
Abstract
Cell death has a fundamental impact on the evolution of degenerative disorders, autoimmune processes, inflammatory diseases, tumor formation and immune surveillance. Over the past couple of decades extensive studies have uncovered novel cell death pathways, which are independent of apoptosis. Among these is necroptosis, a tightly regulated, inflammatory form of cell death. Necroptosis contribute to the pathogenesis of many diseases and in this review, we will focus exclusively on necroptosis in humans. Necroptosis is considered a backup mechanism of apoptosis, but the in vivo appearance of necroptosis indicates that both caspase-mediated and caspase-independent mechanisms control necroptosis. Necroptosis is regulated on multiple levels, from the transcription, to the stability and posttranslational modifications of the necrosome components, to the availability of molecular interaction partners and the localization of receptor-interacting serine/threonine-protein kinase 1 (RIPK1), receptor-interacting serine/threonine-protein kinase 3 (RIPK3) and mixed lineage kinase domain-like protein (MLKL). Accordingly, we classified the role of more than seventy molecules in necroptotic signaling based on consistent in vitro or in vivo evidence to understand the molecular background of necroptosis and to find opportunities where regulating the intensity and the modality of cell death could be exploited in clinical interventions. Necroptosis specific inhibitors are under development, but >20 drugs, already used in the treatment of various diseases, have the potential to regulate necroptosis. By listing necroptosis-modulated human diseases and cataloging the currently available drug-repertoire to modify necroptosis intensity, we hope to kick-start approaches with immediate translational potential. We also indicate where necroptosis regulating capacity should be considered in the current applications of these drugs.
Similar content being viewed by others
Facts
-
Necroptosis is closely associated with the pathogenesis of many human diseases.
-
The in vivo appearance of necroptosis indicates that both caspase-independent and caspase-dependent mechanisms control this cell death pathway.
-
More than 70 human molecules play a role in the regulation of necroptosis.
-
More than 20 approved drugs have the potential to regulate necroptosis.
Open Questions
-
How can we monitor and regulate necroptosis in human diseases?
-
What are the main molecular targets in caspase independent regulatory mechanisms of necroptosis?
-
How effective can the off-label use of already approved drugs in necroptosis-driven diseases be?
Introduction
The development and homeostasis of multicellular organisms depends on the balance between cell proliferation and cell death. In the past few years new regulated cell death pathways have been discovered and classified1. One of these tightly controlled inflammatory cell death pathways – necroptosis – has come to the center of attention because of its known contribution to the pathogenesis of many diseases1,2.
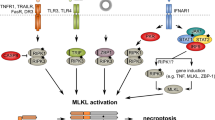
Many death-, pattern recognition-, DNA binding-, adhesion, and dependence-receptors, immune reactions, pathogens and various drugs have been identified as necroptosis triggers1,3. Necroptosis utilizes a signaling pathway requiring the involvement of receptor interacting protein kinase 3 (RIPK3)4, mixed lineage kinase domain-like protein (MLKL)5 and upon stimulation of death receptors (DR)2 RIPK1. RIPK3 oligomerization and its subsequent phosphorylation allows the RIPK3-MLKL interaction and the double phosphorylation of MLKL by RIPK36. After this step, MLKL forms oligomers and translocates to the plasma membrane to execute necroptosis (Fig. 1). Generally, necroptosis requires inhibition of caspases3,7 or the absence of the pro-caspase-8-activating adaptor Fas-associated protein with death domain (FADD)8, demonstrating the crucial role of the apoptotic platform in the negative regulation of necroptosis. Active caspases block necroptosis2 preferentially through the cleavage of RIPK19, RIPK33,10, and cylindromatosis (CYLD) protein11 which acts as the de-ubiqutinase enzyme of RIPK1. During DR-mediated signaling, inhibitors of apoptosis proteins (IAPs) initiate the ubiquitination of RIPK1 and this process favors cell survival12. Blockage of IAPs or the subsequent events of IAP-induced signaling strongly support necroptosis13. Various molecular pathways have been documented as regulators of downstream necroptotic events beside MLKL-mediated membrane rupture, but the complexity of the signaling and regulation network of necroptosis are still not fully understood.

Various extra - or intracellular signals activates the RIPK3 protein directly or through RIPK1. RIPK3-mediated phosphorylation induces MLKL membrane translocation and consequently, ion influx results in necroptosis147. Survival signals through upregulation of IAPs or activation of TAK1 kinase pathway blocks RIPK1-induced signaling and protects cells from unwanted necroptosis. Caspase-8-mediated cleavage of pro-necroptotic RIPK1 and RIPK3 ensures the dominance of immunologically silent apoptosis to immune stimulant necroptosis
The immunological outcome of cell death can be classified as anti-inflammatory or pro-inflammatory and tolerogenic or immunogenic1. Dominance of apoptosis ensures the tolerogenic outcome of cell death under physiological conditions. When apoptosis signaling is blocked, necroptotic pathways are activated and the dying cells have the potential to initiate innate immune responses via production of damage associated molecules (DAMPs) resulting in an inflammatory response14. Signaling in necroptotic cells also supports the cross priming capacity of dendritic cells (DCs)15.
In this review our goal was to understand the molecular background of necroptosis in humans and to find potential points of clinical intervention. We summarized how the expression, posttranslational modification, and localization of necroptotic molecules are regulated and what the interaction partners of the necrosome complex are. Finally, we provide an overview of drugs, which are already used in the clinic and have been shown to affect necroptosis.
Necroptosis involved in human diseases
Currently, necroptosis is mainly documented in various in vivo mice models16,17, but regulated necrosis contributes to the pathogenesis of many human diseases (Table 1). Both up and down-regulation of necroptosis and misregulation of the apoptosis-necroptosis transition which modifies the immunological outcome of cell death contribute to the evolution of degenerative disorders, autoimmune processes, inflammatory diseases or the immune surveillance of tumors.
Some physiological processes such as alteration of glucose level, oxygen deprivation or immune reactions resulted in elevated RIPK3 expression allowing in vivo emergence of necroptosis. Hyperglycemia (35–40 mM glucose) markedly enhanced the expression of RIPK3 in various cell lines and primed cells for necroptosis18,19. Similarly, upregulated expression of RIPK1, RIPK3 and MLKL, and increased RIPK1/3 complex formation have been observed in hypoxic cells20,21,22. At the same time caspase-8 mRNA, functioning as a negative regulator of necroptosis, was reported to be transiently decreased following the deprivation of oxygen and glucose (OGD)23. These processes are also involved in brain injury caused by hypoxia-ischemia and OGD-induced necroptosis24,25. Type I26,27,28 and type II27,29 interferons have been published to induce increased expression of RIPK3, while constitutive IFNβ signaling was demonstrated to increase the intracellular level of MLKL28. CD8+T lymphocytes can trigger both apoptosis and necroptosis, which make these cells capable of killing tumor cells, even those that escaped apoptosis30. T cell-mediated necroptotic cytolysis also plays a role in activation induced cell death, and can be critical in the development of autoimmune reactions31.
Upregulation of necroptosis in human diseases
Necroptosis takes part in the pathogenesis of human neurodegenerative disorders, such as Multiple Sclerosis (MS)32, Alzheimer’s disease (AD)33,34, and Amyotrophic Lateral Sclerosis (ALS)35,36. Defects in the activation of caspase-8 were demonstrated in the pathologic process of MS. Additionally, activated forms of RIPK1, RIPK3 and MLKL were detected in the cortical lesions of human MS samples32. Activated RIPK1 as a marker of necroptosis was also observed in human AD brains correlating positively with Braak stage and negatively with brain mass and cognition33,34. In ALS samples, multiple biochemical hallmarks of necroptosis including increased levels of RIPK1, RIPK3 and MLKL and elevated pRIPK1 and pMLKL were detected in both microglia and oligodendrocytes. Importantly, pMLKL was primarily localized in the white matter, where demyelination was found35. In spinal cord injury strong RIPK3 expression and MLKL phosphorylation were detected37.
In certain cardiovascular diseases, such as chronic heart failure (HF) cell loss and subsequent deterioration of contractile function is associated with elevated expression of RIPK1, RIPK3, and pRIPK3. On the other hand, the expression of caspase-8 was downregulated suggesting activation of necroptosis signaling. MLKL expression did not differ among the control and HF groups; however, pMLKL were present in all HF samples, which is in contrast to the controls where this was almost undetectable38. A genetic variant in the RIP3 promoter region was associated with increased RIPK3 transcription, which contributed to the poor prognosis of HF patients39.
In humans with unstable carotid atherosclerosis, expression of RIPK3 and MLKL was increased, while the phosphorylation of MLKL was detected in advanced atheromas40. In patients with abdominal aorta aneurysm, the tissue showed elevated levels of RIPK1 and RIPK3 proteins41,42. In coronary artery disease higher plasma RIPK3 levels were detected than in controls43.
Regarding gastrointestinal diseases, increased RIPK3 expression was detected in liver biopsies from patients with alcoholic liver disease17, while both RIPK3 and MLKL expression was increased in non-alcoholic fatty liver diseases44,45, as well as elevated MLKL phosphorylation in drug-induced liver injury46. High levels of RIPK3 and MLKL phosphorylation were also detected in the liver biopsies of patients with primary biliary cholangitis, in contrast with its low hepatic expression in healthy controls47. Similarly, increased levels of RIPK3 were documented in the terminal ileum of patients with Crohn’s disease17 and elevated RIPK3 and MLKL levels were observed in inflamed tissues of inflammatory bowel disease (IBD) and allergic colitis patients, whereas the expression of caspase-8 in these tissues was reduced48. The migration of human neutrophils to sites of inflammation was found to activate the RIPK3-MLKL pathway: a strong pMLKL signal was observed in infiltrating tissue neutrophils in samples collected from patients with cutaneous vasculitis, ulcerative colitis, and psoriasis49,50.
Phosphorylation of MLKL molecules was also detected in human acute kidney injury biopsies51, in biopsies taken immediately after excision for transplantation52 and in autosomal dominant polycystic kidney disease53 representing involvement of necroptosis in renal disorders. Antineutrophil cytoplasmic antibody (ANCA) induces neutrophil extracellular traps via necroptosis and causes subsequent endothelial cell damage. ANCA-associated vasculitis exhibited a specific p-MLKL staining in glomerular neutrophils in human kidney biopsies54.
Concerning skin diseases, human biopsy samples obtained from patients with Lichen Planus (LP) and Systemic lupus erythematosus (SLE) confirm the role of necroptosis in their development. RIPK3 and MLKL activation was demonstrated in podocytes in renal biopsies from patients with lupus nephritis55. LP and SLE tissue sections showed enhanced epidermal expression of phosphorylated RIPK356. B cells from SLE patients also significantly displayed high expression levels of necroptosis-related genes57. As we already mentioned, phosphorylation of MLKL in the infiltrated human neutrophils was also found in cutaneous vasculitis and psoriasis49,50. Upregulation of RIPK3, and elevated MLKL phosphorylation were observed in the skin samples from patients with toxic epidermal necrolysis in correlation with unwanted necroptosis and subsequent inflammation58.
Expression of RIPK3 and dynamin-related protein 1 (Drp1) was increased in lung tissue homogenates collected from patients suffering from chronic obstructive pulmonary disease, proving the role of necroptotic cell death in pulmonary diseases59. In Kashin–Beck disease (KBD) necroptosis dominates as a cell death mechanism in the middle zone of cartilage from KBD children60. Necroptotic cell death is involved in the progression of chronic periodontitis, as gingival tissue in patients showed increased levels of RIPK1, RIPK3, and MLKL, as well as increased phosphorylation of MLKL61.
Although RIPK1 is one of the key molecules required for execution of necroptosis, patients with its complete deficiency due to homozygous mutations suffered from recurrent infections, early-onset of IBD and progressive polyarthritis. In vitro, cells with RIPK1 deficiency showed impaired mitogen-activated protein kinase activation and cytokine secretion and were prone to necroptosis62,63.
Role of necroptosis in cancers
An increasing number of studies have been published about the importance of necroptotic cell death in anti-cancer therapies, which have been extensively reviewed in recent papers64,65.
Briefly, both pro- and anti-tumoral effects have been demonstrated following necroptosis in cancer development and progression. The anti-tumoral effect of necroptosis has been shown in many types of cancer in which the expression of RIPK366,67 or MLKL68 was silenced or polymorphisms in their coding genes lead to modified expression of necrosomal components66,69. In general, necroptosis resistance of cancer cells is a common process, and escape from necroptosis was suggested to be a potential hallmark of cancer, similar to the escape from apoptosis64. Additionally, effective anti-cancer agents trigger immunogenic cell death, inducing the killing of the transformed cells and provoking the members of innate and adaptive immune system to attack. Beside the massive release of DAMPs, necroptotic cells create a great possibility to trigger the activation of CD8 + T cells via cross presentation15,70. The dual ability of necroptosis to activate innate and adaptive immunity simultaneously makes this cell death pathway a promising therapeutic target.
However, the tumor-promoting outcome of necroptosis has also been shown. RIPK3 and MLKL expression seems to vary among tissue samples from different subtypes and stages of cancer, and downregulation of necroptosis mediators has also been published in various cancers71,72,73. Upregulated RIPK3 expression is a general phenomenon in tumor necrotic areas playing a critical role in tumor growth and metastasis74. Necroptosis-induced inflammation contributes to tumorigenesis and necroptosis can also lead to an immunosuppressive tumor microenvironment75. The immune-suppressing environment was associated with necroptosis-induced expression of the chemokine attractant CXCL171. It has also been shown that tumor cells induce necroptosis of endothelial cells, which promotes tumor cell extravasation and metastasis76. Thus, we can conclude that necroptosis occurs in different phases during tumorigenesis and plays an ambivalent role in tumor formation.
Molecular mechanisms in the regulation of necroptosis
To understand the molecular background of necroptosis and to find potential points of clinical intervention we summarize below how the expression, the posttranslational modification, and the localization of key necroptotic molecules (RIPK1, RIPK3 and MLKL) are regulated, while also highlighting the interaction partners of the necrosome complex.
Regulation the expression level of necroptotic proteins
RIPK3-RIPK3 homodimerization is sufficient to induce necroptosis; after which, its kinase domain stimulates the activation of RIPK3 through cis-autophosphorylation; a prerequisite step for the recruitment of MLKL77,78,79. Thus, RIPK3 dimerization is probably the most critical point of necroptosis induction. Several lines of evidence support the idea that increased expression of RIPK3 can induce its oligomerization and can initiate necroptosis42,80. RIPK1 dimerization, and accordingly upregulation of RIPK1, facilitates RIPK3 oligomerization, mainly upon death receptor stimuli.
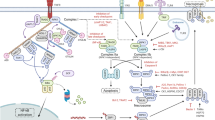
All aspects of necroptotic protein expression are intensely regulated, including their transcriptional activity, the stability of the expressed molecules and their degradation. Specificity protein 1 (Sp1), a zinc-finger transcription factor, directly regulates RIPK3 expression in cancer cells. Knockdown of endogenous Sp1 significantly decreases the transcription of RIPK3, while re-expression of Sp1 restores necroptotic response in vitro81. Induction of necroptosis by interferon gamma (IFN-γ) resulted in elevated levels of RIPK327 and MLKL28,29,82. This effect was found to depend on janus kinase 1 (JAK1) and its substrates: the signal transducer and activator of transcription 1 (STAT1) and interferon regulatory factor (IRF) transcription factors, pinpointing interferon-stimulated gene factor 3 (ISGF3) as a critical promoter83. Bromodomain-containing protein 4 (BRD4), a member of the bromodomain and extraterminal domain (BET) family, has been shown to interact IRF1 and to upregulate MLKL transcription84. Oncogenes such as BRAF and AXL have also been implicated in the regulation of RIPK3 expression67. The activity of RIPK3 promoter is tightly controlled by methylation67,85,86,87 (Fig. 2a). Ubiquitin-like PHD and RING finger domain-containing protein 1 (UHRF1) is essential for the maintenance of the hypermethylation of the RIPK3 promoter and thus contributes to the silencing of RIPK3 expression in quiescent cells.

Sp1 transcription factor increases RIPK3 expression. INFγ-mediated up-regulation of RIPK3 and MKLK level depend on JAK1 kinase, and STAT1 and IRF transcription factors. BRD4 cooperating with IRF1 also increase MLKL transcription. Hypermethylation of the RIPK3 promoter by UHRF1 results in silenced RIPK3 expression. The stability of all RIPK1, RIPK3 and MLKL proteins are increased by HSP90 and CDC37 co-chaperone complex and by FKBP12. The level of both RIPK1 and RIPK3 are down-regulated by caspase-8-mediated cleavage. Cathepsins are also capable of processing RIPK1. A20, CHIP, Optn, PELI1 and Triad3a ubiquitin-ligases mediate K48-linked polyubiquitylation and the subsequent proteasome dependent degradation of: RIPK1, RIPK3 and/or MLKL Upon necroptosis human RIPK1 is autophosphorylated at ser14, ser15, ser161, ser166 and RIPK3 at ser199 and ser227 and ser277. The transient phosphorylation of RIPK1 at ser321 is phosphorylated transiently by TAK1 leads to RIPK1-independent apoptosis and the sustained phosphorylation of RIPK1 by TAK1 at ser321, ser332, ser334 and ser336 induces RIPK1 kinase activation106. IKKα/IKKβ also phosphorylate RIPK1 at ser25 and thereby block RIPK1 activity108,214,215. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) mediates phosphorylation of RIPK1 at ser321 and ser336 and restrains integration of RIPK1 into the cytosolic death complex107,216,217. The phosphorylation at ser89 by a currently unknown kinase inhibits the RIPK1 kinase activity218. Ubiquitylation of RIPK1 at Lys115 by PELI219 or Lys377 by cIAP1, cIAP2 and Parkin220 promotes necroptosis. LUBAC complex and the deubiquitinase CYLD regulates M1 ubiquitination of RIPK1221. Lys363 ubiquitylation of RIPK3 leads to its proteasomal degradation. RIPK3 is responsible for the phosphorylation of MLKL at thr357 and ser358. TAM (Tyro3, Axl, and Mer) family of receptor tyrosine kinases phosphorylate MLKL on Tyr376 to facilitate MLKL oligomerization145. MLKL is also phosphorylated on Ser441 by a still unidentified kinase222. Caspase-8 mediates the cleavage and inactivation of RIPK1 at asp324 and RIPK3 at asp328. O-GlcNAcylation of the RIPK3 at thr467 by OGT prevents necroptosis223. Red names indicate interaction partners of RIPK1, RIPK3, MLKL which activate necroptosis, blue marks necroptosis inhibitors
Following transcriptional regulation multiple processes control the protein level of necrosome components. The heat shock protein 90 (HSP90) and CDC37 co-chaperone complex increases the stability of all RIPK188, RIPK389, and MLKL90 proteins. Consequently, inhibitors of HSP90 facilitated the degradation of these necroptotic components and potently blocked necroptosis91. Protein levels of RIPK1 and RIPK3 also decreased in FK506-binding protein 12 (FKBP12) knockdown cells92.
On the contrary, cells treated with Hsp70 inhibitors underwent cell death, because Hsp70 enhances the stability of necroptosis antagonists, the RIPK1 regulators: cIAP1/2, x-linked inhibitor of apoptosis protein (XIAP), and the cellular FLICE-like inhibitor protein (cFLIP)93.
The expression of necroptotic molecules are downregulated by cleavage and proteosomal degradation. The most well-known inhibitor of necroptosis, caspase-8 cleaves both RIPK19, RIPK394, and the necroptosis promoting deubiquitinase CYLD proteins11. In macrophages, cathepsins were also reported to be capable of processing RIPK1, which resulted in significant decrease in necroptotic cell death95.
Several ubiquitin-ligases mediate K48-linked polyubiquitylation and the subsequent proteasome dependent degradation of necroptotic molecules: RIPK1 is regulated by A2096, carboxyl terminus of Hsp70-interacting protein (CHIP; also known as STUB1)97, optineurin (Optn)35, Triad3a98, RIPK3 by CHIP97, Optn35, E3 ubiquitin ligase Pellino 1 (PELI1)99, and MLKL by Optn (Table 2)35. Knock down of any of these K48 ubiquitin-ligases increased the sensitivity of necroptosis in both in vitro and in vivo studies. (Fig. 2b).
Posttranslational modifications in the regulation of necroptosis
Accumulating evidence suggests that cell death pathways are finely tuned by posttranslational modifications, such as ubiquitination and phosphorylation. Multiple excellent recent reviews go into extensive detail about the role of these processes in necroptosis100, therefore we only provide a brief overview of these processes below. These pathways are mentioned in the tables and figures of this manuscript in the interest of providing a comprehensive visual guide to these processes as well (Fig. 2c).
The necrosome is formed due to the phosphorylation driven assembly of RIPK1, RIPK3, and MLKL4,80,101. However several phosphorylation steps have been published to inhibit necroptosis, chief among them the transforming growth factor beta-activated kinase 1 (TAK1) complex, which is the most important hub for these necroptosis-dampening signals102,103. Various protein complexes are assembled along TNFR signaling; namely the survival (complex I), the apoptotic (complex IIa and IIb) and the necroptosis inducer (complex IIc) complexes. Upon activation TNFR recruits TRADD, RIPK1, TRAF2, TRAF5 proteins. The gathered E3 ubiquitin ligases, cIAP-1 and cIAP-2 molecules, and the linear ubiquitin chain assembly complex LUBAC (consisting of HOIP, HOIL-1L and Sharpin)104 polyubiquitinates RIPK1, and modified RIPK1 can now act as a scaffold for TAK1 and the IKK complex105 which molecules in many ways block RIPK1-mediated cell death pathways, and thus the formation of complex II:106,107,108 These mechanism are: (1) By inducing the activation of NFκB and MAPK signaling pathways and thereby increasing the transcription of several survival molecules such as cIAP1/2109 and FLIP110 (2) by blocking the binding of cell death related molecules to RIPK1111 and (3) by phosphorylating RIPK1106,108.
Interaction partners of necrosome components
The activity of necrosome components are also mediated by molecular interactions (Fig. 2d). Three molecules, aurora kinase A (AURKA), PPM1b, and HSP90 have been recently identified as binding partners of RIPK390,91,112,113 and/ or RIPK191,112 in resting cells. AURKA112 and PPM1b113 act as local inhibitors against spontaneous necroptosis, since their silencing induces necroptosis. PPM1b as a phosphatase prevents RIPK3 autophosphorylation in resting cells113. AURKA together with its downstream target, Glycogen synthase kinase 3β (GSK3β) regulates the formation of RIPK1-RIPK3 and RIPK3-MLKL complexes112. Silencing or blocking of AURKA, or inhibitors of GSK3β result in necroptosis without any other stimuli. Phosphorylation of GSK3β at Ser9 suppresses necroptosis through interfering with the formation of RIPK3-MLKL complex, however the direct targets of GSK3β still have not been identified. The third molecule which associates with RIPK3 in resting cells, HSP90, is required for proper activation of necroptosis. Formation of the HSP90–CDC37 complex is necessary for RIPK1–RIPK3 interaction, thus it mediates RIPK3 activation during necroptosis. Unsurprisingly HSP90 inhibitors can block TNF-induced systemic inflammatory response syndrome (SIRS) in rats91. Additionally, membrane tethered mucins have been shown to interact with RIPK1 to block necroptosis in human bronchial epithelial cells in vitro114.
The nuclear retinoic acid receptor gamma (RARγ) is released from the nucleus to initiate the formation of cell death signaling complexes by mediating RIPK1 dissociation from TNFR when cIAP activity is blocked. In vitro silencing of RARγ inhibited necroptosis and in vivo results also confirmed that RARγ was essential for TNF-induced RIPK1-initiated apoptosis and necroptosis (Table 2)115.
Although RIPK1 initiates RIPK3 activation during death receptor driven necroptosis, it plays an ambivalent role in the regulation of RIPK3 aggregation. Under special circumstances instead of activation, RIPK1 acts to suppress the spontaneous activation of RIPK3 by TIR-domain-containing adapter-inducing interferon-β (TRIF)116 or DNA-dependent activator of IFN-regulatory factors (DAI; also known as ZBP1)78,117. RIPK3 oligomerization is able to seed a RHIM dependent oligomer and this process is both sufficient and a necessary step in necroptosis. RHIM domains of RIPK1 intrinsically inhibit RHIM-mediated RIPK3 aggregation by competing with the RHIM domain of TRIF or DAI; conversely death domain-driven RIPK1 oligomerization results in RIPK3 aggregation and necroptosis. In vivo results also reveal a kinase-independent function for RIPK1 in inhibiting necroptosis. Caspase-8/RIPK1 double-knockout animals die shortly after birth, however, additional ablation of RIPK3 to make caspase-8/RIPK1/RIPK3 triple knockouts rescues the viability of these animals117,118,119,120. These data undoubtedly prove the anti-necroptotic activity of RIPK1 under special conditions78.
MLKL association with RIPK3 is also suppressed by a constitutive interaction of MLKL with a competitive inhibitor, TRAF2, in resting cells. TRAF2 deubiquitination by CYLD promotes the dissociation of TRAF2 from MLKL and allows necroptosis121. Two other molecules inhibit cell death by blocking MLKL association with pro-necroptotic components: Repulsive guidance molecule b (RGMb) inhibits MLKL membrane translocation or membrane binding122 and Redox regulator thioredoxin-1 (TRX1) blocks MLKL disulfide bond formation, and through it the critical polymerization of MLKL123.
Various molecules have been published to act as downstream targets of RIPK3 and others to regulate MLKL localization and/or activation. RIPK3 constitutes an important upstream kinase of death associated protein (Daxx), triggering its nuclear export. The Akt/mTOR pathway124,125,126, and Ca2+/calmodulin-dependent protein kinase II (CaMKII)127 are also active effectors of downstream necroptotic signaling. Accordingly, several models suggest that effects on these signaling routes modify necroptotic intensity. Poly [ADP-ribose] polymerase 1 (PARP-1)128 (debated in ref. 129,130) and phosphoglycerate mutase family member 5 (PGAM5)131 (debated in ref. 132) have been documented as cell type specific regulators of downstream necroptotic events (Table 2).
Glucose metabolism and ROS production in necroptosis
Reactive oxygen species (ROS) have long been considered to contribute to necroptosis49,133,134,135. Oxidation of specific cysteine residues in RIPK1 by ROS activates RIPK1 autophosphorylation. A positive feedback loop is generated because silencing of RIPK1 or RIPK3 reduces ROS production. RIPK1 autophosphorylation is also promoted by mitochondrial ROS and is essential for RIPK3 recruitment into the necrosome. However, necroptosis could occur without ROS induction in some cell lines135,136.
Metabolic enzymes − human liver glycogen phosphorylase (PYGL), glutamate-ammonia ligase (GLUL), glutamate dehydrogenase 1 (GLUD1) − increase pyruvate production from glycogen or play a role in glutamine catabolism. These enzymes are activated by RIPK3, resulting in enhancement of aerobic respiration and thus likely contribute to TNF-induced ROS production80. Pyruvate dehydrogenase complex (PDC) converts pyruvate to acetyl-CoA, and triggers the entrance of metabolic flux into the tricarboxylic acid cycle. Activated RIPK3 in the necrosome enhances PDC activity by phosphorylating the PDC E3 at T135 and plays a major role in increasing aerobic respiration. Based on in vitro studies, activation of these enzymes has additive effects to aerobic respiration and ROS production (Table 2)80,134.
Intracellular localization of necrosome components
The intracellular localization of necrosome components seems to be crucial in the regulation of necroptosis. The RHIM domain of RIPK1 and RIPK3 mediates the assembly of heterodimeric filamentous structures, and the amyloid-like aggregation of RIPK1/RIPK3 complexes79. Compromised cluster formation correlated with decreased programmed necrosis. MLKL has also been reported to form SDS-resistant, disulfide bond-dependent polymers during necroptosis and it has been shown that these MLKL polymers were independent of RIPK1/RIPK3 fibers137.
MLKL translocation to the cell membrane is an obligatory step in necroptotic signaling. Phosphatidyl-inositol phosphates (PIPs) as critical binders of MLKL are required for plasma membrane targeting of MLKL and subsequent membrane permeabilization in necroptosis138,139. Highly phosphorylated inositol products, but not weakly phosphorylated precursors are able to displace the MLKL auto-inhibitory brace region, which is a necessary event for late plasma membrane breakdown and cell death140,141. Accordingly, necroptosis requires inositol polyphosphate-specific kinase activity and in cells containing mutant IP kinases, MLKL failed to oligomerize and localize to membranes despite proper RIPK3-dependent phosphorylation142. Deletion of inositol polyphosphate multikinase (IPMK), inositol-tetrakisphosphate 1-kinase (ITPK1)142 or inositol pentakisphosphate 2-kinase (IPPK)143 inhibited necroptosis. Connected to this, phosphatidylinositol transfer protein alpha (PITPα) interacts with MLKL which facilitates MLKL oligomerization and plasma membrane translocation144. Following membrane localization TAM (Tyro3, Axl, and Mer) family of receptor tyrosine kinases phosphorylate MLKL to protmote MLKL oligomerizatin and necroptosis145. Beside their direct pore forming ability, membrane-localized MLKL regulates transient receptor potential cation channel, subfamily M, member 7 (TRPM7), a non-voltage-sensitive ion channel, for the mediation of Ca2+ influx146,147.
Once MLKL is membrane associated, all the endosomal sorting complexes required for transport III machinery (ESCRT-III), flotillin-mediated endocytosis and ALIX-syntenin-1-mediated exocytosis act to sustain survival of the cell. The ESCRT-III-driven plasma membrane repair machinery limits the duration of the loss of plasma membrane integrity upon MLKL activation52,148, while endo- and exocytosis removes phospho-MLKL from the plasma mebrane149. MLKL also forms a complex with multiple membrane metalloproteinases upon necroptotic stimulus. A disintegrin and metalloproteinase (ADAM)-enzymes are activated to mediate the shedding of cell-surface proteins in response to necroptotic stimuli and through this process also play a key role in promoting necroptosis, but only in adherent cells (Table 2)150.
RIPK1151, RIPK3152,153, and MLKL154,155 have all been reported to localize to the nucleus and these translocations preceded necroptotic death154. RIPK3 and MLKL have been shown to became activated in the nucleus, and after their cooperative nuclear export, they contribute to cytosolic necrosome formation155. Following the interaction of RIPK3 and MLKL, the translocation of this complex to mitochondria-associated membranes has also been demonstrated and this relocation was found to be essential for necroptosis signaling156. The intracellular trafficking of necrosomes is regulated by the TNF-induced guanine nucleotide-binding protein γ 10 (Gγ10) – Src signaling pathway157, however, RIPK1/RIPK3 kinase activity has no direct interaction with Gγ10 or on Src kinase.
Drugs to regulate necroptosis intensity
In vitro studies prefer to use caspase inhibitors to activate necroptosis, however we still do not fully understand how necroptosis is activated under physiological conditions. The in vivo appearance of necroptosis indicates that in addition to caspase-mediated processes various caspase independent regulatory mechanisms control necroptosis. Drugs affecting either the expression or the activity of necroptosis mediators, or that modify the indirect regulators of necroptosis may have therapeutic potential (Tables 3 and 4).
Regulation the expression level of necrosome components
Drugs that control the promoters of RIPK3 or MLKL or modify the stability and degradation of these molecules can regulate necroptosis sensitivity. Interferons27,29, hypomethylating agents such as decitabine (5-aza-2′-deoxycytidine) and 5-azacytidine (used in Myelodysplastic syndromes and AML)87, histone deacetylase inhibitor valproic acid158 (VPA), anti-fungal miconazole159, traditional Chinese medicine drugs (shikonin160,161, resibufogenin162, bufalin163, youdujing164, emodin165), and components found in different plants (matrine166, genipine167, lycorine168, quercetin169, curcumol170, Bulnesia sarmientoi171) were all found to upregulate the expression of RIPK1 or RIPK3.
On the other hand, various inhibitors of the HSP90 have been documented to downregulate necroptosis (Kongensin A172, G-TPP173, geldanamycin174, gamitrinib10, DHQ3175 and 17-demethoxy-reblastatin175). Cyclosporine A176, Diacerein177 (Used in Europe and Asia to treat joint diseases), immunosuppressive and antiproliferative Rapamycin178 and traditional Chinese medicine such as patchouli alcohol179 have been also documented to reduce the expression of principal necroptotic mediators. Ex-527180 (which completed a phase II clinical trial in Huntington disease) regulates necroptosis through the inhibition of Sirt1 deacetylase.
Regulation the activity of necrosome components
Beside the expression of necrosome components, the activity of these enzymes is also modified by various drugs. Promising specific inhibitors are currently being developed for the central molecules of necroptosis. RIPK1, RIPK3, and MLKL (reviewed in refs. 181,182) which may interfere with unwanted cell death and subsequent inflammation. Multiple second mitochondria-derived activator of caspase (SMAC) mimetics and TAK-1 (reviewed in ref. 183,184) inhibitors are being tested in clinical trials to activate necroptosis for therapeutical intervention, by restoring the sensitivity of apoptosis-resistant tumors to cell death. Since these drugs are reviewed elsewhere, we focus on currently available necroptosis regulators.
Drugs currently used for the treatment of different forms of tumors display anti-necroptotic activity (Dabrafenib185,186, Sorafenib187,188, Pazopanib189, Ponatinib189, and Carfilzomib190) as does the anti-epilepsy drug Phenhydan191. Phenytoin51 (a clinically used anti-convulsant) or herbal components such us wogonin192 and aucubin193 inhibit RIPK1 activity. All these drugs provide immediate translational potential to dampen necroptosis-driven tissue degradation. Presumably, these drugs will be additive to the above-mentioned necroptosis inhibitors which downregulate the expression of necrosome components.
On the other hand, radiation194, or chemotherapeutic agents such as anthracyclines and oxaliplatin195, cisplatin196,197, 5-fluorouracil198 or the pan-BCL-2 inhibitor Obatoclax199 (several phase two trials have been completed), traditional Chinese medicines such as resibufogenin162 (also tested in phase II of a clinical trial on pancreatic cancer), aucubin193, tanshinone200 or neoalbaconol201 have been documented to upregulate necroptosis. Based on current results, these drugs regulate the activity, and not the expression of necroptotic component. As a mono-therapy these group of necroptosis regulators could be ineffective in tumors that downregulate the level of RIPK3 or MLKL, but these medicines may increase the effect of the above listed mediators in combination therapy following the restoration of RIPK1 or RIPK3 expression in cancer cells.
Regulation the signaling of necroptotic pathway
Some drugs regulate necroptosis by modulating the level or activity of partner molecules of the necrosome. For example, VPA induces the release of SMAC from mitochondria thereby upregulating necroptosis similarly to the widely tested SMAC mimetics. Dimethyl fumarate (DMF202, which is currently used in relapsing-remitting multiple sclerosis) induces necroptosis via downregulation of the negative regulators of necroptosis such as IAPs and cFLIPs. Aurora kinase inhibitors have been shown to directly induce necroptosis and stimulated intra-tumoral phosphorylation of MLKL203. Drugs antagonizing Trx1function as necroptosis inducers. PX-12123 (completed phase I of a clinical trial on advanced metastatic cancer) and DMF204 target TRX1 and have been shown to sensitize tumor cells to necroptosis.
Various drugs activate necroptosis via regulation of downstream components of necroptosis. Adiponectin receptor agonists205 (tested in various clinical trials), DMF202, neoablaconol206 induce ROS production. Lithium126 (clinically used for treating bipolar disorders) facilitates AKT-mTOR-mediated necroptosis, while dasatinib (used drug in CML) induces HMGB1-mediated necroptosis.
Necroptosis can be inactivated via the regulation of interacting partners of the necrosome or by downstream components, as well. The proteasome inhibitor Bortezomib207 (used in Multiple Myeloma treatment) and a HDAC inhibitor Vorinostat208 (approved for the treatment of Cutaneous T cell lymphoma) have been demonstrated to inhibit necroptosis through the upregulation of necroptosis inhibitors, sequentially stabilizing IAPs or increasing FLIP expression.
Various ROS scavengers have been implicated in the modulation of necroptosis209,210. Dexmedetomidine (used in moderate sedation) inhibits HMGB1 production211. Melatonin212 (used for jetlag sleep disorder) blocks PGAM5, while P110 is a selective inhibitor of Drp1213, therefore, these two drugs dampen the intensity of necroptosis via a well-documented PGAM-Drp1 pathway.
While there are no drugs on the market directly approved to regulate necroptosis, various medicines have the potential to both up and downregulate necroptosis, and to interact different levels of necroptosis signaling. Necroptosis has fundamental roles in various human diseases which makes it rational to try and apply the necroptosis regulator drugs in these syndromes.
References
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Holler, N. et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 (2000).
Vanden Berghe, T., Kaiser, W. J., Bertrand, M. J. & Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell Oncol. 2, e975093 (2015).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009).
Zhao, J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl Acad. Sci. USA 109, 5322–5327 (2012).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Oberst, A. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011).
Zhang, H. et al. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471, 373–376 (2012).
Lin, Y., Devin, A., Rodriguez, Y. & Liu, Z. G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 (1999).
Ch’en, I. L., Tsau, J. S., Molkentin, J. D., Komatsu, M. & Hedrick, S. M. Mechanisms of necroptosis in T cells. J. Exp. Med. 208, 633–641 (2011).
O’Donnell, M. A. et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 13, 1437–1442 (2011).
Park, S. M., Yoon, J. B. & Lee, T. H. Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett. 566, 151–156 (2004).
Moulin, M. et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 31, 1679–1691 (2012).
Kaczmarek, A., Vandenabeele, P. & Krysko, D. V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013).
Yatim, N. et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 350, 328–334 (2015).
Wallach, D., Kang, T. B., Yang, S. H. & Kovalenko, A. The in vivo significance of necroptosis: lessons from exploration of caspase-8 function. Cytokine Growth Factor Rev. 25, 157–165 (2014).
Jouan-Lanhouet, S. et al. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev. Biol. 35, 2–13 (2014).
Lin, J. et al. Exogenous hydrogen sulfide protects human umbilical vein endothelial cells against high glucoseinduced injury by inhibiting the necroptosis pathway. Int. J. Mol. Med. 41, 1477–1486 (2018).
Liang, W. et al. A novel damage mechanism: Contribution of the interaction between necroptosis and ROS to high glucose-induced injury and inflammation in H9c2 cardiac cells. Int J. Mol. Med. 40, 201–208 (2017).
Huang, C. Y., Kuo, W. T., Huang, Y. C., Lee, T. C. & Yu, L. C. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 4, e622 (2013).
Yang, X. S. et al. Hypoxia-inducible factor-1 alpha is involved in RIP-induced necroptosis caused by in vitro and in vivo ischemic brain injury. Sci. Rep. 7, 5818 (2017).
Zhou, Y. et al. The degradation of mixed lineage kinase domain-like protein promotes neuroprotection after ischemic brain injury. Oncotarget 8, 68393–68401 (2017).
Vieira, M. et al. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol. Dis. 68, 26–36 (2014).
McCaig, W. D. et al. Hyperglycemia potentiates a shift from apoptosis to RIP1-dependent necroptosis. Cell Death Disco. 4, 55 (2018).
LaRocca, T. J., Sosunov, S. A., Shakerley, N. L., Ten, V. S. & Ratner, A. J. Hyperglycemic conditions prime cells for RIP1-dependent necroptosis. J. Biol. Chem. 291, 13753–13761 (2016).
Li, Y. et al. Type I IFN operates pyroptosis and necroptosis during multidrug-resistant A. baumannii infection. Cell Death Differ. 25, 1304–1318 (2018).
Thapa, R. J. et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl Acad. Sci. USA 110, E3109–E3118 (2013).
Sarhan, J. et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ, https://doi.org/10.1038/s41418-018-0122-7 (2018).
Gunther, C. et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Invest 126, 4346–4360 (2016).
Ivanova, O. K. et al. CD3(+) CD8(+) NKG2D(+) T lymphocytes induce apoptosis and necroptosis in HLA-negative cells via FasL-Fas interaction. J. Cell Biochem. 118, 3359–3366 (2017).
Kesarwani, P. et al. Blocking TCR restimulation induced necroptosis in adoptively transferred T cells improves tumor control. Oncotarget 7, 69371–69383 (2016).
Ofengeim, D. et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849 (2015).
Yuan, J., Amin, P. & Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2019).
Ofengeim, D. et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 114, E8788–E8797 (2017).
Ito, Y. et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608 (2016).
Xu, D. et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in. Aging Cell 174, 1477–1491 e1419 (2018).
Fan, H. et al. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol. Neurodegener. 11, 14 (2016).
Szobi, A. et al. Analysis of necroptotic proteins in failing human hearts. J. Transl. Med. 15, 86 (2017).
Hu, D. et al. A common variant of RIP3 promoter region is associated with poor prognosis in heart failure patients by influencing SOX17 binding. J. Cell Mol. Med., https://doi.org/10.1111/jcmm.14408 (2019).
Karunakaran, D. et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2, e1600224 (2016).
Gupta, K., Phan, N., Wang, Q. & Liu, B. Necroptosis in cardiovascular disease - a new therapeutic target. J. Mol. Cell Cardiol. 118, 26–35 (2018).
Wang, Q. et al. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ. Res. 116, 600–611 (2015).
Hu, X. M. et al. Plasma levels of receptor interacting protein kinase-3 correlated with coronary artery disease. Chin. Med. J. (Engl), https://doi.org/10.1097/CM9.0000000000000225 (2019).
Saeed, W. K. et al. Decrease in fat de novo synthesis and chemokine ligand expression in non-alcoholic fatty liver disease caused by inhibition of mixed lineage kinase domain-like pseudokinase. J. Gastroenterol. Hepatol., https://doi.org/10.1111/jgh.14740 (2019).
Afonso, M. B. et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 129, 721–739 (2015).
Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014).
Afonso, M. B. et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis. 7, e2390 (2016).
Pierdomenico, M. et al. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am. J. Gastroenterol. 109, 279–287 (2014).
Wang, X., He, Z., Liu, H., Yousefi, S. & Simon, H. U. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J. Immunol. 197, 4090–4100 (2016).
Wang, X., Yousefi, S. & Simon, H. U. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 9, 111 (2018).
von Massenhausen, A. et al. Phenytoin inhibits necroptosis. Cell Death Dis. 9, 359 (2018).
Gong, Y. N. et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300 e216 (2017).
Yang, B. et al. Interleukin-1 receptor activation aggravates autosomal dominant polycystic kidney disease by modulating regulated necrosis. Am. J. Physiol. Renal Physiol., https://doi.org/10.1152/ajprenal.00104.2019 (2019).
Schreiber, A. et al. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl Acad. Sci. USA 114, E9618–E9625 (2017).
Guo, C. et al. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J. Autoimmun., https://doi.org/10.1016/j.jaut.2019.05.014 (2019).
Lauffer, F. et al. Type I immune response induces keratinocyte necroptosis and is associated with interface dermatitis. J. Invest. Dermatol. 138, 1785–1794 (2018).
Fan, H. et al. Activation-induced necroptosis contributes to B-cell lymphopenia in active systemic lupus erythematosus. Cell Death Dis. 5, e1416 (2014).
Kim, S. K. et al. Upregulated RIP3 Expression Potentiates MLKL Phosphorylation-Mediated Programmed Necrosis in Toxic Epidermal Necrolysis. J. Invest Dermatol 135, 2021–2030 (2015).
Mizumura, K. et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Invest 124, 3987–4003 (2014).
Zhang, Y. et al. Death of chondrocytes in Kashin-Beck disease: Apoptosis, necrosis or necroptosis? Int J. Exp. Pathol. 99, 312–322 (2018).
Shi, J. et al. Loss of periodontal ligament fibroblasts by RIPK3-MLKL-mediated necroptosis in the progress of chronic periodontitis. Sci. Rep. 9, 2902 (2019).
Cuchet-Lourenco, D. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018).
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 116, 970–975 (2019).
Qin, X., Ma, D., Tan, Y. X., Wang, H. Y. & Cai, Z. The role of necroptosis in cancer: A double-edged sword? Biochim Biophys. Acta Rev. Cancer 1871, 259–266 (2019).
Messmer, M. N., Snyder, A. G. & Oberst, A. Comparing the effects of different cell death programs in tumor progression and immunotherapy. Cell Death Differ. 26, 115–129 (2019).
Conev, N. V. et al. RIPK3 expression as a potential predictive and prognostic marker in metastatic colon cancer. Clin. Invest Med 42, E31–E38 (2019).
Dziedzic, S. A. et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat. Cell Biol. 20, 58–68 (2018).
Hu, B. et al. Prognostic and clinicopathological significance of MLKL expression in cancer patients: a meta-analysis. BMC Cancer 18, 736 (2018).
Cerhan, J. R. et al. Genetic variation in 1253 immune and inflammation genes and risk of non-Hodgkin lymphoma. Blood 110, 4455–4463 (2007).
Snyder, A. G. et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol 4, https://doi.org/10.1126/sciimmunol.aaw2004 (2019).
Seifert, L. et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249 (2016).
Ertao, Z. et al. Prognostic value of mixed lineage kinase domain-like protein expression in the survival of patients with gastric caner. Tumour Biol. 37, 13679–13685 (2016).
He, L., Peng, K., Liu, Y., Xiong, J. & Zhu, F. F. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther. 6, 1539–1543 (2013).
Jiao, D. et al. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res 28, 868–870 (2018).
Wang, W. et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 34, 757–774 e757 (2018).
Strilic, B. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218 (2016).
Wu, X. N. et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21, 1709–1720 (2014).
Orozco, S. et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21, 1511–1521 (2014).
Li, J. et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012).
Zhang, D. W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009).
Yang, C. et al. Regulation of RIP3 by the transcription factor Sp1 and the epigenetic regulator UHRF1 modulates cancer cell necroptosis. Cell Death Dis. 8, e3084 (2017).
Knuth, A. K. et al. Interferons Transcriptionally Up-Regulate MLKL Expression in Cancer Cells. Neoplasia 21, 74–81 (2019).
McComb, S. et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc. Natl. Acad. Sci. USA 111, E3206–E3213 (2014).
Xiong, Y. et al. The bromodomain protein BRD4 positively regulates necroptosis via modulating MLKL expression. Cell Death Differ, https://doi.org/10.1038/s41418-018-0262-9 (2019).
Morgan, M. J. & Kim, Y. S. The serine threonine kinase RIP3: lost and found. BMB Rep. 48, 303–312 (2015).
Yang, Z. et al. 2-HG Inhibits Necroptosis by Stimulating DNMT1-Dependent Hypermethylation of the RIP3 Promoter. Cell Rep. 19, 1846–1857 (2017).
Koo, G. B. et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 25, 707–725 (2015).
Lewis, J. et al. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J. Biol. Chem. 275, 10519–10526 (2000).
Wang, Z. et al. Inhibition of HSP90alpha protects cultured neurons from oxygen-glucose deprivation induced necroptosis by decreasing RIP3 expression. J. Cell Physiol. 233, 4864–4884 (2018).
Zhao, X. M. et al. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 7, e2089 (2016).
Li, D. et al. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc. Natl. Acad. Sci. USA 112, 5017–5022 (2015).
Wang, Z., Feng, J., Yu, J. & Chen, G. FKBP12 mediates necroptosis by initiating RIPK1/RIPK3/MLKL signal transduction in response to TNF receptor 1 ligation. J Cell Sci, https://doi.org/10.1242/jcs.227777 (2019).
Srinivasan, S. R. et al. Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades. Mol. Cancer Res 16, 58–68 (2018).
Feng, S. et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 19, 2056–2067 (2007).
McComb, S. et al. Cathepsins limit macrophage necroptosis through cleavage of Rip1 kinase. J. Immunol. 192, 5671–5678 (2014).
Wertz, I. E. et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699 (2004).
Seo, J. et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 18, 291–302 (2016).
Alturki, N. A. et al. Triad3a induces the degradation of early necrosome to limit RipK1-dependent cytokine production and necroptosis. Cell Death Dis. 9, 592 (2018).
Choi, S. W. et al. PELI1 Selectively Targets Kinase-Active RIP3 for Ubiquitylation-Dependent Proteasomal Degradation. Mol. Cell 70, 920–935 e927 (2018).
Liu, X. et al. Post-translational modifications as key regulators of TNF-induced necroptosis. Cell Death Dis. 7, e2293 (2016).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Vanlangenakker, N. et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 18, 656–665 (2011).
Dondelinger, Y. et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20, 1381–1392 (2013).
Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Geng, J. F. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, ARTN 359 https://doi.org/10.1038/s41467-017-00406-w (2017).
Jaco, I. et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710 e695 (2017).
Dondelinger, Y. et al. NF-kappaB-independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015).
Wang, C. Y., Mayo, M. W., Korneluk, R. G., Goeddel, D. V. & Baldwin, A. S. Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281, 1680–1683 (1998).
Micheau, O., Lens, S., Gaide, O., Alevizopoulos, K. & Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell Biol. 21, 5299–5305 (2001).
Guo, X. et al. TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints. Cell Death Dis. 7, e2381 (2016).
Xie, Y. et al. Inhibition of aurora Kinase A induces necroptosis in pancreatic carcinoma. Gastroenterology 153, 1429–1443 e1425 (2017).
Chen, W. et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat. Cell Biol. 17, 434–444 (2015).
Zhang, H., Ji, J., Liu, Q. & Xu, S. MUC1 downregulation promotes TNF-alpha-induced necroptosis in human bronchial epithelial cells via regulation of the RIPK1/RIPK3 pathway. J. Cell Physiol., https://doi.org/10.1002/jcp.28148 (2019).
Xu, Q. et al. The cytoplasmic nuclear receptor RARgamma controls RIP1 initiated cell death when cIAP activity is inhibited. Nat. Commun. 8, 425 (2017).
Kaiser, W. J. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 (2013).
Lin, J. et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128 (2016).
Dillon, C. P. et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202 (2014).
Rickard, J. A. et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 (2014).
Kaiser, W. J. et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl Acad. Sci. USA 111, 7753–7758 (2014).
Petersen, S. L. et al. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 22, 1846–1857 (2015).
Liu, W. et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc. Natl Acad. Sci. USA 115, E1475–E1484 (2018).
Reynoso, E. et al. Thioredoxin-1 actively maintains the pseudokinase MLKL in a reduced state to suppress disulfide bond-dependent MLKL polymer formation and necroptosis. J. Biol. Chem. 292, 17514–17524 (2017).
Liu, Q. et al. Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis. 5, e1084 (2014).
Radonjic-Hoesli, S. et al. Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counterregulated by autophagy. J. Allergy Clin. Immunol. 140, 1632–1642 (2017).
Wang, Y., Zhang, Q., Wang, B., Li, P. & Liu, P. LiCl treatment induces programmed cell death of Schwannoma cells through AKT- and MTOR-mediated necroptosis. Neurochem. Res. 42, 2363–2371 (2017).
Zhang, T. et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 22, 175–182 (2016).
Sosna, J. et al. Differences and similarities in TRAIL- and tumor necrosis factor-mediated necroptotic signaling in cancer cells. Mol. Cell Biol. 36, 2626–2644 (2016).
Sosna, J. et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol. Life Sci. 71, 331–348 (2014).
Jang, K. H., Jang, T., Son, E., Choi, S. & Kim, E. Kinase-independent role of nuclear RIPK1 in regulating parthanatos through physical interaction with PARP1 upon oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 1865, 132–141 (2018).
Wang, Z., Jiang, H., Chen, S., Du, F. & Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 (2012).
Remijsen, Q. et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 5, e1004 (2014).
Chen, X. et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121 (2014).
Yang, Z. et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 20, 186–197 (2018).
Schenk, B. & Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 34, 5796–5806 (2015).
Tait, S. W. et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 5, 878–885 (2013).
Liu, S. et al. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl Acad. Sci. USA 114, E7450–E7459 (2017).
McNamara, D. E., Quarato, G., Guy, C. S., Green, D. R. & Moldoveanu, T. Characterization of MLKL-mediated plasma membrane rupture in necroptosis. J. Vis. Exp., https://doi.org/10.3791/58088 (2018).
Quarato, G. et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 61, 589–601 (2016).
Ros, U. et al. Necroptosis execution is mediated by plasma membrane nanopores independent of calcium. Cell Rep. 19, 175–187 (2017).
Dondelinger, Y. et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981 (2014).
Dovey, C. M. et al. MLKL requires the inositol phosphate code to execute necroptosis. Mol. Cell 70, 936–948 e937 (2018).
McNamara, D. E. et al. Direct activation of human MLKL by a select repertoire of inositol phosphate metabolites. Cell Chem. Biol., https://doi.org/10.1016/j.chembiol.2019.03.010 (2019).
Jing, L. et al. MLKL-PITPalpha signaling-mediated necroptosis contributes to cisplatin-triggered cell death in lung cancer A549 cells. Cancer Lett. 414, 136–146 (2018).
Najafov, A. et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol. Cell, https://doi.org/10.1016/j.molcel.2019.05.022 (2019).
Cai, Z. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16, 55–65 (2014).
Xia, B. et al. MLKL forms cation channels. Cell Res. 26, 517–528 (2016).
Tonnus, W., Gembardt, F., Hugo, C. & Linkermann, A. Die later with ESCRT! Oncotarget 8, 41790–41791 (2017).
Fan, W. et al. Flotillin-mediated endocytosis and ALIX-syntenin-1-mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci. Signal. 12, https://doi.org/10.1126/scisignal.aaw3423 (2019).
Cai, Z. et al. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 26, 886–900 (2016).
Janssens, S., Tinel, A., Lippens, S. & Tschopp, J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell 123, 1079–1092 (2005).
Li, M., Feng, S. & Wu, M. Multiple roles for nuclear localization signal (NLS, aa 442-472) of receptor interacting protein 3 (RIP3). Biochem. Biophys. Res. Commun. 372, 850–855 (2008).
Yang, Y., Ma, J., Chen, Y. & Wu, M. Nucleocytoplasmic shuttling of receptor-interacting protein 3 (RIP3): identification of novel nuclear export and import signals in RIP3. J. Biol. Chem. 279, 38820–38829 (2004).
Yoon, S., Bogdanov, K., Kovalenko, A. & Wallach, D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 23, 253–260 (2016).
Weber, K., Roelandt, R., Bruggeman, I., Estornes, Y. & Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 1, 6 (2018).
Chen, W. et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 288, 16247–16261 (2013).
Li, L. et al. The Gbetagamma-Src signaling pathway regulates TNF-induced necroptosis via control of necrosome translocation. Cell Res. 24, 417–432 (2014).
Bollino, D., Balan, I. & Aurelian, L. Valproic acid induces neuronal cell death through a novel calpain-dependent necroptosis pathway. J. Neurochem. 133, 174–186 (2015).
Chengzhu, W. U. et al. Miconazole triggers various forms of cell death in human breast cancer MDA-MB-231 cells. Pharmazie 74, 290–294 (2019).
Kim, H. J. et al. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J. Transl. Med. 15, 123 (2017).
Fu, Z. et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 13, 580 (2013).
Han, Q. et al. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J. Transl. Med. 16, 201 (2018).
Li, Y., Tian, X., Liu, X. & Gong, P. Bufalin inhibits human breast cancer tumorigenesis by inducing cell death through the ROS-mediated RIP1/RIP3/PARP-1 pathways. Carcinogenesis 39, 700–707 (2018).
Chen, X., Hu, X., Liu, L., Liang, X. & Xiao, J. Extracts derived from a traditional Chinese herbal formula triggers necroptosis in ectocervical Ect1/E6E7 cells through activation of RIP1 kinase. J. Ethnopharmacol. 239, 111922 (2019).
Zhou, J. et al. Emodin induced necroptosis in the glioma cell line U251 via the TNF-alpha/RIP1/RIP3 pathway. Invest. New Drugs, https://doi.org/10.1007/s10637-019-00764-w (2019).
Xu, B. et al. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Disco. 3, 16096 (2017).
Seo, M. J., Hong, J. M., Kim, S. J. & Lee, S. M. Genipin protects d-galactosamine and lipopolysaccharide-induced hepatic injury through suppression of the necroptosis-mediated inflammasome signaling. Eur. J. Pharm. 812, 128–137 (2017).
Luo, Y. et al. Lycorine induces programmed necrosis in the multiple myeloma cell line ARH-77. Tumour Biol. 36, 2937–2945 (2015).
Khorsandi, L. et al. Quercetin induces apoptosis and necroptosis in MCF-7 breast cancer cells. Bratisl. Lek. Listy 118, 123–128 (2017).
Jia, Y. et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 19, 375–387 (2018).
Wang, H. L. et al. Bulnesia sarmientoi supercritical fluid extract exhibits necroptotic effects and anti-metastatic activity on lung cancer cells. Molecules 23, https://doi.org/10.3390/molecules23123304 (2018).
Li, D. et al. Natural product Kongensin A is a non-canonical HSP90 inhibitor that blocks RIP3-dependent necroptosis. Cell Chem. Biol. 23, 257–266 (2016).
Yan, C. et al. The targeted inhibition of mitochondrial Hsp90 overcomes the apoptosis resistance conferred by Bcl-2 in Hep3B cells via necroptosis. Toxicol. Appl. Pharm. 266, 9–18 (2013).
Chen, W. W. et al. RIP1 mediates the protection of geldanamycin on neuronal injury induced by oxygen-glucose deprivation combined with zVAD in primary cortical neurons. J. Neurochem. 120, 70–77 (2012).
Zhang, Z. et al. Non-benzoquinone geldanamycin analogs trigger various forms of death in human breast cancer cells. J. Exp. Clin. Cancer Res. 35, 149 (2016).
Fakharnia, F., Khodagholi, F., Dargahi, L. & Ahmadiani, A. Prevention of cyclophilin D-mediated mPTP opening using cyclosporine-A alleviates the elevation of necroptosis, autophagy and apoptosis-related markers following global cerebral ischemia-reperfusion. J. Mol. Neurosci. 61, 52–60 (2017).
Abd-Ellatif, R. N. et al. Diacerein protects against glycerol-induced acute kidney injury: modulating oxidative stress, inflammation, apoptosis and necroptosis. Chem. Biol. Interact. 306, 47–53 (2019).
Ding, J. et al. Rapamycin Inhibited Photoreceptor Necroptosis and Protected the Retina by Activation of Autophagy in Experimental Retinal Detachment. Curr. Eye Res., 1–7, https://doi.org/10.1080/02713683.2019.1588331 (2019).
Qu, C. et al. Patchouli alcohol ameliorates dextran sodium sulfate-induced experimental colitis and suppresses tryptophan catabolism. Pharm. Res. 121, 70–82 (2017).
Nikseresht, S., Khodagholi, F. & Ahmadiani, A. Protective effects of ex-527 on cerebral ischemia-reperfusion injury through necroptosis signaling pathway attenuation. J. Cell Physiol. 234, 1816–1826 (2019).
Chen, X. et al. Identification of TAK-632 and its analogues as potent inhibitors of necroptosis by targeting RIPK1 and RIPK3. Br. J. Pharmacol., https://doi.org/10.1111/bph.14653 (2019).
Degterev, A. & Linkermann, A. Generation of small molecules to interfere with regulated necrosis. Cell Mol. Life Sci. 73, 2251–2267 (2016).
Fulda, S. Smac mimetics to therapeutically target IAP proteins in cancer. Int Rev. Cell Mol. Biol. 330, 157–169 (2017).
Kilty, I. & Jones, L. H. TAK1 selective inhibition: state of the art and future opportunities. Future Med. Chem. 7, 23–33 (2015).
Li, J. X. et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 5, e1278 (2014).
Cruz, S. A., Qin, Z., Stewart, A. F. R. & Chen, H. H. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen. Res. 13, 252–256 (2018).
Martens, S. et al. Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell Death Dis. 8, e2904 (2017).
Feldmann, F., Schenk, B., Martens, S., Vandenabeele, P. & Fulda, S. Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget 8, 68208–68220 (2017).
Fauster, A. et al. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 6, e1767 (2015).
Ali, M. & Mocarski, E. S. Proteasome inhibition blocks necroptosis by attenuating death complex aggregation. Cell Death Dis. 9, 346 (2018).
Moerke, C. et al. The anticonvulsive Phenhydan((R)) suppresses extrinsic cell death. Cell Death Differ., https://doi.org/10.1038/s41418-018-0232-2 (2018).
Meng, X. M. et al. Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab Invest. 98, 79–94 (2018).
Wang, J. et al. The protective effect of aucubin from eucommia ulmoides against status epilepticus by inducing autophagy and inhibiting necroptosis. Am. J. Chin. Med. 45, 557–573 (2017).
Nehs, M. A. et al. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 150, 1032–1039 (2011).
Yang, H. et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 5, e1149673 (2016).
Xu, Y. et al. Cisplatin-induced necroptosis in TNFalpha dependent and independent pathways. Cell Signal 31, 112–123 (2017).
Choi, M. J. et al. Cisplatin-induced ototoxicity in rats is driven by RIP3-dependent necroptosis. Cells 8, https://doi.org/10.3390/cells8050409 (2019).
Oliver Metzig, M. et al. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-alpha-dependent necroptosis driven by RIP1 kinase and NF-kappaB. Oncogene 35, 3399–3409 (2016).
Basit, F., Cristofanon, S. & Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 20, 1161–1173 (2013).
Lin, C. Y. et al. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Disco. 2, 16065 (2016).
Deng, Q. et al. Neoalbaconol induces energy depletion and multiple cell death in cancer cells by targeting PDK1-PI3-K/Akt signaling pathway. Cell Death Dis. 4, e804 (2013).
Xie, X. et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br. J. Pharm. 172, 3929–3943 (2015).
Martens, S. et al. RIPK1-dependent cell death: a novel target of the Aurora kinase inhibitor Tozasertib (VX-680). Cell Death Dis. 9, 211 (2018).
Schroeder, A. et al. Targeting Thioredoxin-1 by dimethyl fumarate induces ripoptosome-mediated cell death. Sci. Rep. 7, 43168 (2017).
Akimoto, M., Maruyama, R., Kawabata, Y., Tajima, Y. & Takenaga, K. Antidiabetic adiponectin receptor agonist AdipoRon suppresses tumour growth of pancreatic cancer by inducing RIPK1/ERK-dependent necroptosis. Cell Death Dis. 9, 804 (2018).
Yu, X. et al. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFalpha and ROS production. Oncotarget 6, 1995–2008 (2015).
Zhang, Y. et al. Proteasome inhibitor PS-341 limits macrophage necroptosis by promoting cIAPs-mediated inhibition of RIP1 and RIP3 activation. Biochem. Biophys. Res. Commun. 477, 761–767 (2016).
Wang, D. et al. The histone deacetylase inhibitor vorinostat prevents TNFalpha-induced necroptosis by regulating multiple signaling pathways. Apoptosis 18, 1348–1362 (2013).
Cho, Y. S. The role of necroptosis in the treatment of diseases. BMB Rep. 51, 219–224 (2018).
Florean, C., Song, S., Dicato, M. & Diederich, M. Redox biology of regulated cell death in cancer: A focus on necroptosis and ferroptosis. Free Radic. Biol. Med. 134, 177–189 (2019).
Chen, J. et al. Dexmedetomidine preconditioning protects cardiomyocytes against hypoxia/reoxygenation-induced necroptosis by inhibiting HMGB1-mediated inflammation. Cardiovasc Drugs Ther. 33, 45–54 (2019).
Zhou, H. et al. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J. Pineal Res. 65, e12503 (2018).
Luo, F., Herrup, K., Qi, X. & Yang, Y. Inhibition of Drp1 hyper-activation is protective in animal models of experimental multiple sclerosis. Exp. Neurol. 292, 21–34 (2017).
Koppe, C. et al. IkappaB kinasealpha/beta control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell-death mediator receptor-interacting protein kinase 1. Hepatology 64, 1217–1231 (2016).
Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 10, 1729 (2019).
Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247 (2017).
Menon, M. B. et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259 (2017).
McQuade, T., Cho, Y. & Chan, F. K. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 456, 409–415 (2013).
Wang, H. et al. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proc. Natl Acad. Sci. USA 114, 11944–11949 (2017).
Wang, Y., Shan, B., Liang, Y., Wei, H. & Yuan, J. Parkin regulates NF-kappaB by mediating site-specific ubiquitination of RIPK1. Cell Death Dis. 9, 732 (2018).
Wei, R. et al. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev. 31, 1162–1176 (2017).
Ying, Z. et al. Mixed lineage kinase domain-like protein MLKL breaks down myelin following nerve injury. Mol. Cell 72, 457–468 e455 (2018).
Li, X. et al. O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity, https://doi.org/10.1016/j.immuni.2019.01.007 (2019).
Cougnoux, A. et al. Necroptosis in Niemann-Pick disease, type C1: a potential therapeutic target. Cell Death Dis. 7, e2147 (2016).
Onizawa, M. et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627 (2015).
Vanlangenakker, N., Bertrand, M. J., Bogaert, P., Vandenabeele, P. & Vanden Berghe, T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2, e230 (2011).
Newton, K. et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576 (2016).
Amin, P. et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc. Natl Acad. Sci. USA 115, E5944–E5953 (2018).
Karch, J. et al. Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS ONE 10, e0130520 (2015).
Tischner, D., Woess, C., Ottina, E. & Villunger, A. Bcl-2-regulated cell death signalling in the prevention of autoimmunity. Cell Death Dis. 1, e48 (2010).
Qu, Y. et al. RIPK3 interactions with MLKL and CaMKII mediate oligodendrocytes death in the developing brain. Cell Death Dis. 8, e2629 (2017).
Zamaraev, A. V., Kopeina, G. S., Buchbinder, J. H., Zhivotovsky, B. & Lavrik, I. N. Caspase-2 is a negative regulator of necroptosis. Int. J. Biochem. Cell Biol. 102, 101–108 (2018).
Chan, F. K. et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 278, 51613–51621 (2003).
Kaiser, W. J. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011).
Alvarez-Diaz, S. et al. The Pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity 45, 513–526 (2016).
Lu, J. V. et al. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc. Natl Acad. Sci. USA 108, 15312–15317 (2011).
Linkermann, A. et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA 110, 12024–12029 (2013).
Roca, F. J. & Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153, 521–534 (2013).
Hitomi, J. et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 (2008).
Wang, L., Du, F. & Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
Bonnet, M. C. et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35, 572–582 (2011).
Moquin, D. M., McQuade, T. & Chan, F. K. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 8, e76841 (2013).
Draber, P. et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 13, 2258–2272 (2015).
Lee, Y. S. et al. Daxx is a key downstream component of receptor interacting protein kinase 3 mediating retinal ischemic cell death. FEBS Lett. 587, 266–271 (2013).
Yoon, S., Kovalenko, A., Bogdanov, K. & Wallach, D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47, 51–65 e57 (2017).
Zhang, H. et al. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471, 373–376 (2011).
Dillon, C. P. et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 1, 401–407 (2012).
Zhang, X. et al. MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep. 16, 3247–3259 (2016).
Zhao, Q. et al. RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in fadd-deficient mice. Cell Rep. 19, 798–808 (2017).
Petersen, S. L. et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12, 445–456 (2007).
Lafont, E. et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 36, 1147–1166 (2017).
Feoktistova, M., Geserick, P., Panayotova-Dimitrova, D. & Leverkus, M. Pick your poison: the Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 11, 460–467 (2012).
Feoktistova, M. et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011).
Hughes, M. A. et al. Co-operative and hierarchical binding of c-FLIP and Caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell 61, 834–849 (2016).
Tortola, L. et al. The tumor suppressor Hace1 is a critical regulator of TNFR1-mediated cell fate. Cell Rep. 15, 1481–1492 (2016).
Jacobsen, A. V. et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 7, e2051 (2016).
Sosna, J. et al. The proteases HtrA2/Omi and UCH-L1 regulate TNF-induced necroptosis. Cell Commun. Signal 11, 76 (2013).
Zhang, C. et al. Inhibition of HtrA2 alleviated dextran sulfate sodium (DSS)-induced colitis by preventing necroptosis of intestinal epithelial cells. Cell Death Dis. 10, 344 (2019).
Bertrand, M. J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
Anderton, H., Rickard, J. A., Varigos, G. A., Lalaoui, N. & Silke, J. Inhibitor of apoptosis proteins (IAPs) Limit RIPK1-mediated skin inflammation. J. Invest. Dermatol. 137, 2371–2379 (2017).
Bertrand, M. J. et al. cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1-4). PLoS ONE 6, e22356 (2011).
Dondelinger, Y., Darding, M., Bertrand, M. J. & Walczak, H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol. Life Sci. 73, 2165–2176 (2016).
Annibaldi, A. et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol. Cell 69, 566–580 e565 (2018).
Yabal, M. et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 7, 1796–1808 (2014).
de Almagro, M. C., Goncharov, T., Newton, K. & Vucic, D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis. 6, e1800 (2015).
Legarda-Addison, D., Hase, H., O’Donnell, M. A. & Ting, A. T. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. 16, 1279–1288 (2009).
O’Donnell, M. A., Hase, H., Legarda, D. & Ting, A. T. NEMO inhibits programmed necrosis in an NFkappaB-independent manner by restraining RIP1. PLoS ONE 7, e41238 (2012).
Vlantis, K. et al. NEMO prevents RIP Kinase 1-mediated epithelial cell death and chronic intestinal inflammation by NF-kappaB-dependent and -independent functions. Immunity 44, 553–567 (2016).
Nenci, A. et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561 (2007).
Cekay, M. J. et al. Smac mimetics and type II interferon synergistically induce necroptosis in various cancer cell lines. Cancer Lett. 410, 228–237 (2017).
Thapa, R. J. et al. NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol. Cell Biol. 31, 2934–2946 (2011).
Taraborrelli, L. et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 9, 3910 (2018).
Peltzer, N. et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 557, 112–117 (2018).
Seymour, R. E. et al. Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun. 8, 416–421 (2007).
Rickard, J. A. et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife 3, https://doi.org/10.7554/eLife.03464 (2014).
Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844 (2009).
Lee, E. W. et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat. Commun. 3, 978 (2012).
Heger, K. et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559, 120–124 (2018).
Dionisio, P. E. A., Oliveira, S. R., Amaral, J. & Rodrigues, C. M. P. Loss of microglial parkin inhibits necroptosis and contributes to neuroinflammation. Mol. Neurobiol., https://doi.org/10.1007/s12035-018-1264-9 (2018).
Xu, X. et al. The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res 1343, 206–212 (2010).
Jouan-Lanhouet, S. et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 19, 2003–2014 (2012).
Xu, C. et al. Embryonic lethality and host immunity of RelA-deficient mice are mediated by both apoptosis and necroptosis. J. Immunol. 200, 271–285 (2018).
Zhao, J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl Acad. Sci. USA 109, 5322–5327 (2012).
Wagner, S. A., Satpathy, S., Beli, P. & Choudhary, C. SPATA2 links CYLD to the TNF-alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 35, 1868–1884 (2016).
Kupka, S. et al. SPATA2-mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep. 16, 2271–2280 (2016).
Mihaly, S. R., Morioka, S., Ninomiya-Tsuji, J. & Takaesu, G. Activated macrophage survival is coordinated by TAK1 binding proteins. PLoS ONE 9, e94982 (2014).
Broglie, P., Matsumoto, K., Akira, S., Brautigan, D. L. & Ninomiya-Tsuji, J. Transforming growth factor beta-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. J. Biol. Chem. 285, 2333–2339 (2010).
Mihaly, S. R., Ninomiya-Tsuji, J. & Morioka, S. TAK1 control of cell death. Cell Death Differ. 21, 1667–1676 (2014).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Karl, I. et al. TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell Death Dis. 5, e1444 (2014).
Guo, X. et al. Cardioprotective role of tumor necrosis factor receptor-associated factor 2 by suppressing apoptosis and necroptosis. Circulation 136, 729–742 (2017).
Li, J. et al. TRAF2 protects against cerebral ischemia-induced brain injury by suppressing necroptosis. Cell Death Dis. 10, 328 (2019).
He, S., Liang, Y., Shao, F. & Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl Acad. Sci. USA 108, 20054–20059 (2011).
Meylan, E. et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 5, 503–507 (2004).
Dasari, S. & Tchounwou, P. B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharm. 740, 364–378 (2014).
Sun, Y. et al. Down-regulation of RIP3 potentiates cisplatin chemoresistance by triggering HSP90-ERK pathway mediated DNA repair in esophageal squamous cell carcinoma. Cancer Lett. 418, 97–108 (2018).
Xu, Z. et al. High-mobility group box 1 protein-mediated necroptosis contributes to dasatinib-induced cardiotoxicity. Toxicol. Lett. 296, 39–47 (2018).
Stephen, L. J. Drug treatment of epilepsy in elderly people: focus on valproic Acid. Drugs Aging 20, 141–152 (2003).
Jung, S. et al. Anticancer activity of gomisin J from Schisandra chinensis fruit. Oncol. Rep. 41, 711–717 (2019).
Chen, C. et al. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am. J. Transl. Res. 9, 5507–5517 (2017).
Acknowledgements
The European Regional Development Fund GINOP-2.3.2-15-2016-00005, the National Research, Development and Innovation Office – NKFIH, 125224 and 128034 as well as the New National Excellence Program (ÚNKP-18-3) are acknowledged for financial support of this work. AGS is recipient of the János Bolyai Research Scholarship of the Hungarian Academy of Sciences, and by the New National Excellence Program of the Ministry of Human Capacities (ÚNKP-19-4-DE-142).”
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Oberst
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Molnár, T., Mázló, A., Tslaf, V. et al. Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis 10, 860 (2019). https://doi.org/10.1038/s41419-019-2094-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-2094-z
This article is cited by
-
The broad impact of cell death genes on the human disease phenome
Cell Death & Disease (2024)
-
Chlorpyrifos induces apoptosis and necroptosis via the activation of CYP450s pathway mediated by nuclear receptors in LMH cells
Environmental Science and Pollution Research (2023)
-
Necroptosis in the developing brain: role in neurodevelopmental disorders
Metabolic Brain Disease (2023)
-
Types of necroinflammation, the effect of cell death modalities on sterile inflammation
Cell Death & Disease (2022)
-
UVA induces retinal photoreceptor cell death via receptor interacting protein 3 kinase mediated necroptosis
Cell Death Discovery (2022)
