 (GSK3) and accumulation of β-catenin induced by mechanical stimulation144. Until recently, it was revealed that exosomes inhibited GCs-induced osteocyte apoptosis through activating miR21-PTEN-AKT signaling pathway145. Based on these results, it is proposed that β-catenin accumulation is required for osteocytes to maintain the mechanical properties. Of course, localization of β-catenin within caveolae is also essential for osteocytes to fully activate the ERKs pathway and promote the survival of osteocytes. Meanwhile, mechanical stimulus-triggered ERK activation, in turn, is also a precondition for accumulation of β-catenin by a bidirectional regulation loop145. Nonetheless, the complex cross-talk mechanism among PI3K/AKT pathway, β-catenin signaling, cAMP/PKA pathway, and ERKs signalings need to be further investigated.
Sema3A-Nrp1-sGC-PKG pathway
Sema3A, as a potent axon guidance molecule, is known to increase skeleton mass by suppression of skeleton resorption and enhancement of skeleton formation, suggesting that Sema3A also functions as a crucial osteoprotective role146. However, it needs the further study to explicit what the relationship between Sema3A and the neural system is. Until recently, the evidence indicates that Sema3A, derived from osteoblast lineage cells and independent of neural system, has a fundamental role in regulating the interaction of bone cells including osteoclasts, osteoblasts, and osteocytes, as well as controlling bone mass in a paracrine and autocrine manner in the postnatal period77. Also, it is clearly demonstrated in aged mice model that osteocytes are a major source of Sema3A, which specifically regulates the survival of mature osteocytes, as evidenced by the result that the survival of osteocytic IDG-SW3 cells, but not the survival of undifferentiated cell, was enhanced by treatment with Sema3A77. Of note in this regard, polymorphisms in PLXNA2, which encodes Plexin-A2 and participates in a receptor complex for Sema3A together with neuropilin-1 (Nrp1), are correlated with lower bone mineral density (BMD) and higher fracture risk in post-menopausal women147, indicating Sema3A serves as a crucial anti-apoptotic factor in maintaining the osteocytic vitality and skeleton strength upon Es (Table 2). Consistent with this evidence, it was found that Es could inhibit the secretion of microRNAs (miR-497 and miR-195), which acts on untranslated region (UTR) of Sema3A gene to reduce the production of Sema3A, therefore elevating the expression level of Sema3A77. As previously reported, Gucy1a1 is well known to encode the subunit of soluble guanylate cyclase (sGC), and to be only secreted in IDG-SW3 osteocytes and primary osteocyte-enriched population. Importantly, cGMP/NO-stimulated initiation of AKT and AKT/cGMP/PKG-dependent activation of BAD is implicated in estrogen-mediated pro-survival of osteocytes148. Likewise, Sema3A, as a member of semaphorin family proteins, activates sGC-cGMP signaling to protect osteocytes from apoptosis (Fig. 4), and consequently ameliorates bone loss after OVX146. Specifically, when Sema3A or sGC activator in the sGC-PKG pathway is triggered and downstream signalosomes including AKT, ERKs, and VASP are activated, the pro-apoptotic protein cleaved-Caspase-3 was diminished but restored by the inhibition of PKG77. Taken together, Sema3A-sGC-cGMP signaling might be shared in mediating bone homeostasis upon Es deficiency and aging, thereby providing a new approach for the therapy of resorption-related bone disease through targeting Sema3A to promote the survival of osteocytes.
The role of apoptotic osteocytes in osteoclastogenesis-triggered bone loss
To date, many studies have been addressing the question whether there is a causative relationship between osteocyte apoptosis and osteoclast-associated bone diseases such as osteoporosis and fragility fractures. The most prevalent notion refers to apoptotic osteocytes as the trigger of osteoclastogenesis and ensuing bone loss36,52,71. This is manifested by the fact that osteocyte apoptosis and ensuing resorption-related bone loss in response to fatigue loading9 and OVX71 were abolished by the application of a pan-caspase inhibitor. Accordingly, it has been established that the prevalence of apoptotic osteocytes is a causative determinant for enhanced osteoclastogenesis and ensuing bone loss9,36,52,71,149,150,151,152. Strikingly, multiple endocrine neoplasia type 1 (Men1) expression was significantly decreased in Men1Runx2Cre mice, which was obtained by crossing Men1tm1.1Zqw mice to Tg (Runx2-iCre) 1Jtuc mice. Meanwhile, it was exhibited by micro-computed tomography (Micro CT) that the obvious bone loss occurred in the distal femur of female Men1Runx2Cre mice, resembling osteoporosis153, which implying Men1 has a crucial role in preserving bone integrity and suppressing the development of osteoporosis. As described above, apoptotic osteocytes in inflammatory conditions were found to elevate the secretion of the pro-osteoclastogenic and pro-inflammatory biomarkers such as TNF-α, IL-6, IL-11, RANKL, HMGB1112,150, as well as intercellular cell adhesion molecule-1 (ICAM-1)152, VEGF-A, IGF-193, and DMP-1154, all of which have a crucial role in bone destruction and resorption (Fig. 5). Currently, accumulated evidence indicate that osteocyte apoptosis induced under pathologies (e.g., unloading/disuse, aging) initiates the increased expression level of NF-κB, thereby triggering RANK-induced osteoclastogenesis and bone resorption52,99. Aging-induced osteocytes apoptosis was demonstrated to promote bone loss through RANKL activation and sclerostin upregulation due to increased mitochondrial uncoupling and superoxide production (e.g., ROS)46. Based on above these findings, it is indicated that there exists an intrinsic and complex cross-talk mechanism between apoptotic osteocytes and osteoclastogenesis, while it is not fully deciphered and is still debatable. Of note, it was found that apoptotic osteocytes could serve not only as a direct role but also as an indirect role (i.g., by signaling to neighbor vital osteocytes) in regulating the migration and differentiation of osteoclasts precursor155 (Fig. 5). Indeed, it has been shown in an in vitro study that once mixed with osteocyte apoptotic bodies (OABs), mononuclear osteoclast precursors (OPs) could give rise to the preserving of OPs amounts and elevated the number and actions of TRACP+ cells. In contrast, whether in vivo or in vitro, apoptotic bodies (ABs) derived from osteoblasts exhibited no activity of osteoclastogenesis150. Notably, deletion of RANKL in osteocytes of Tnfsf11flox/∆ mice crossed with Dmp1-Cre mice (Tnfsf11flox/∆; Dmp1-Cre) diminished the activity of osteocytes in triggering the osteoclastogenesis, whereas it had no effects on osteoblasts, which indicates that RANKL derived from osteocyte might have an important role in triggering the osteoclastogenesis in vivo156. This is in consistent with the findings that HMGB1, released during osteocyte apoptosis, directly triggers osteoclastogenesis via activation of RAGE, and consequently increases pro-osteoclastogenic cytokine release (e.g., RANKL) from apoptotic osteocytes93. Until recently, it has been demonstrated that while the differentiation and formation of osteoclasts were not inhibited by Cx43def CM with immunodepletion of HMGB1, it yet was suppressed by Cx43def CM treated with an HMGB1 neutralizing antibody and then followed by immunodepletion of HMGB1, which indicates that the direct role of HMGB1, but not its indirect role, enhances apoptotic osteocytes-triggered osteoclastogenesis157. Similarly, Cx43def osteocytic cells are found to undergo accelerated apoptosis and ensuing increased release of HMGB1, which directly stimulates pro-osteoclastogenic signal release from Cx43def osteocytes41. Of interest, while Cx43def osteocytes gave rise to the elevated number of empty lacunae, this situation was only positioned in sits of the cortical skeleton with dying osteocytes, implying that signaling biomarkers from dying or apoptotic osteocytes, but not that from healthy or vital osteocytes, are necessary to promote the differentiation and formation of osteoclasts41. Inconsistent with this notion, some evidence suggest that apoptotic osteocytes do not directly initiate osteoclastogenesis, but indirectly trigger that via the neighboring and viable osteocytes, as evidenced by the fact that removal of Cx43 in Cx43∆Ot mice, compared with Cx43fl/fl mice, significantly elevated the number of apoptotic osteocytes concomitant with targeted recruitment of osteoclasts, however, disconnection of mechanical signals transduction between the dying or apoptotic osteocytes (Cx43def) and the nearby healthy osteocytes could give rise to the increased expression levels of pro-osteoclastogenic biomarkers (e.g., RANKL) in vital osteocytes158. Mechanically, the expression level of RANKL was reported to be increased in viable and neighboring osteocytes upon osteocyte apoptosis, which, in turn, stimulates the differentiation and formation of osteoclasts, as well as subsequent focal bone resorption52,159. In this line, a report by Cheung WY et al.160 has shown that apoptotic osteocytes could activate the P2X7-receptor and pannexin-1 (Panx1) in skeleton upon fatigue to stimulate the production of RANKL in nearby vital osteocytes. Meanwhile, it was emphasized that ATP could is a pivotal mediator in regulating the process as described above. In addition, it was demonstrated that while the process of skeleton remodeling was stimulated by apoptotic osteocytes upon microdamage, the pro-osteoclastogenic and pro-inflammatory biomarkers (e.g., TNF-α, IL-6, IL-11, RANKL, HMGB1, etc) were mainly expressed in the nearby healthy osteocytes. Of interest, both apoptotic osteocytes and neighboring vital osteocytes in response to microdamage are positioned in a temporally and spatially determined model, which is in the line with the natural feature of targeted skeleton remodeling under the environment of microdamage4. Whether initiated by directly or indirectly, or even both, osteoclastogenesis-triggered bone resorption and bone loss due to unloading/disuse or sex steroids deficiency have been shown to, at least in part, be suppressed pharmacologically1,149. Intriguingly, it is also proposed that suppression of osteocyte apoptosis eliminated the expression level of RANKL derived from osteocytes, whereas, it did not prevent the resorption-related bone loss triggered by unloading/disuse, implying osteocyte apoptosis is independent of osteoclastogenesis-associated bone resorption process161. It also is evidenced by the result from osteocytic response to particles that inhibition of Caspase-3 induced a pro-osteoclastogenic effect through upregulation of the RANKL/OPG ratio, meanwhile, it also evoked the osteocytic osteolysis with the elevation of both carbonicanhydrase 2 (CA2) and cathepsin K (CTSK) expression162. This is likely due to different apoptotic mechanism upon various pathological agents. Until recently, it was reported that the expression and activity of matrix metalloproteinase 14 (MMP14) are elevated in the transgenic mice model (caPTH1ROt), rather than in cKOPTH1ROt mice model (i.g., conditional knockout of PTH1R in osteocytes). As a result, activity of MMP14 increased the expression of soluble RANKL, which subsequently stimulated the differentiation of osteoclasts and ensuing bone resorption. Instead, while the activity of MMP14 was inhibited pharmacologically, the high level of bone remodeling and bone resorption was reduced in the caPTH1ROt mice model or mice exposed to chronic PTH elevation, which indicates that the resorption-associated bone loss seems to have no relations with osteocyte apoptosis163.

The direct and indirect role of osteocyte apoptosis in osteoclastogenesis, eventually, results in increased bone loss and bone fragility. During sustained exposure to apoptotic stimuli, masses of osteocytes may undergo apoptosis, concomitant with the unleashing of the pro-inflammatory and pro-osteoclastogenic biomarkers from osteocytes and osteoclasts cytokines including RANKL, HMGB1, TNF-ɑ, IL-6, etc, which in turn promote osteocytic apoptosis by an amplifying loop mechanism.
Here, it should be emphasized that apoptosis of osteoblasts was also shown to be directly induced under some pathological agents, such as hypoxia164, Es deficiency165, aging166, and GCs167, etc, which enhanced the reduction of the number of osteocyte processes, eventually leading to increased bone resorption and decreased BMD. However, autophagy or apoptosis of osteocytes, as the most abundant cells in bone, could have a more important role in osteoclastogenesis-triggered bone loss, compared with apoptosis of osteoblasts. Given the above, it is required to further investigate the exact role of osteocyte apoptosis, hopefully providing a more comprehensive explanation for various osteoclast-associated bone disorders.
Conclusions and future perspectives
Indifferent from apoptosis, ferroptosis is not implicated in the activation of caspases, but depends on iron to enhance lipid peroxidation and initiate cell death. Indeed, it was suggested that the injury in bone marrow stromal cells resulted from chemotherapy could be regulated by ferroptosis, which offers an amenable target to decrease the side effects of ferroptosis initiaters168. In addition, it has also been demonstrated both in human and murine model that pyroptosis, a novel programmed cell death, has a pivotal role in inflammatory-related bone diseases with bone loss, such as osteomyelitis169 and diabetes mellitus-associated periodontitis170, which may be due to inhibition of proliferation and differentiation of osteoblasts. Nevertheless, whether the mechanism of ferroptosis or pyroptosis could occur in osteocytes remains ill-defined.
Currently, clinical anabolic drugs, such as teriparatide (PTH1-34)171, tetracycline172, and bisphosphonate173, etc, are illustrated to contribute to enhance bone formation. Nevertheless, both these drugs produce inevitable side effects due to lack of target-cell specificity of delivery systems. With a surge of material science and nanomedicine, several specific osteoblast-targeting delivery systems, such as aptamer-functionalized lipid nanoparticles (LNPs)174 and Ser-Asp-Ser-Ser-Asp-polyurethane (SDSSD-PU)175, have been updated from a tissue level to a cellular level. Though these delivery systems were shown to enhance the bone microarchitecture and elevate the bone mass in vitro, their safety and effectiveness in vivo remain a challenging issue. Most importantly, because of short of insight into mechanisms of therapy, the subsequent translational practice is also hindered. Of note, while osteocytes are indicated to master the process of bone metabolism by theirs interplay with osteoblasts and osteoclasts, osteocyte-targeting tools, as an anabolic strategy, have been rare. Until recently, it was reported by Qiao et al.176 that a PH-responsive osteocyte-targeting theranostic system, zoledronic acid (ZA)-anchored Gd-doped mesoporous silica-coated upconversion nanoparticles (UCMSs) loaded with plumbagin (PL) and polyacrylic acid (PAA), facilitated to accurately diagnose and effectively treat early bone metastasis of breast cancer. Thereby, targeted drugs that are transformed to only reach and act on osteocytes without affecting other bone tissue cells should also be highly desirable, as an anabolic strategy, to treat resorption-related bone diseases including osteoporosis and fragility fractures.
Taken together, vital osteocytes have been shown to be of paramount importance in preserving the mechanosensation and mechanoconduction of bone. Admittedly, some key anti-apoptotic biomarkers (e.g., Cx43, PGE2 FGF7, CCL7) have a crucial role in protecting osteocytic LCN and promoting intercellular communication. However, in the context of homeostasis unbalance under various pathological agents, key pro-apoptotic biomarkers (e.g., ROS, BNIP3, BAX, DMP1) could master the process of osteocytic apoptosis via activating intrinsic and complex signaling pathways. Once a bulk of apoptotic osteocytes occur, osteoclastogenesis may be initiated, consequently triggering and promoting the increased bone loss and bone fragility.
In addition, the lack of understanding about the actions of osteocytes cells death in human bone pathologies also constrains the attractive and proper actions of pharmacotherapeutics of targeting apoptosis. Accordingly, the potential cross-talk mechanisms whereby osteocyte apoptosis occurs in various states of pathologies and subsequently initiates osteoclastogenesis need to be more clearly established in future preclinical models for translation to human biology. Of course, it is important that other potential mechanisms should also be considered as osteocyte apoptosis and ensuing osteoclastogenesis, hopefully resulting in a more complete explanation for bone disorders under key pathological conditions.
References
Plotkin, L. I. Apoptotic osteocytes and the control of targeted bone resorption. Curr. Osteoporos. Rep. 12, 121–126 (2014).
Tsujimoto, Y. & Shimizu, S. Another way to die: autophagic programmed cell death. Cell Death Differ. 12, 1528–1534 (2015).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Kennedy, O. D. et al. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 50, 1115–1122 (2012).
Aguirre, J. I. et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 21, 605–615 (2006).
Kogianni, G. et al. Fas/CD95 is associated with glucocorticoid-induced osteocyte apoptosis. Life Sci. 75, 2879–2895 (2004).
Lirani-Galvao, A. P. et al. Low-intensity electrical stimulation counteracts the effects of ovariectomy on bone tissue of rats: effects on bone microarchitecture, viability of osteocytes, and nitric oxide expression. Calcif. Tissue Int. 84, 502–509 (2009).
Morita, M. et al. Elevation of pro-inflammatory cytokine levels following anti-resorptive drug treatment is required for osteonecrosis development in infectious osteomyelitis. Sci. Rep. 7, 46322 (2017).
Tatsumi, S. et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 5, 464–475 (2007).
Bonewald, L. F. The amazing osteocyte. J. Bone Miner. Res. 26, 229–238 (2011).
Klein-Nulend, J., Bakker, A. D., Bacabac, G., Vatsa, A. & Weinbaum, S. Mechanosensation and transduction in osteocytes. Bone 54, 182–190 (2013).
Deepak, V., Kayastha, P. & McNamara, L. M. Estrogen deficiency attenuates fluid flow-induced [Ca(2+)]i oscillations and mechanoresponsiveness of MLO-Y4 osteocytes. FASEB J. 31, 3027–3039 (2017).
van Hove, R. P. et al. Osteocyte morphology in human tibiae of different bone pathologies with different bone mineral density — Is there a role for mechanosensing? Bone 45, 321–329 (2009).
Milovanovic, P. et al. Osteocytic canalicular networks: morphological implications for altered mechanosensitivity. ACS Nano. 7, 7542–7551 (2013).
Qiu, S., Rao, D. S., Fyhrie, D. P., Palnitkar, S. & Parfitt, A. M. The morphological association between microcracks and osteocyte lacunae in human cortical bone. Bone 37, 10–15 (2005).
Busse, B. et al. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell. 9, 1065–1075 (2010).
Fimia, G. M. & Piacentini, M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell. Mol. Life. Sci. 67, 1581–1588 (2010).
Mukhopadhyay, S., Panda, P. K., Sinha, N., Das, D. N. & Bhutia, S. K. Autophagy and apoptosis: where do they meet? Apoptosis 19, 555–566 (2014).
Marino, G., Niso-Santano, M., Baehrecke, E. H. & Kroemer, G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cel. Biol. 15, 81–94 (2014).
Mizushima, N., Levine, B., Cuervo, A. M. & Klionsky, D. J. Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 (2008).
Piemontese, M. et al. Low bone mass and changes in the osteocyte network in mice lacking autophagy in the osteoblast lineage. Sci. Rep. 6, 24262 (2016).
Zahm, A. M., Bohensky, J., Adams, C. S., Shapiro, I. M. & Srinivas, V. Bone cell autophagy is regulated by environmental factors. Cells Tissues Organs 194, 274–278 (2011).
Yao, W., Dai, W., Jiang, J. X. & Lane, N. E. Glucocorticoids and osteocyte autophagy. Bone 54, 279–284 (2013).
Filomeni, G., De Zio, D. & Cecconi, F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388 (2015).
Maiuri, M. C. & Kroemer, G. Autophagy in stress and disease. Cell Death Differ. 22, 365–366 (2015).
Monastyrska, I., Rieter, E., Klionsky, D. J. & Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. Camb. Philos. Soc. 84, 431–448 (2009).
Zhu, Y. et al. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell. 1, 468–477 (2010).
Florencio-Silva, R., Sasso, G. R. S., Sasso-Cerric, E., Simõesa, M. J. & Cerricy, P. S. Effects of estrogen status in osteocyte autophagy and its relation to osteocyte viability in alveolar process of ovariectomized rats. Biomed. Pharmacother. 98, 406–415 (2018).
Wang, K. et al. A novel osteogenic cell line that differentiates into GFP-tagged osteocytes and forms mineral with a bone-like lacunocanalicular structure. J. Bone Miner. Res. 34, 979–995 (2019).
Bernhardt, A., Wolf, S., Weiser, E., Vater, C. & Gelinsky, M. An improved method to isolate primary human osteocytes from bone. Biomed. Technol. 65, 107–111 (2020).
Hemmatian, H., Bakker, A. D., Klein-Nulend, J. & van Lenthe, G. H. Aging, osteocytes, and mechanotransduction. Cur. Osteoporos. Rep. 15, 401–411 (2017).
Galea, G. L. et al. Old age and the associated impairment of bones’ adaptation to loading are associated with transcriptomic changes in cellular metabolism, cell-matrix interactions and the cell cycle. Gene 599, 36–52 (2017).
Nishida, T., Kubota, S., Yokoi, H., Mukoyama, M. & Takigawa, M. Roles of matricellular CCN2 deposited by osteocytes in osteoclastogenesis and osteoblast differentiation. Sci. Rep. 9, 10913 (2019).
Bach-Gansmo, F. L. et al. Osteocyte lacunar properties and cortical microstructure in human iliac crest as a function of age and sex. Bone 91, 11–19 (2016).
Lai, X. et al. The dependences of osteocyte network on bone compartment, age, and disease. Bone Res. 3, 15009 (2015).
Jilka, R. L. & O’Brien, C. A. The role of osteocytes in age-related bone loss. Curr. Osteoporos. Rep. 14, 16–25 (2016).
Plotkin, L. I. & Bellido, T. Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat. Rev. Endocrinol. 12, 593–605 (2016).
Noble, B. S. et al. Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone. Am. J. Physiol. Cell. Physiol. 284, C934–C943 (2003).
Javaheri, B. & Pitsillides, A. A. Aging and mechanoadaptive responsiveness of bone. Curr. Osteoporos. Rep. 17, 560–569 (2019).
Chastin, S. F., Mandrichenko, O., Helbostadt, J. L. & Skelton, D. A. Associations between objectively- measured sedentary behaviour and physical activity with bone mineral density in adults and older adults, the NHANES study. Bone 64, 254–262 (2014).
Davis, H. M. et al. Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell 16, 551–563 (2017).
Talbot, J. et al. Connexin43 intercellular communication drives the early differentiation of human bone marrow stromal cells into osteoblasts. J. Cell Physiol. 233, 946–957 (2018).
Xu, H. et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. J. Bone Miner. Res. 30, 436–448 (2015).
Joiner, D. M., Tayim, R. J., McElderry, J. D., Morris, M. D. & Goldstein, S. A. Aged male rats regenerate cortical bone with reduced osteocyte density and reduced secretion of nitric oxide after mechanical stimulation. Calcif. Tissue Int. 94, 484–494 (2014).
Figueiredo, P. A., Powers, S. K., Ferreira, R. M., Appell, H. J. & Duarte, J. A. Aging impairs skeletal muscle mitochondrial bioenergetic function. J. Gerontol. A Biol. Sci. Med. Sci. 64, 21–33 (2009).
Kang, C., Chung, E., Diffee, G. & Ji, L. L. Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: role of PGC-1alpha. Exp. Gerontol. 48, 1343–1350 (2013).
Kobayashi, K. et al. Mitochondrial superoxide in osteocytes perturbs canalicular networks in the setting of age-related osteoporosis. Sci. Rep. 5, 9148 (2015).
Almeida, M. et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 282, 27285–27297 (2007).
Korotchkina, L. G. et al. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging 2, 344–352 (2010).
Lloyd, S. A. et al. Interdependence of muscle atrophy and bone loss induced by mechanical unloading. J. Bone Miner. Res. 29, 1118–1130 (2014).
Mann, V., Huber, C., Kogianni, G., Jones, D. & Noble, B. The influence of mechanical stimulation on osteocyte apoptosis and bone viability in human trabecular bone. J. Musculoskelet. Neuronal. Interact. 6, 408–417 (2006).
Cabahug-Zuckerman, P. et al. Osteocyte apoptosis caused by hindlimb unloading is required to trigger osteocyte RANKL production and subsequent resorption of cortical and trabecular bone in mice femurs. J. Bone Miner. Res. 31, 1356–1365 (2016).
Lin, C. et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J. Bone Miner. Res. 24, 1651–1661 (2009).
Tan, S. D., Bakker, A. D., Semeins, C. M., Kuijpers-Jagtman, A. M. & Klein-Nulend, J. Inhibition of osteocyte apoptosis by fluid flow is mediated by nitric oxide. Biochem. Biophys. Res. Commun. 369, 1150–1154 (2008).
Plotkin, L. I. et al. Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases, and ERKs. Am. J. Physiol. Cell. Physiol. 289, C633–C643 (2005).
Kitase, Y. et al. Mechanical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the beta-catenin and PKA pathways. J. Bone Miner. Res. 25, 2657–2668 (2010).
Storlino, G. et al. Irisin prevents disuse-induced osteocyte apoptosis. J. Bone Miner. Res. 35, 766–775 (2020).
Kim, H. et al. Irisin mediates effects on bone and fat via alphaV integrin receptors. Cell 178, 507–508 (2019).
Liu, X. Y. et al. FGF-7 dictates osteocyte cell processes through Beta-catenin transduction. Sci. Rep. 8, 14792 (2018).
Hinoi, E. et al. Positive regulation of osteoclastic differentiation by growth differentiation factor 15 upregulated in osteocytic cells under hypoxia. J. Bone Miner. Res. 27, 938–949 (2012).
van Breukelen, F., Krumschnabel, G. & Podrabsky, J. E. Vertebrate cell death in energy-limited conditions and how to avoid it: what we might learn from mammalian hibernators and other stress- tolerant vertebrates. Apoptosis 15, 386–399 (2010).
Montesi, M. et al. Hypoxia mediates osteocyte ORP150 expression and cell death in vitro. Mol. Med. Rep. 14, 4248–4254 (2016).
Guo, D. et al. Identification of osteocyte-selective proteins. Proteomics 10, 3688–3698 (2010).
Burr, D. B. Stress concentrations and bone microdamage: John Currey’s contributions to understanding the initiation and arrest of cracks in bone. Bone 127, 517–525 (2019).
Liu, X. et al. Spatiotemporal distribution of linear microcracks and diffuse microdamage following daily bouts of fatigue loading of rat ulnae. J. Orthop. Res. 37, 2112–2121 (2019).
Verborgt, O., Tatton, N. A., Majeska, R. J. & Schaffler, M. B. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J. Bone Miner. Res. 17, 907–914 (2002).
Hagan, M. L. et al. Decreased pericellular matrix production and selection for enhanced cell membrane repair may impair osteocyte responses to mechanical loading in the aging skeleton. Aging Cell. 19, e13056 (2020).
Hagan, M. L. et al. Inhibition of osteocyte membrane repair activity via dietary Vitamin E deprivation impairs osteocyte survival. Calcif. Tissue Int. 104, 224–234 (2019).
Hoshi, K. et al. Compressive force-produced CCN2 induces osteocyte apoptosis through ERK1/2 pathway. J. Bone Miner. Res. 29, 1244–1257 (2014).
Almeida, M. et al. Estrogens and androgens in skeletal physiology and pathophysiology. Physiol. Rev. 97, 135–187 (2017).
Emerton, K. B. et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone 46, 577–583 (2010).
Almeida, M., Han, L., Ambrogini, E., Bartell, S. M. & Manolagas, S. C. Oxidative stress stimulates apoptosis and activates NF-kappaB in osteoblastic cells via a PKCbeta/p66shc signaling cascade: counter regulation by estrogens or androgens. Mol. Endocrinol. 24, 2030–2037 (2010).
Fu, J. et al. miR-199a-3p is involved in estrogen-mediated autophagy through the IGF-1/mTOR pathway in osteocyte-like MLO-Y4 cells. J. Cell Physiol. 233, 2292–2303 (2018).
Qi, M. et al. Autophagy maintains the function of bone marrow mesenchymal stem cells to prevent estrogen deficiency-induced osteoporosis. Theranostics 7, 4498–4516 (2017).
Wei, Y. & Huang, J. Role of estrogen and its receptors mediated-autophagy in cell fate and human diseases. J. Steroid Biochem. Mol. Biol. 191, 105380 (2019).
de Souza Faloni, A. P. et al. Jaw and long bone marrows have a different osteoclastogenic potential. Calcif. Tissue Int. 88, 63–74 (2011).
Hayashi, M. et al. Autoregulation of osteocyte sema3A orchestrates estrogen action and counteracts bone aging. Cell Metab. 29, 627–637 (2019).
Wang, T., Yu, X. & He, C. Pro-inflammatory cytokines: cellular and molecular drug targets for glucocorticoid-induced-osteoporosis via osteocyte. Curr. Drug. Targets 20, 1–15 (2019).
Fowler, T. W. et al. Glucocorticoid suppression of osteocyte perilacunar remodeling is associated with subchondral bone degeneration in osteonecrosis. Sci. Rep. 7, 44618 (2017).
Shen, G. et al. Autophagy as a target for glucocorticoid-induced osteoporosis therapy. Cell. Mol. Life. Sci. 75, 2683–2693 (2018).
Jia, J. et al. Glucocorticoid dose determines osteocyte cell fate. FASEB J. 25, 3366–3376 (2011).
Plotkin, L. I., Manolagas, S. C. & Bellido, T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival: evidence for inside-out signaling leading to anoikis. J. Biol. Chem. 282, 24120–24130 (2007).
Kitase, Y. et al. CCL7 is a protective factor secreted by mechanically loaded osteocytes. J. Dent. Res. 93, 1108–1115 (2014).
Zhu, L. et al. Parathyroid hormone (PTH) induces autophagy to protect osteocyte cell survival from dexamethasone damage. Med. Sci. Monit. 23, 4034–4040 (2017).
Jia, Y. B. et al. Inhibitory effects of vitamin E on osteocyte apoptosis and DNA oxidative damage in bone marrow hemopoietic cells at early stage of steroid-induced femoral head necrosis. Mol. Med. Rep. 15, 1585–1592 (2017).
Agrawal, M. et al. Bone, inflammation, and inflammatory bowel disease. Cur. Osteoporos. Rep. 9, 251–257 (2011).
Abrahami, D. et al. Dipeptidyl peptidase-4 inhibitors and incidence of inflammatory bowel disease among patients with type 2 diabetes: population based cohort study. BMJ 360, k872 (2018).
Mazzaferro, S. et al. Bone, inflammation and the bone marrow niche in chronic kidney disease: what do we know? Nephrol. Dial. Transplant. 33, 2092–2100 (2018).
Allison, D. J. & Ditor, D. S. Immune dysfunction and chronic inflammation following spinal cord injury. Spinal Cord. 53, 14–18 (2015).
Metzger, C. E., Narayanan, A., Zawieja, D. C. & Bloomfield, S. A. Inflammatory bowel disease in a rodent model alters osteocyte protein levels controlling bone turnover. J. Bone Miner. Res. 32, 802–813 (2017).
Narayanan, S. A., Metzger, C. E., Bloomfield, S. A. & Zawieja, D. C. Inflammation induced lymphatic rchitecture and bone turnover changes are ameliorated by irisin treatment in chronic inflammatory bowel disease. FASEB J. 32, 4848–4861 (2018).
Ahuja, S. S. et al. CD40 ligand blocks apoptosis induced by tumor necrosis factor-alpha, glucocorticoids, and etoposide in osteoblasts and the osteocyte-like cell line murine long bone osteocyte-Y4. Endocrinology 144, 1761–1769 (2003).
Chen, H. et al. Advanced glycation end products induced IL-6 and VEGF-A production and apoptosis in osteocyte-like MLO-Y4 cells by activating RAGE and ERK1/2, P38 and STAT3 signalling pathways. Int. Immunopharmacol. 52, 143–149 (2017).
Zhang, C. et al. FOXO1 mediates advanced glycation end products induced mouse osteocyte-like MLO- Y4 cell apoptosis and dysfunctions. J. Diabetes Res. 2019, 6757428 (2019).
Li JKarim, M. A., Che, H., Geng, Q. & Miao, D. Deletion of p16 prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Am. J. Transl. Res. 12, 672–683 (2020).
Takeno, A. et al. Activation of AMP-activated protein kinase protects against homocysteine-induced apoptosis of osteocytic MLO-Y4 cells by regulating the expressions of NADPH oxidase 1 (Nox1 and Nox2). Bone 77, 135–141 (2015).
Brown, D. I. & Griendling, K. K. Nox proteins in signal transduction. Free Radic. Biol. Med. 47, 1239–1253 (2009).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 31, 1920–1929 (2016).
Piemontese, M. et al. Old age causes de novo intracortical bone remodeling and porosity in mice. Jci. Insight 2, 93771 (2016).
Zhai, Y. K. et al. Icariin stimulates the osteogenic differentiation of rat bone marrow stromal cells via activating the PI3K-AKT-eNOS-NO-cGMP-PKG. Bone 66, 189–198 (2014).
Khan, M. P. et al. Pathophysiological mechanism of bone Loss in type 2 diabetes involves inverse regulation of osteoblast function by PGC-1alpha and skeletal muscle atrogenes: adipoR1 as a potential target for reversing diabetes-induced osteopenia. Diabetes 64, 2609–2623 (2015).
Baldelli, S., Aquilano, K. & Ciriolo, M. R. PGC-1alpha buffers ROS-mediated removal of mitochondria during myogenesis. Cell. Death. Dis. 5, e1515 (2014).
Son, M. et al. Age dependent accumulation patterns of advanced glycation end product receptor (RAGE) ligands and binding intensities between RAGE and its ligands differ in the liver, kidney, and skeletal muscle. Immun. Ageing 14, 12 (2017).
Sorci, G., Riuzzi, F., Giambanco, I. & Donato, R. RAGE in tissue homeostasis, repair and regeneration. Biochim. Biophys. Acta 1833, 101–109 (2013).
Cheng, Y. Z. et al. Irbesartan attenuates advanced glycation end products-mediated damage in diabetes- associated osteoporosis through the AGEs/RAGE pathway. Life. Sci. 205, 184–192 (2018).
Ramasamy, R., Yan, S. F. & Schmidt, A. M. Advanced glycation endproducts: from precursors to RAGE: round and round we go. Amino. Acids 42, 1151–1161 (2012).
Tanaka, K., Yamaguchi, T., Kanazawa, I. & Sugimoto, T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte- like MLO-Y4-A2 cells. Biochem. Biophys. Res. Commun. 461, 193–199 (2015).
Notsu, M. et al. Advanced glycation end products (AGEs) increases apoptosis and the expression of sclerostin by stimulating TGF-beta expression and secretion in osteocyte-like MLO-Y4-A2 cells. Calcif. Tissue Int. 100, 402–411 (2017).
Yu, Y., Tang, D. & Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 6, 93 (2015).
Feng, L. et al. HMGB1 promotes the secretion of multiple cytokines and potentiates the osteogenic differentiation of mesenchymal stem cells through the Ras/MAPK signaling pathway. Exp. Ther. Med. 12, 3941–3947 (2016).
Yang, H., Wang, H. & Andersson, U. Targeting inflammation driven by HMGB1. Front. Immunol. 11, 484 (2020).
Zhou, Z. et al. HMGB1 regulates RANKL-induced osteoclastogenesis in a manner dependent on RAGE. J. Bone Miner. Res. 23, 1084–1096 (2008).
Yoshida, T., Flegler, A., Kozlov, A. & Stern, P. H. Direct inhibitory and indirect stimulatory effects of RAGE ligand S100 on sRANKLinduced osteoclastogenesis. J. Cell. Biochem. 107, 917–925 (2009).
Davalos, A. R. et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell. Biol. 201, 613–629 (2013).
Pesce Viglietti, A. I. et al. Brucella abortus invasion of osteocytes modulates vonnexin 43 and integrin expression and induces osteoclastogenesis via receptor activator of NF-kappaB ligand and tumor necrosis factor alpha secretion. Infect. Immun. 84, 11–20 (2016).
Yang, R. et al. Autophagy plays a protective role in tumor necrosis factor-alpha-induced apoptosis of bone marrow-derived mesenchymal stem cells. Stem. Cells Dev. 25, 788–797 (2016).
Schmidt, M. et al. Role of the CD95/CD95 ligand system in glucocorticoid-induced monocyte apoptosis. J. Immunol. 166, 1344–1351 (2001).
Wang, L. et al. Osteoblast-induced osteoclast apoptosis by FAS ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ. 22, 1654–1664 (2015).
Boya, P., Reggiori, F. & Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell. Biol. 15, 713–720 (2013).
Fimia, G. M., Kroemer, G. & Piacentini, M. Molecular mechanisms of selective autophagy. Cell. Death. Differ. 20, 1–2 (2013).
Shapiro, I. M., Layfield, R., Lotz, M., Settembre, C. & Whitehouse, C. Boning up on autophagy: the role of autophagy in skeletal biology. Autophagy 10, 7–19 (2014).
Yurube, T., Ito, M., Kakiuchi, Y., Kuroda, R. & Kakutani, K. Autophagy and mTOR signaling during intervertebral disc aging and degeneration. Jor. Spine 3, e1082 (2020).
Onal, M. et al. Suppression of autophagy in osteocytes mimics skeletal aging. J. Biol. Chem. 288, 17432–17440 (2013).
Chen, K., Yang, Y. H., Jiang, S. D. & Jiang, L. S. Decreased activity of osteocyte autophagy with aging may contribute to the bone loss in senile population. Histochem. Cell. Biol. 142, 285–295 (2014).
Greenhill, C. Bone: autophagy regulates bone growth in mice. Nat. Rev. Endocrinol. 12, 4 (2016).
Wang, T., Liu, X. & He, C. Glucocorticoid-induced autophagy and apoptosis in bone. Apoptosis 25, 157–168 (2020).
Djavaheri-Mergny, M., Maiuri, M. C. & Kroemer, G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 29, 1717–1719 (2010).
Powell, W. F. Jr. et al. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol. 209, 21–32 (2011).
Ansari, N. et al. Autocrine and paracrine regulation of the murine skeleton by osteocyte-derived parathyroid hormone-related protein. J. Bone Miner. Res. 33, 137–153 (2018).
Chandra, A. et al. PTH1-34 alleviates radiotherapy-induced local bone loss by improving osteoblast and osteocyte survival. Bone 67, 33–40 (2014).
Sugiyama, T. et al. Mechanical loading enhances the anabolic effects of intermittent parathyroid hormone (1-34) on trabecular and cortical bone in mice. Bone 43, 238–248 (2008).
Shibamoto, A. et al. Effect of high-frequency loading and parathyroid hormone administration on peri-implant bone healing and osseointegration. Int. J. Oral. Sci. 10, 6 (2018).
Maycas, M. et al. Role of the parathyroid hormone type 1 receptor (PTH1R) as a mechanosensor in osteocyte survival. J. Bone Miner. Res. 30, 1231–1244 (2015).
Bellido, T., Saini, V. & Pajevic, P. D. Effects of PTH on osteocyte function. Bone 54, 250–257 (2013).
Yavropoulou, M. P., Michopoulos, A. & Yovos, J. G. PTH and PTHR1 in osteocytes. New insights into old partners. Hormones 16, 150–160 (2017).
Liedert, A., Kaspar, D., Blakytny, R., Claes, L. & Ignatius, A. Signal transduction pathways involved in mechanotransduction in bone cells. Biochem. Biophys. Res. Commun. 349, 1–5 (2006).
Rhee, Y. et al. PTH Receptor Signaling in Osteocytes governs periosteal bone formation and intracortical remodeling. J. Bone Miner. Res. 26, 1035–1046 (2011).
Bivi, N., Lezcano, V., Romanello, M., Bellido, T. & Plotkin, L. I. Connexin43 interacts with barrestin: a prerequisite for osteoblast survival induced by parathyroid hormone. J. Cell. Biochem. 112, 2920–2930 (2011).
Ugland, H., Naderi, S., Brech, A., Collas, P. & Blomhoff, H. K. cAMP induces autophagy via a novel pathway involving ERK, cyclin E and Beclin 1. Autophagy 7, 1199–1211 (2011).
de Castro, L. F., Maycas, M., Bravo, B., Esbrit, P. & Gortazar, A. VEGF receptor 2 (VEGFR2) activation is essential for osteocyte survival induced by mechanotransduction. J. Cell. Physiol. 230, 278–285 (2015).
Clarkin, C. E. & Gerstenfeld, L. C. VEGF and bone cell signalling: an essential vessel for communication? Cell. Biochem. Funct. 31, 1–11 (2013).
Domazetovic, V. et al. Entroges inhibits starvation-induced apoptosis in osteocytes by a redox-independent process involving association of JNK and glutathione S-transferase P-1. FEBS Open. Bio. 7, 705–718 (2017).
Plotkin, L. I., Bivi, N. & Bellido, T. A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone 49, 122–127 (2011).
Gortazar, A. R., Martin-Millan, M., Bravo, B., Plotkin, L. I. & Bellido, T. Crosstalk between caveolin-1/extracellular signal-regulated kinase (ERK) and beta-catenin survival pathways in osteocyte mechanotransduction. J. Biol. Chem. 288, 8168–8175 (2013).
Kuang, M. J. et al. Exosomes derived from Wharton’s jelly of human umbilical cord mesenchymal stem cells reduce osteocyte apoptosis in glucocorticoid-induced osteonecrosis of the femoral head in rats via the miR-21-PTEN-AKT signalling pathway. Int. J. Biol. Sci. 15, 1861–1871 (2019).
Hayashi, M. et al. Osteoprotection by semaphorin 3A. Nature 485, 69–74 (2012).
Hwang, J. Y. et al. Association of PLXNA2 polymorphisms with vertebral fracture risk and bone mineral density in postmenopausal Korean population. Osteoporos. Int. 17, 1592–1601 (2006).
Marathe, N., Rangaswami, H., Zhuang, S., Boss, G. R. & Pilz, R. B. Pro-survival effects of 17beta- estradiol on osteocytes are mediated by nitricoxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J. Biol. Chem. 287, 978–988 (2012).
Chai, S., Wan, L., Wang, J. L., Huang, J. C. & Huang, H. X. Gushukang inhibits osteocyte apoptosis and enhances BMP-2/Smads signaling pathway in ovariectomized rats. Phytomedicine 64, 153063 (2019).
Kogianni, G., Mann, V. & Noble, B. S. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J. Bone Miner. Res. 23, 915–927 (2008).
Cardoso, L. et al. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J. Bone Miner. Res. 24, 597–605 (2009).
Cheung, W. Y., Simmons, C. A. & You, L. Osteocyte apoptosis regulates osteoclast precursor adhesion via osteocytic IL-6 secretion and endothelial ICAM-1 expression. Bone 50, 104–110 (2012).
Liu, P. et al. Loss of menin in osteoblast lineage affects osteocyte-osteoclast crosstalk causing osteoporosis. Cell Death Differ. 24, 672–682 (2017).
He, F. et al. Irradiation-induced osteocyte damage promotes HMGB1-mediated osteoclastogenesis in vitro. J. Cell Physiol. 234, 17314–17325 (2019).
Al-Dujaili, S. A. et al. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J. Cell. Biochem. 112, 2412–2423 (2011).
Nakashima, T. et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 (2011).
Davis, H. M. et al. High mobility group box 1 protein regulates osteoclastogenesis through direct actions on osteocytes and osteoclasts in vitro. J. Cell. Biochem. 120, 16741–16749 (2019).
Bivi, N. et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 27, 374–389 (2012).
Kennedy, O. D., Laudier, D. M., Majeskam, R. J., Sun, H. B. & Schaffler, M. B. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone 64, 132–137 (2014).
Cheung, W. Y. et al. Pannexin-1 and p2X7-receptor are required for apoptotic osteocytes in fatigued bone to trigger RANKL production in neighboring bystander osteocytes. J. Bone Miner. Res. 31, 890–899 (2016).
Plotkin, L. I. et al. Inhibition of osteocyte apoptosis prevents the increase in osteocytic receptor activator of nuclear factor kappaB ligand (RANKL) but it does not stop bone resorption or the loss of bone induced by unloading. J. Biol. Chem. 290, 18934–18942 (2015).
Ormsby, R. T. et al. Osteocytes respond to particles of clinically-relevant conventional and cross-linked polyethylene and metal alloys by up-regulation of resorptive and inflammatory pathways. Acta Biomater. 87, 296–306 (2019).
Delgado-Calle, J. et al. MMP14 is a novel target of PTH signaling in osteocytes that controls resorption by regulating soluble RANKL production. Faseb. J. 32, 2878–2890 (2018).
Yang, C. N. et al. Simvastatin alleviates bone resorption in apical periodontitis possibly by inhibition of mitophagy-related osteoblast apoptosis. Int. Endod. J. 52, 676–688 (2019).
Zhang, Y. et al. PSMC6 promotes osteoblast apoptosis through inhibiting PI3K/AKT signaling pathway activation in ovariectomy-induced osteoporosis mouse model. J. Cell Physiol. 235, 5511–5524 (2020).
Moriishi, T. et al. Overexpression of BCLXL in osteoblasts inhibits osteoblast apoptosis and increases bone volume and strength. J. Bone Miner. Res. 31, 1366–1380 (2016).
Deng, S. et al. Dexamethasone induces osteoblast apoptosis through ROS-PI3K/AKT/GSK3β signaling pathway. Biomed. Pharmacother. 110, 602–608 (2019).
Song, X. et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochem. Biophys. Res. Commun. 480, 443–449 (2016).
Zhu, X. et al. Inhibition of pyroptosis attenuates staphylococcus aureus-induced bone injury in traumatic osteomyelitis. Ann. Transl. Med. 7, 170 (2019).
Yang, L., Liu, J., Shan, Q., Geng, G. & Shao, P. High glucose inhibits proliferation and differentiation of osteoblast in alveolar bone by inducing pyroptosis. Biochem. Biophys. Res. Commun. 522, 471–478 (2020).
Daddona, P. E., Matriano, J. A., Mandema, J. & Maa, Y. F. Parathyroid hormone (1–34)-coated micro needle patch system: clinical pharmacokinetics and pharmacodynamics for treatment of osteoporosis. Pharm. Res. 28, 159–165 (2011).
Neale, J. R. et al. Bone selective effect of an estradiol conjugate with a novel tetracycline-derived bone-targeting agent. Bioorg. Med. Chem. Lett. 19, 680–683 (2009).
Hirabayashi, H. & Fujisaki, J. Bone-specific drug delivery systems. Clin. Pharmacokinet. 42, 1319–1330 (2003).
Liang, C. et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. Nat. Med. 21, 288–294 (2015).
Sun, Y. et al. Osteoblast-targeting-peptide modified nanoparticle for siRNA/microRNA delivery. ACS Nano. 10, 5759–5768 (2016).
Qiao, H. et al. Targeting osteocytes to attenuate early breast cancer bone metastasis by theranostic upconversion nanoparticles with responsive plumbagin release. ACS Nano. 11, 7259–7273 (2017).
Acknowledgements
This work was funded by the youth medical talents funding subject from the 13th five-year plan on science and education strengthening health project of Jiangsu province [Grant No. QNRC2016355 to Dr Yan-fen Wang and Grant No. QNRC2016356 to Dr. Jiang-ying Ru]; the key research and development (social development) funding project of Yangzhou city to Dr. Jiang-ying Ru [Grant No. YZ2018086]; the social development funding project of Yangzhou city to Dr Yan-fen Wang [Grant No. YZ2019055]; the special funding subject from the 13th five-year plan on science and education strengthening health project of Yangzhou city [Grant No. ZDRC201878 to Dr. Yan-fen Wang and Grant No. ZDRC201879 to Dr. Jiang-ying Ru].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Y. Shi
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ru, Jy., Wang, Yf. Osteocyte apoptosis: the roles and key molecular mechanisms in resorption-related bone diseases. Cell Death Dis 11, 846 (2020). https://doi.org/10.1038/s41419-020-03059-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03059-8
This article is cited by
-
Glucocorticoid-induced osteoporosis—from molecular mechanism to clinical practice
Drugs & Therapy Perspectives (2024)
-
Targeting strategies for bone diseases: signaling pathways and clinical studies
Signal Transduction and Targeted Therapy (2023)
-
Quercetin potentiates the anti-osteoporotic effects of alendronate through modulation of autophagy and apoptosis mechanisms in ovariectomy-induced bone loss rat model
Molecular Biology Reports (2023)
-
Assembling the Puzzle Pieces. Insights for in Vitro Bone Remodeling
Stem Cell Reviews and Reports (2023)
-
Fluoride Exposure Provokes Mitochondria-Mediated Apoptosis and Increases Mitophagy in Osteocytes via Increasing ROS Production
Biological Trace Element Research (2023)
