
Abstract
Soft tissue sarcomas (STS) are a heterogeneous group of malignancies predominantly affecting children and young adults. Despite improvements in multimodal therapies, 5-year survival rates are only 50% and new treatment options in STS are urgently needed. To develop a rational combination therapy for the treatment of STS we focused on ABT-199 (Venetoclax), a BCL-2 specific BH3-mimetic, in combination with the proteasome inhibitor bortezomib (BZB). Simultaneous inhibition of BCL-2 and the proteasome resulted in strongly synergistic apoptosis induction. Mechanistically, ABT-199 mainly affected the multidomain effector BAX by liberating it from BCL-2 inhibition. The combination with BZB additionally resulted in the accumulation of BOK, a BAX/BAK homologue, and of the BH3-only protein NOXA, which inhibits the anti-apoptotic protein MCL-1. Thus, the combination of ABT-199 and BZB sensitizes STS cells to apoptosis by simultaneously releasing several defined apoptotic restraints. This synergistic mechanism of action was verified by CRISPR/Cas9 knock-out, showing that both BAX and NOXA are crucial for ABT-199/BZB-induced apoptosis. Noteworthy, efficient induction of apoptosis by ABT-199/BZB was not affected by the p53 status and invariably detected in cell lines and patient-derived tumor cells of several sarcoma types, including rhabdomyo-, leiomyo-, lipo-, chondro-, osteo-, or synovial sarcomas. Hence, we propose the combination of ABT-199 and BZB as a promising strategy for the treatment of STS, which should warrant further clinical investigation.
Similar content being viewed by others


Introduction
Soft tissue sarcomas (STS) comprise a heterogeneous group of malignancies of mesenchymal origin with more than 100 different histologic subtypes and entities1. Sarcomagenesis arises from various alterations, such as fusion proteins resulting from genomic rearrangements, mutations in tumor-suppressor and cell cycle regulatory genes, and DNA copy-number variations. Sarcomas are traditionally classified in two major genetic groups: the first group includes sarcomas with simple karyotypes and specific genetic alterations like EWS-ATF1 fusions in Ewing sarcomas, KIT mutations in GISTs, or PAX3-FKHR fusions in alveolar rhabdomyosarcomas. The second group is represented by complex genetic aberrations associated with impaired p53 checkpoint function, dysfunctional DNA repair by homologous or nonhomologous end joining, and an increase of unrepaired double-strand breaks. Complex karyotypes in STS are frequently related to BRAF, RAS (H-, K-, and N-), PTEN, RB1, BRCA2, APC, and PIK3CA mutations2.
Due to the genetic heterogeneity of STS, especially in a metastatic disease, therapeutic options are limited and the average 5-year survival rate, regardless of the type of sarcoma, is only 50%3. A commonly used chemotherapeutic treatment option especially for young patients is doxorubicin that shows a rather steep dose-response curve4. Because resistance to monotherapies frequently occurs in sarcomas, the combined treatment with doxorubicin and ifosfamide or additional anti-cancer drugs is thought to overcome drug resistance. In several clinical trials such combination therapies showed promising improvements in progression-free survival (PFS). However, the 5-year survival rate was only marginally affected5 and, due to its high toxicity, doxorubicin in combination with ifosfamide is a rational treatment option only in young and fit patients. Other combinations of doxorubicin, e.g., with the HSP-90 inhibitor 17-DMAG and the histone deacetylase inhibitor vorinostat, have been shown to synergistically increase cell death in cultured small cell sarcomas6. Efficacy of vorinostat in the treatment of sarcoma has also been tested together with the proteasome inhibitor bortezomib (BZB), but despite good tolerability patients did not respond in a clinical phase II trial7. In line, although proteasome inhibition was recently shown to be effective in the treatment of Ewing sarcoma8, BZB was ineffective in other STS9. Thus, efficient treatment of sarcomas still remains an unmet clinical need10.
A relatively new class of targeted anti-cancer drugs that are efficient especially in the treatment of hematopoietic malignancies are BH3-mimetics/BCL-2 inhibitors11. BH3-mimetics are small molecule drugs that - like BH3-only proteins (e.g. BAD, BID, BIM, NOXA, and PUMA) – specifically bind to anti-apoptotic proteins of the BCL-2 family (BCL-2, BCL-xL, BCL-w, MCL-1, and A1). The BCL-2 inhibitors occupy the hydrophobic cleft in anti-apoptotic proteins that physiologically accommodates the BH3-domain of pro-apoptotic proteins. Thereby, BH3-mimetics prevent binding of pro-apoptotic BH3-only or multidomain effector proteins (BAX, BAK, and BOK). Ultimately, the apoptosis effectors are no longer counteracted by anti-apoptotic proteins, and active conformers of BAX, BAK, and BOK are free to oligomerize and permeabilize the mitochondrial outer membrane. Permeabilization releases cytochrome c into the cytosol and initiates activation of cell-death-specific proteases, the caspases, that establish apoptotic demise of the cell12.
Like BH3-only proteins, the pro-apoptotic multidomain effector proteins specifically interact with anti-apoptotic BCL-2 proteins. Although interaction of BOK with anti-apoptotic proteins is debated13,14, the binding preferences of BAK and BAX are largely established. BAK preferentially binds to MCL-1 and BCL-xL15, while BAX binds to BCL-2, BCL-xL, and BCL-w16. Thus, the prototypic and broad BH3-mimetic ABT-737 can induce cell death in various tumor cell types, but has dose-limiting side effects such as thrombocytopenia17. A successor of ABT-737 is the orally available BH3-mimetic ABT-199 (Venetoclax) that shows enhanced specificity for BCL-2. ABT-199 induces cell death in several hematopoietic malignancies and is also tested in other cancer entities. In line with its specific binding to BCL-2, ABT-199 tends to be more efficient in multiple myeloma (MM) with high expression of BCL-2 and less effective in MCL-1 overexpressing MM18. This limitation might be overcome by combination of ABT-199 with the newly developed MCL-1 specific inhibitors, such as A-1210477 and others19,20.
Despite sequence homology to the pore-forming effector proteins BAX and BAK, the protein BOK has only recently been verified as a bona fide pro-apoptotic multidomain protein of the BCL-2 family21. BOK, similar to BAX and BAK, is an apoptosis effector that mediates mitochondrial outer membrane permeabilization (MOMP)21,22. The regulation of BOK-induced apoptosis is rather unconventional, because BOK shows “intrinsic instability” or “metastability”, meaning that BOK spontaneously changes its conformation from an inactive to an active state23. How active BOK is prevented from apoptosis induction is rather unclear. Although inhibition of BOK by anti-apoptotic BCL-2 proteins, such as MCL-1, has been repeatedly shown13,14,21, a recent publication proposed that BOK does neither interact with anti-apoptotic BCL-2 proteins nor requires activation by BH3-only proteins22. Rather, BOK activity is controlled at the level of protein stability and is prevented from constitutive apoptosis induction by the AMFR/gp78 E3 ubiquitin ligase complex that targets BOK to proteasomal degradation. Consequently, inhibition of BOK degradation by the proteasome inhibitor MG132 was shown to stabilize BOK and induce apoptosis, while knock-down of BOK attenuates MG132-induced apoptosis22. Interestingly, proteasome inhibitors have also been reported to induce expression of the pro-apoptotic BH3-only protein NOXA24. NOXA specifically binds and inactivates the anti-apoptotic proteins MCL-1 and A125 that in turn predominantly counteract the effectors BAK26,27,28 and allegedly BOK13,14.
Intrigued by proteasome inhibitor mediated stabilization of BOK22 and the reported induction of NOXA expression29, we hypothesized that the combined inhibition of the proteasome and anti-apoptotic BCL-2 proteins by BH3-mimetics should synergize in apoptosis induction. Indeed, we show here that the combined application of ABT-199 and the proteasome inhibitor BZB synergistically induces apoptosis in STS cell lines and primary tumor-derived cells irrespective of their tissue origin. As the underlying mechanism we identified that the drug combination not only inhibited BCL-2 but also enhanced expression of BOK and the MCL-1 antagonist NOXA. As a net result, BCL-2 and MCL-1 are simultaneously liberated from their prebound pro-apoptotic BCL-2 proteins. In line with the proposed mechanism, CRISPR/Cas9-mediated knock-out of BAX, but not BAK or BOK, significantly reduced apoptosis induction by ABT-199/BZB. Moreover, the additional knock-down of NOXA in BAX-deficient cells most efficiently blocked apoptosis induction by ABT-199/BZB. We propose that the combined treatment with ABT-199/BZB is a promising strategy to improve the therapy of various STS types independent of their tissue origin.
Results
Tumor cells frequently overexpress anti-apoptotic BCL-2 proteins and have therefore become “BCL-2 addicted”30. BCL-2 addicted cells are prone to apoptosis induction by small-molecule inhibitors of anti-apoptotic BCL-2 proteins, such as ABT-19931. In addition to directly blocking anti-apoptotic proteins, tumor cells can also be sensitized toward apoptosis by shifting the equilibrium of BCL-2 proteins32 towards pro-apoptotic members, such as BH3-only or multidomain effector proteins. Because expression of the BH3-only protein NOXA as well as the pro-apoptotic effector BOK22 is enhanced by proteasome inhibition, we hypothesized that reducing the anti-apoptotic capacity by ABT-199 and simultaneously enhancing the pro-apoptotic activity of NOXA and BOK by the proteasome inhibitor BZB might be a rational strategy in STS.
ABT-199/BZB synergistically reduce viability
Initially, we performed dose-response viability assays in RD (rhabdomyosarcoma) and SW982 (synovial sarcoma) cells with mutant and wildtype p53, respectively. Cell viability of RD and SW982 cells was assayed after 48 h of incubation with various concentrations of ABT-199 and/or BZB. In both cell lines single drug treatments led to a moderate decrease in cell viability (Fig. 1a). The combination of BZB and ABT-199 was however strongly synergistic, as also confirmed by calculation of the combination index. Importantly, similar synergistic activity was seen in primary tumor cells derived from patients with rhabdomyosarcoma (P-RMS) and synovial sarcoma (P-SS), respectively (Fig. 1b). Also, microscopic evaluation confirmed that neither ABT-199 nor BZB alone elicited significant toxicity, while the combination of both drugs acted in concert to decrease cell viability (Fig. 1c).

a, b RD, SW982, and corresponding patient-derived tumor cells were incubated with the indicated concentrations of ABT-199 and/or BZB for 48 h. Cell viability was assessed by CellTiter-Glo Luminescent assay and normalized to the DMSO vehicle control. Drug interaction and synergisms were analyzed by calculation of the combination index (CI) using CompuSyn software. c Microscopic assessment of RD and SW982 shows only marginal numbers of floating (dead) cells 24 h after incubation with 5 nM BZB or 15 µM ABT-199 alone. In contrast, ABT-199/BZB massively induces cell death. d RD and SW982 cells were incubated in the absence (ctrl) or presence of 5 nM BZB, 15 µM ABT-199, or both for 8 h in the presence of Q-VD-OPh, before expression of the indicated proteins was analyzed by western blotting. Asterisk represents identical GAPDH blot because identical membrane was probed.
To investigate the mechanism of the combined BCL-2 protein and proteasome inhibition in more detail, we analyzed expression of BOK, NOXA, and p53 by western blotting after incubation of cells with ABT-199 and/or BZB (Fig. 1d). p53 mutant RD cells revealed a high constitutive expression of p53, while p53 was upregulated by the proteasome inhibitor in p53wt SW982 cells. BZB augmented expression of BOK and NOXA as well as of the NOXA-inhibitory protein MCL-1 that is acceptedly degraded by the proteasome33,34 (Fig. 1d). Intriguingly, also ABT-199 treatment alone led to the accumulation of BOK, NOXA, and MCL-1. Moreover, although more pronounced in SW982 and to a lesser extent also in RD cells, ABT-199/BZB strongly augmented the expression of BOK, NOXA, and MCL-1 (Fig. 1d).
ABT-199/BZB induce apoptotic cell death
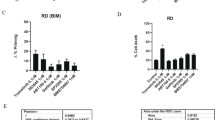
Intrigued by the efficient cell death induction by ABT-199/BZB, we thought to analyze the mode of cell death in more detail. RD and SW982 cells and corresponding primary tumor cells, i.e. P-RMS and P-SS, were treated with ABT-199 and BZB alone or in combination for 24 h. Subsequently, cell nuclei were stained with propidium iodide and relative DNA content was analyzed by flow cytometry; cells with hypodiploid DNA (subG1) were assumed apoptotic35. In these analyses <22% of hypodiploid cells were detected after incubation with BZB or ABT-199 alone in each sarcoma cell line and tumor-derived cells. In contrast, simultaneous incubation with ABT-199/BZB-induced apoptosis in 54–76% of the cells (Fig. 2a, b).

a, b RD and SW982 cells were incubated in the presence of 5 nM BZB and 15 µM ABT-199 alone or in combination. After 48 h the percentage of cells with hypodiploid DNA (subG1) was assessed by flow cytometry. c, d RD and SW982 cells were incubated with 5 nM BZB and/or 15 µM ABT-199 for 24 h. Subsequently, exposure of phosphatidylserine (Annexin V) and loss of mitochondrial membrane potential (TMRM) were analyzed by flow cytometry. e SW982 cells were incubated without or with ABT-199/BZB for 24 h. Immunofluorescence microscopy shows activation/clustering of the effectors BAX, BAK, and BOK (green) in response to ABT-199/BZB.
These results are in line with the proposed hypothesis that inhibition of BCL-2 and the proteasome synergistically induce cell death. To corroborate that cell death is apoptotic we analyzed exposure of phosphatidylserine (PS) and loss of ΔΨm, two established hallmarks of apoptosis, by staining cells with fluorescence-labeled Annexin V or TMRM. In line with the previous results, <20% of RD and SW982 cells exposed PS after incubation with ABT-199 or BZB alone, while >50% (RD) and >90% (SW982) Annexin V+ cells were detected after incubation with the drug combination (Fig. 2c and Supplemental Fig. 2A). Compared to the single drug treatments, the proportion of cells exhibiting reduced mitochondrial membrane potential was also significantly higher after simultaneous incubation with ABT-199 and BZB, i.e. 77% in RD and 87% in SW982 cells (Fig. 2d, Supplemental Fig. 2B). Thus, the combination of ABT-199 and BZB induces cell death by apoptosis, as evidenced by DNA fragmentation, exposure of PS and loss of mitochondrial membrane potential. Finally, fluorescence microscopy revealed clustering and, hence, activation of BAX, BAK, and BOK after incubation with ABT-199/BZB (Fig. 2e) that was not detected in cells incubated with BZB or ABT-199 alone (Supplemental Fig. 1).
Specific synergism of BZB with BCL-2 inhibition
To delineate the role of anti-apoptotic BCL-2 proteins in ABT-199/BZB-induced apoptosis, we further utilized recently developed BCL-xL and MCL-1 specific inhibitors. RD and SW982 cells were incubated with the BCL-xL inhibitor A-115546336 or the MCL-1 specific inhibitor A-121047720 alone or in combination with BZB. After 24 h cells were flow cytometrically analyzed for apoptosis induction. Similar to ABT-199, A-1155463, and A-1210477, when applied alone, elicited only marginal exposure of PS or loss of mitochondrial membrane potential in ~20% of RD or SW982 cells (Fig. 3a–d). However, compared to ABT-199, the combination of the BCL-xL inhibitor A-1155463 and BZB resulted in reduced Annexin V+ and TMRMlow cells, with <45% of RD and SW982 being apoptotic (Fig. 3a, b). The MCL-1 inhibitor A-1210477 and BZB even less efficiently induced apoptosis with <20% of cells positive for PS exposure (Fig. 3c) and <25% of cells showing low ΔΨm (Fig. 3d). Hence, the synergism of BZB and ABT-199 is clearly based on specific inhibition of BCL-2 rather than of BCL-xL or MCL-1.

RD and SW982 cells were incubated with 5 nM BZB and/or 15 µM of the BCL-xL inhibitor A-1155463 (a, b) or 15 µM of the MCL-1 inhibitor A-1210477 (c, d). After 24 h, exposure of phosphatidylserine (a, c) and loss of mitochondrial membrane potential (b, d) were analyzed by flow cytometric staining with Annexin V and TMRM, respectively.
BAX is a key effector of ABT-199/BZB-induced apoptosis
Though these data identify BCL-2 rather than BCL-xL or MCL-1 as the anti-apoptotic BCL-2 protein that is most relevant for the synergism with proteasome inhibition, we next explored the identity of the pro-apoptotic effector BCL-2 protein responsible for the release of cytochrome c from mitochondria. In view of the complex interplay of BCL-2 family proteins and the redundant function of effector proteins in MOMP, it is likely that the synergism is based on a set rather than one single effector protein. To elucidate the relevance of the known effectors BAX, BAK, and BOK, we generated SW982 cell lines devoid of each of these proteins by CRISPR/Cas9-mediated KO technology. As verified by western blotting (Supplemental Fig. 3), we selected clonal cell lines with individual knock-outs of BAX, BAK, or BOK, which retained similar expression of other BCL-2 family proteins. These cell lines were incubated with ABT-199/BZB and analyzed for apoptosis by flow cytometry. ABT-199/BZB-induced Annexin V+ (Fig. 4a, b) and TMRMlow (Fig. 4c, d) cells, which was similar in the parental wt, BAK or BOK knock-out SW982 cells. Interestingly, however, in SW982/BAXKO cells apoptosis induction and loss of mitochondrial membrane potential by the drug combination were most strongly reduced (Fig. 4a, c). Thus, the synergistic apoptosis induction by inhibition of BCL-2 largely depends on BAX rather than on BAK or BOK. This is in line with the weak sensitization of BZB-challenged cells by inhibitors of BCL-xL or MCL-1 (Fig. 3) as well as the fact of BCL-2 being the main BAX antagonist28.

SW982 knock-out cells deficient in the effector proteins BAX, BAK, or BOK were generated by CRISPR/Cas9 technology and incubated with 5 nM BZB and/or 15 µM ABT-199 for 24 h. Apoptosis induction was evaluated by flow cytometric measurements of phosphatidylserine exposure using Annexin V (a, b) or of the mitochondrial membrane potential using TMRM (c, d).
NOXA contributes to synergistic cell death induction by ABT-199/BZB
Data so far shows that the synergistic activity of ABT-199/BZB largely relies on the BCL-2/BAX axis. Because BZB enhances expression of NOXA that inhibits the BAK/BOK antagonist MCL-1, we further aimed to pinpoint the role of NOXA in the synergistic apoptosis induction. Hence, we transfected SW982/wt and different KO cell lines with siRNA to knock-down expression of NOXA and subsequently analyzed apoptosis induction by ABT-199/BZB. In wt, BAKKO and BOKKO SW982 cells knock-down of NOXA had little impact on apoptosis induction, and ~80% of the cells were apoptotic in each cell line after incubation with ABT-199/BZB (Supplemental Fig. 4A, B). In contrast, in SW982/BAXKO cells knock-down of NOXA diminished cell death induction to 40% as compared to 80% in SW982/wt cells (Fig. 5a, c). Similar results were obtained in an inverse approach by knocking down BAX in SW982/NOXAKO cells (Fig. 5a, c and Supplemental Fig. 4C). Thus, the data corroborates the hypothesis that NOXA, by blocking MCL-1, targets a BCL-2/BAX-independent axis of apoptosis regulation. In combination, inhibition of BCL-2 and NOXA together are instrumental for synergistic cell death induction by ABT-199/BZB.

SW982/wt and SW982/BAXKO or SW982/NOXAKO were transfected with siRNA-NOXA or siRNA-BAX and incubated with 5 nM BZB and/or 15 µM ABT-199. Apoptosis induction was evaluated after 24 h by measuring a phosphatidylserine exposure (Annexin V-APC+) and c loss of mitochondrial membrane potential (TMRMlow). b, d Relative reduction of phosphatidylserine exposure (b) and loss of mitochondrial membrane potential (d) by siRNA-mediated knock-down of NOXA and BAX in wt and BAK-, BOK-, or BAX-deficient SW982 cells in response to treatment with ABT-199/BZB.
ABT-199/BZB is effective in various sarcoma cell lines and tumor-derived cells
Although data in RD and SW982 cells show that ABT-199 and BZB synergistically induce cell death regardless of the status of p53, there are typically varying genetic aberrations in soft tissue sarcomas. To account for the genetic heterogeneity, we analyzed additional STS cell lines for their response to ABT-199/BZB. We found that combined application of ABT-199/BZB effectively and in most cases synergistically reduced viability of STS cell lines, i.e., SW1353 (chondrosarcoma; CI: 0.78), SK-LMS (leiomyosarcoma; CI: 0.44), Sa-OS (osteosarcoma; CI: 0.83), and SW872 (liposarcoma; CI: 0.92; Fig. 6a–d). These data indicate that the synergistic action of ABT-199/BZB is largely independent of the genetic background of sarcoma cell lines (Supplemental Table 1). Moreover, synergistic cell death induction was found in patient-derived primary cells of different STS types (Fig. 6e–h). In line with the reduced viability, we detected significantly more hypodiploid (subG1) cells in each of the cell lines (Fig. 7a) and primary tumor cells (Fig. 7b) in response to incubation with ABT-199/BZB. Thus, these findings suggest that the combined use of ABT-199 and BZB is a promising strategy for the treatment of soft tissue sarcomas, irrespective of their tissue origin and p53 status.

a–d Sarcoma cell lines and e–h primary tumor-derived cells of different STS types were incubated in the presence of the indicated concentrations of BZB, ABT-199, or both. After 48 h cell viability was analyzed by CellTiter-Glo Luminescent cell viability assay and normalized to the DMSO control. Viability curves, IC50, and CI values were calculated using GraphPad Prism and CompuSyn. BZB and ABT-199 synergism is indicated by CI values <1. P-CS primary chondrosarcoma, P-LMS primary leiomyosarcoma, P-OS primary osteosarcoma, P-LS primary liposarcoma.

Cell lines and primary tumor-derived cells of different STS types (Fig. 6) were additionally treated with 5 nM BZB in the presence or absence of 15 µM ABT-199 for 48 h. After 48 h flow cytometric staining with propidium iodide was used to determine the percentage of cells with hypodiploid DNA (subG1).
Discussion
The mitochondrial apoptosis signaling pathway is regulated by BCL-2 family proteins. The BCL-2 multidomain effector proteins BAX, BAK, and BOK mediate permeabilization of the mitochondrial outer membrane by pore formation. The pore formation by BAX and BAK relies on an activating conformational change that might occur spontaneously15,37,38 or after transient interaction with BH3-only proteins39,40. The active conformers are then either inhibited by anti-apoptotic BCL-2 proteins or accumulate in pore-forming multimers. In contrast to BAX and BAK, BOK shows intrinsic instability41 and a significant proportion of BOK proposedly is in its active conformation. Thus, to prevent constitutive apoptosis induction, BOK is broadly controlled by proteasomal degradation22. Also, expression of the MCL-1 inhibiting pro-apoptotic BH3-only protein NOXA is enhanced by inhibition of the proteasome42,43.
Given this information we hypothesized that the combined action of ABT-199 and BZB, i.e., enhanced expression of the MCL-1 antagonist NOXA and stabilization of the effector BOK together with simultaneous inhibition of anti-apoptotic BCL-2, exacerbates apoptosis by releasing restraints on BAX, BAK, and BOK. In line with this hypothesis and the proteasomal degradation of BOK22, we show enhanced expression of NOXA and BOK in RD and SW982 cells incubated with ABT-199/BZB. The expression of NOXA and BOK (Fig. 1d) and efficient apoptosis induction by the combined presence of ABT-199/BZB is therefore well compatible with the above-mentioned hypothesis. Furthermore, we provide evidence that this drug combination efficiently induces cell death in several STS cell lines (RD, SW982, SW1353, SK-LMS, Sa-OS, and SW872). Because these STS cell lines differ in their genetic alterations and tissue origin, we propose that the therapeutic strategy is more widely applicable in the treatment of advanced STS. This proposition is corroborated by effective apoptosis induction in cells derived from STS tumor samples: irrespective of their genetic alterations also cells derived from patients effectively undergo apoptosis in response to ABT-199/BZB.
BZB had already been tested earlier in the treatment of STS in a dose escalation study but was found to be largely ineffective9. Similarly, the combined treatment of BZB with histone deacetylase inhibitor vorinostat, while being well tolerated, mostly failed to show responses in a phase I study in advanced malignancies. In contrast, we here combined BZB with the BCL-2 inhibitor ABT-199. ABT-199 is effective in the treatment of hematopoietic malignancies11. Both drugs are in clinical use and well tolerated, suggesting that the combination of both drugs is a suitable therapeutic option for the treatment of STS. Especially in view of the limited treatment options for STS our findings warrant further clinical investigation. Therefore, it is conceivable that the combination of ABT-199/BZB is also applicable in patients that cannot receive high-dose doxorubicin-based therapy.
CRISPR/Cas9-mediated knock-out of BAX reveals that BAX is most relevant for synergistic action of ABT-199/BZB. Also, as compared to ABT-199, the specific inhibition of BCL-xL or MCL-1 does not synergize with BZB. This is in line with ABT-199 targeting the BCL-2/BAX-axis while BZB independently targets the MCL-1/BAK&BOK-axis by enhancing expression of NOXA and BOK (Supplemental Fig. 5).
ABT-199 is effective in CLL, AML, non-Hodgkin lymphoma11,44,45,46, and likely in multiple myeloma with t11;14 translocation18. Interestingly, acquired resistance to ABT-199 in AML cell lines is overcome by the MCL-1 specific inhibitor VU661013, which synergizes with ABT-199 in apoptosis induction47. The synergistic activity of ABT-199 and VU661013 is well compatible with the here proposed mechanism for the synergism of ABT-199 and BZB. Proteasome inhibition acceptedly enhances expression of the MCL-1 inhibiting BH3-only protein NOXA42,48. Originally, NOXA (PMAIP1) has been described as a p53-induced BH3-only protein49, which is in line with stronger induction of NOXA expression in p53-proficient SW982 compared to p53-defective RD cells. Regardless of the specific mechanism of enhanced NOXA expression, e.g. enhanced p53-dependent or independent50 transcription or reduced proteasomal degradation42,43, the net result is inhibition of anti-apoptotic MCL-1. In summary, the proposed strategy inactivates the BAX antagonist BCL-2 and the BAK antagonist MCL-1. Thus, the anti-apoptotic capacity of cells to restrain BAX and BAK is largely reduced.
Additionally, BZB-mediated block of BOK degradation shifts the BCL-2 rheostat even further towards cell death. The shift towards apoptosis still prevails even when considering the BZB-induced increase of MCL-1 expression, because also expression of NOXA is enhanced. In this scenario apoptosis is augmented irrespective of whether BOK interacts with anti-apoptotic BCL-2 proteins13,14 or not22,51. Although interaction of BOK with anti-apoptotic BCL-2 proteins is debated, stabilization of BOK and MCL-1 might even be caused upstream of the proteasome by interaction of BOK with MCL-1. Indeed, BOK and MCL-1 have been proposed to interact at least functionally and constitute a regulatory feedback52. Non-proteasomal regulation of BOK expression by mRNA stability, as proposed by Oyneagucha et al.52, was also published by Fernandez-Marrero et al.53. Thus, there is increasing evidence that there are levels of regulation for the expression of BOK other than proteasomal degradation.
In this study we propose the combination of BZB and ABT-199 for the treatment of patients with soft tissue sarcoma. The data in the present study is well in line with analog approaches to treat patients with relapsed/refractory multiple myeloma (MM). A phase Ib trial (NCT1794507) and an ongoing phase III trial (NCT02755597) indicate promising results for the combined treatment of MM patients with ABT-199 and BZB54,55. In another trial (NCT01794520), the benefit from ABT-199/BZB for relapsed/refractory MM patients was associated with high BCL-2 expression (i.e. BCL-2/BCL-xL or BCL-2/MCL-1 ratio): 12 out of the 14 patients with t(11:14) translocation, which correlates with enhanced BCL-2 expression, showed a positive response18. In general, the combination of specific BCL-2 inhibitors with other approved anti-cancer drugs repeatedly proved a successful strategy in various tumor models56,57). The data presented here suggests that BCL-2 inhibition by BH3-mimetics is also a valid option for the treatment of solid tumors, especially in combination with established and effective drugs56,58,59,60,61,62.
Materials and methods
Tumor-derived cells and STS cell lines
Patients from the University Tübingen Center for Soft Tissue Sarcomas and Bone Tumors who underwent surgical resection of primary or metastatic tumors were included in this study (Supplementary Table 2). All patients had given written informed consent to the scientific analysis of tissue samples, the study was approved by the ethics committee of the University of Tübingen. Fresh tumor samples were obtained after surgical resection. Primary sarcoma samples were mechanically dissociated into 1–2 mm3 tissue fragments, washed, and cultured in standard media containing 1% Antibiotic-Antimycotic (Gibco, Life Technologies, Darmstadt, Germany). All cells and cell lines were cultured in media (DMEM, DMEM/F12, or MEM; Gibco) supplemented with 10% fetal calf serum (FCS; Biochrom) and 1% Antibiotic-Antimycotic. MEM was additionally supplemented with 1% Na-pyruvate and 1% glutamine. Human STS cell lines including SW982, SK-LMS-1, SW872, RH30/RD, SW1353, and Sa-OS were authenticated by STR-profiling at the DSMZ and ATCC and cultured in medium as stated in Supplemental Table 2. Cells were harvested after incubation in trypsin/EDTA solution, centrifuged at 600 × g for 5 min and processed for subsequent analysis as described.
Viability assay
Cell viability was assessed using CellTiter-Glo Assay (Promega, Mannheim, Germany) according to the manufacturer’s instructions. Briefly, 4000 cells per well were seeded in white 96-well plates with plastic bottom (Invitrogen, Karlsbad, Germany). After 2 h BZB (0.5–10 nM; Selleckchem, Munich, Germany) and ABT-199 (1.5–30 μM; Selleckchem) were added. Cell viability was analyzed in quadruples after 48 h.
Flow cytometry
Annexin V and TMRM staining
Cells were harvested using trypsin/EDTA, resuspended in supernatant and washed in ice-cold PBS. Then, pellets were resuspended in 300 μL Annexin V-binding buffer (PBS, 2.5 mM CaCl2) supplemented with Annexin V-APC (1:100) and incubated for 20 min. Subsequently, samples were analyzed using a FACS Lyric flow cytometer. To assess the mitochondrial membrane potential, cell pellets were resuspended in PBS supplemented with 2% FCS and 50 nM of the potentiometric dye TMRM. Cells were incubated at 37 °C for 20 min and fluorescence was analyzed using a FACS Lyric flow cytometer. The proportion of TMRMlow and Annexin V+ cells was calculated using FACS Suite software.
Nicoletti assay
Cells were harvested by trypsination, washed in ice-cold PBS, and resuspended in ice-cold hypotonic buffer N (0.1% sodium citrate, 0.1% Triton X-100, and 50 µg/mL propidium iodide) as described63. After 30 min of incubation at 4 °C relative cellular DNA content was detected using a Becton Dickinson FACS Calibur flow cytometer (Becton Dickinson, Heidelberg, Germany) and Cell Quest Software. Cells showing relative DNA content <2N were assumed apoptotic.
Antibodies
The following antibodies were used: BAX (Cell Signaling, #2772), BAK (Cell Signaling, #3814), BOK (Abcam, #186745), BCL-2 (Cell Signaling, #15071), MCL-1 (Cell Signaling, #5453), NOXA (Merck, #OP180), p53 (Santa Cruz, #sc-126), GAPDH (Cell Signaling, #2118), TOM20 (Becton Dickinson, #612278). Secondary anti-mouse (#7076S), and anti-rabbit (#7074S) horseradish-coupled antibodies were from Cell Signaling. Chicken anti-rabbit Alexafluor488 and goat anti-mouse DyeLight 649 were from Thermo Fisher Scientific (Waltham, MA, USA) and Abcam (Berlin, Germany), respectively.
Western blot
Cells were harvested by scraping, washed in ice-cold PBS, and lysed in 150 μL/10 cm2 lysis buffer (50 mM Tris-HCl pH 7.6, 250 mM NaCl, 0.1% Triton X-100, and 5 mM EDTA) supplemented with protease and phosphatase inhibitors (complete and PhosphoSTOP, Roche, Basel, Switzerland). Samples were sonified (Diagenode, Liège, Belgium) and cleared by centrifugation (15 min, 4 °C, 14000 × g). Relative protein content of supernatants was assessed using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein (typically 30 μg) were separated by SDS-PAGE and blotted (Biometra FastblotTM, Analytic Jena, Jena, Germany) onto nitrocellulose membrane (0.1 μm; GE Healthcare, Munich, Germany) by semi-dry blotting (1 mA/cm2, 1 h). Primary antibody was applied in PBST (0.1% Tween-20) overnight at 4 °C. Membranes were washed thrice 10 min incubated with horseradish-coupled secondary antibody (1:2000) for 2 h at room temperature. After washing, ECL solution was applied (SuperSignal™ West Dura, Thermo Fisher Scientific) and specific bands were detected using a Stella gel documentation system (Raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany).
Fluorescence microscopy
SW982 cells were seeded on coverslips (#1) in 12-well plates at a density of 5 × 104/well. The next day, medium was removed and cells were incubated in medium supplemented with 10 µM Q-VD-OPh and the indicated drugs. After 24 h, cells were fixed in PBS/4% formaldehyde for 30 min on ice, washed in PBS, and incubated overnight at 4 °C with the following primary antibodies in PBS/4% BSA/0.05% Saponin: mouse anti-TOM20 (clone 29, BD Biosciences), anti-BAK-NT (06–536, Merck Millipore), anti-BAX-NT (06–499, Merck Millipore), or anti-BOK (EPR15331, Abcam). Samples were washed in PBS and incubated with fluorescence-labeled secondary antibodies (chicken anti-rabbit Alexafluor488, Life Technologies and goat anti-mouse DyeLight649, Abcam) for 4 h. Then, cells were washed in PBS supplemented with 100 ng/mL DAPI for 5 min, washed twice in PBS and mounted using DAKO fluorescent mounting medium (Life Technologies). Fluorescence microscopy images were taken using a Leica TCS SP8 confocal laser scanning microscope. Images (512 × 512 pixel) were taken using excitation at 405 nm, 488 nm, and 647 nm at 600 Hz scan speed with line average and identical laser intensity/amplification of detectors for each series.
CRISPR/Cas9 Knock-out
For lentivirus production 1 × 107 HEK2983FT cells/flask were transfected with 10 μg pMD2.G, 15 μg psPAX2, and 20 μg plentiCRISPRv2 containing the desired guideRNA64 using PEI (polyethylenimine 40,000; Warrington, PA, USA) reagent (1 mg/mL PEI in 25 mM HEPES pH 7.5 and 150 mM NaCl). Virus containing supernatant was collected 48 h and 72 h after transfection and concentrated by ultracentrifugation. SW982 cells were transduced with 10 μL concentrated virus overnight and then cultured with 1 μg/mL puromycin (Thermo Fisher Scientific) for 48 h. For the generation of knock-out cell lines deficient in BAK, BOK, BAX, or NOXA, clones were isolated and protein expression was analyzed by western blotting.
RNA interference
0.6 × 105 cells/well were seeded in 12-well plates 24 h prior to transfection. Then, cells were transfected with ON-TARGET Plus Smartpool siRNAs targeting BAX or NOXA or non-targeted (NT) Smartpool ON-TARGET plus control siRNA (Horizon Discovery, Waterbeach, UK) using Dharmafect Ι reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. After 24 h cells were incubated with ABT-199 and/or BZB for additional 24 h. Then, cells were harvested and analyzed by flow cytometry. For verification of knock-down efficacy, cells were lysed and assessed through western blot analysis as mentioned previously.
Statistical analysis
Continuous variables are presented as mean ± SD and categorical variables are given by number and percentages. The statistical significance of differences was analyzed using Student’s t test. All statistical tests were considered significant when p < 0.05. Statistical analyses were computed using GraphPad Prism (v5.04) or Microsoft Excel (Professional Plus 2019).
References
Hoang, N. T., Acevedo, L. A., Mann, M. J. & Tolani, B. A review of soft-tissue sarcomas: translation of biological advances into treatment measures. Cancer Manag. Res. 10, 1089–1114 (2018).
Movva, S. et al. Multi-platform profiling of over 2000 sarcomas: identification of biomarkers and novel therapeutic targets. Oncotarget 6, 12234–12247 (2015).
Weitz, J., Antonescu, C. R. & Brennan, M. F. Localized extremity soft tissue sarcoma: improved knowledge with unchanged survival over time. J. Clin. Oncol. 21, 2719–2725 (2003).
In, G. K., Hu, J. S. & Tseng, W. W. Treatment of advanced, metastatic soft tissue sarcoma: latest evidence and clinical considerations. Ther. Adv. Med. Oncol. 9, 533–550 (2017).
Tap, W. D. et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 18, 1089–1103 (2017).
Dumont, S. N. et al. Targeted polytherapy in small cell sarcoma and its association with doxorubicin. Mol. Oncol. 8, 1458–1468 (2014).
Attia, S. et al. A phase II consortium trial of vorinostat and bortezomib for advanced soft tissue sarcomas. J. Clin. Oncol. 29, 10075–10075 (2011).
Shukla, N. et al. Proteasome addiction defined in ewing sarcoma is effectively targeted by a novel class of 19S proteasome inhibitors. Cancer Res. 76, 4525–4534 (2016).
Maki, R. G. et al. A multicenter Phase II study of bortezomib in recurrent or metastatic sarcomas. Cancer 103, 1431–1438 (2005).
Taylor, B. S. et al. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 11, 541–557 (2011).
Roberts, A. W. & Huang, D. Targeting BCL2 with BH3 mimetics: basic science and clinical application of venetoclax in chronic lymphocytic leukemia and related B cell malignancies. Clin. Pharmacol. Ther. 101, 89–98 (2017).
Fulda, S., Galluzzi, L. & Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464 (2010).
Hsu, S. Y., Kaipia, A., McGee, E., Lomeli, M. & Hsueh, A. J. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc. Natl Acad. Sci. USA 94, 12401–12406 (1997).
Stehle, D. et al. Contribution of BH3-domain and Transmembrane-domain to the activity and interaction of the pore-forming Bcl-2 proteins Bok, Bak, and Bax. Sci. Rep. 8, 12434 (2018).
Willis, S. N. et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19, 1294–1305 (2005).
Fletcher, J. I. et al. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl Acad. Sci. USA 105, 18081–18087 (2008).
Adams, J. M. & Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27–36 (2018).
Kumar, S. et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 130, 2401–2409 (2017).
Bruncko, M. et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J. Med. Chem. 58, 2180–2194 (2015).
Leverson, J. D. et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 6, e1590 (2015).
Einsele-Scholz, S. et al. Bok is a genuine multi-BH-domain protein that triggers apoptosis in the absence of Bax and Bak. J. Cell Sci. 129, 3054 (2016).
Llambi, F. et al. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165, 421–433 (2016).
Moldoveanu, T. & Zheng, J. H. Metastability, an emerging concept governing BOK-mediated apoptosis initiation. Oncotarget 9, 30944–30945 (2018).
Fennell, D. A., Chacko, A. & Mutti, L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene 27, 1189–1197 (2008).
Dutta, S. et al. Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J. Mol. Biol. 398, 747–762 (2010).
Ku, B., Liang, C., Jung, J. U. & Oh, B. H. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 21, 627–641 (2011).
Zhang, S. & Link, A. J. Bcl-2 family interactome analysis using bacterial surface display. Integr. Biol. 3, 823–831 (2011).
Kale, J., Osterlund, E. J. & Andrews, D. W. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 25, 65–80 (2018).
Kubiczkova, L., Pour, L., Sedlarikova, L., Hajek, R. & Sevcikova, S. Proteasome inhibitors - molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 18, 947–961 (2014).
Certo, M. et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365 (2006).
Scheffold, A., Jebaraj, B. M. C. & Stilgenbauer, S. Venetoclax: targeting BCL2 in hematological cancers. Recent Results Cancer Res. 212, 215–242 (2018).
Korsmeyer, S. J., Shutter, J. R., Veis, D. J., Merry, D. E. & Oltvai, Z. N. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin. Cancer Biol. 4, 327–332 (1993).
Adams, K. W. & Cooper, G. M. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J. Biol. Chem. 282, 6192–6200 (2007).
Iglesias-Serret, D., Pique, M., Gil, J., Pons, G. & Lopez, J. M. Transcriptional and translational control of Mcl-1 during apoptosis. Arch. Biochem. Biophys. 417, 141–152 (2003).
Nicoletti, I., Migliorati, G., Pagliacci, M. C., Grignani, F. & Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139, 271–279 (1991).
Tao, Z. F. et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med. Chem. Lett. 5, 1088–1093 (2014).
Edlich, F. et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116 (2011).
Willis, S. N. et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856–859 (2007).
Czabotar, P. E., Lessene, G., Strasser, A. & Adams, J. M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 (2014).
Lovell, J. F. et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084 (2008).
Zheng, J. H. et al. Intrinsic instability of BOK enables membrane permeabilization in apoptosis. Cell Rep. 23, 2083–2094. e6 (2018).
Baou, M. et al. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica 95, 1510–1518 (2010).
Craxton, A. et al. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell Death Differ. 19, 1424–1434 (2012).
Davids, M. S. et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-hodgkin lymphoma. J. Clin. Oncol. 35, 826–833 (2017).
Konopleva, M. et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 6, 1106–1117 (2016).
Stilgenbauer, S. et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 17, 768–778 (2016).
Ramsey, H. E. et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 8, 1566–1581 (2018).
Hagenbuchner, J. et al. The anti-apoptotic protein BCL2L1/Bcl-xL is neutralized by pro-apoptotic PMAIP1/Noxa in neuroblastoma, thereby determining bortezomib sensitivity independent of prosurvival MCL1 expression. J. Biol. Chem. 285, 6904–6912 (2010).
Oda, E. et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 (2000).
Gutekunst, M. et al. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 73, 1460–1469 (2013).
Echeverry, N. et al. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 20, 785–799 (2013).
Onyeagucha, B. et al. Novel post-transcriptional and post-translational regulation of pro-apoptotic protein BOK and anti-apoptotic protein Mcl-1 determine the fate of breast cancer cells to survive or die. Oncotarget 8, 85984–85996 (2017).
Fernandez-Marrero, Y., Bachmann, D., Lauber, E. & Kaufmann, T. Negative regulation of BOK expression by recruitment of TRIM28 to regulatory elements in its 3’ untranslated region. iScience 9, 461–474 (2018).
Matulis, S. M. et al. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia 30, 1086–1093 (2016).
Moreau, P. et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 130, 2392–2400 (2017).
Inoue-Yamauchi, A. et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat. Commun. 8, 16078 (2017).
Montero, J. & Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 25, 56–64 (2018).
Carter, R. J. et al. Exploring the potential of BH3 mimetic therapy in squamous cell carcinoma of the head and neck. Cell Death Dis. 10, 912 (2019).
Deng, J. et al. Bruton’s tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia 31, 2075–2084 (2017).
Kim, E. Y., Jung, J. Y., Kim, A., Chang, Y. S. & Kim, S. K. ABT-737 synergizes with cisplatin bypassing aberration of apoptotic pathway in non-small cell lung cancer. Neoplasia 19, 354–363 (2017).
Kuroda, J. et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc. Natl Acad. Sci. USA 103, 14907–14912 (2006).
Panayotopoulou, E. G. et al. Targeting of apoptotic pathways by SMAC or BH3 mimetics distinctly sensitizes paclitaxel-resistant triple negative breast cancer cells. Oncotarget 8, 45088–45104 (2017).
Essmann, F., Engels, I. H., Totzke, G., Schulze-Osthoff, K. & Janicke, R. U. Apoptosis resistance of MCF-7 breast carcinoma cells to ionizing radiation is independent of p53 and cell cycle control but caused by the lack of caspase-3 and a caffeine-inhibitable event. Cancer Res. 64, 7065–7072 (2004).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Acknowledgements
We thank Kerstin Willecke for expert technical assistance. This study was supported by grants from the Robert Bosch Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by H.U. Simon
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muenchow, A., Weller, S., Hinterleitner, C. et al. The BCL-2 selective inhibitor ABT-199 sensitizes soft tissue sarcomas to proteasome inhibition by a concerted mechanism requiring BAX and NOXA. Cell Death Dis 11, 701 (2020). https://doi.org/10.1038/s41419-020-02910-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-02910-2
