
Abstract
Protein phosphatase magnesium-dependent 1 delta (PPM1D) terminates cell response to genotoxic stress by negatively regulating the tumor suppressor p53 and other targets at chromatin. Mutations in the exon 6 of the PPM1D result in production of a highly stable, C-terminally truncated PPM1D. These gain-of-function PPM1D mutations are present in various human cancers but their role in tumorigenesis remains unresolved. Here we show that truncated PPM1D impairs activation of the cell cycle checkpoints in human non-transformed RPE cells and allows proliferation in the presence of DNA damage. Next, we developed a mouse model by introducing a truncating mutation in the PPM1D locus and tested contribution of the oncogenic PPM1DT allele to colon tumorigenesis. We found that p53 pathway was suppressed in colon stem cells harboring PPM1DT resulting in proliferation advantage under genotoxic stress condition. In addition, truncated PPM1D promoted tumor growth in the colon in Apcmin mice and diminished survival. Moreover, tumor organoids derived from colon of the ApcminPpm1dT/+ mice were less sensitive to 5-fluorouracil when compared to ApcminPpm1d+/+and the sensitivity to 5-fluorouracil was restored by inhibition of PPM1D. Finally, we screened colorectal cancer patients and identified recurrent somatic PPM1D mutations in a fraction of colon adenocarcinomas that are p53 proficient and show defects in mismatch DNA repair. In summary, we provide the first in vivo evidence that truncated PPM1D can promote tumor growth and modulate sensitivity to chemotherapy.
Similar content being viewed by others


Introduction
Genome instability is one of the hallmarks of cancer1. In the presence of DNA damage, healthy cells protect their genome integrity by arresting progression through the cell cycle and by activation of specialized DNA repair pathways2,3. Double-strand DNA breaks (such as those generated by ionizing radiation, IR) and replication stress (for example induced by oncogenes) activate ataxia telangiectasia-mutated kinase (ATM) and ataxia telangiectasia and Rad3-related kinase (ATR), respectively. Subsequently, phosphorylation and ubiquitination-dependent recruitment of various proteins [such as TP53-binding protein 1 (53BP1) and Breast cancer type-1 susceptibility protein (BRCA1)] to the flanking chromatin enables DNA repair by non-homologous end joining or homologous recombination. In addition, ATM/ATR jointly activate tumor suppressor protein p53 triggering expression of its target genes that control cell fate decisions, including duration of the cell cycle arrest, senescence, or programmed cell death4. Functional DNA damage response (DDR) prevents proliferation of cells in the presence of DNA damage and thus represents an intrinsic barrier preventing oncogenic transformation5,6. Conversely, molecular defects in DDR as well as inactivating mutations in the TP53 gene (coding for p53 protein) lead to genome instability, promote tumor development and can affect the therapeutic response2,4,7.
Protein phosphatase magnesium-dependent 1 delta (PPM1D; called also Wip1) is a negative regulator of p53 that allows timely termination of the G2 checkpoint8,9,10. Loss of Ppm1d protected mice from development of MMTV-Erb2-driven mammary tumors, Eµ-myc-induced B-cell lymphomas and Apcmin-driven intestinal adenocarcinoma11,12,13,14. Inactivation of Ppm1d increased p53-, checkpoint kinase 2 (CHK2)-, and growth arrest and DNA damage gene 45 alpha (GADD45A)-dependent apoptosis of the intestinal stem cells (ISCs) and prevented their transformation into tumor-initiating stem cells12,13. Conversely, amplification of the PPM1D locus (17q23.2) leading to overexpression of PPM1D phosphatase was observed in about 10% of human breast cancers and several other cancer types15,16,17. Typically, overexpression of PPM1D occurs in p53-proficient tumors suggesting that suppression of the p53 pathway is the major role of the phosphatase during oncogenesis15. In addition to amplification of the PPM1D locus, nonsense mutations in exon 6 of PPM1D leading to production of the C-terminally truncated protein were recently reported in human cancers18,19,20,21. Since the C-terminal truncation does not affect enzymatic activity of PPM1D nor its subcellular distribution, truncated PPM1D protein can access its physiological substrates at chromatin18. In particular, heterozygous truncating mutations in the PPM1D are present in several p53-proficient cancer cell lines (including U2OS and HCT116 cells) and disable activation of the G1 checkpoint18. Gain-of-function phenotype of the truncated PPM1D is caused by abnormally prolonged protein half-life due to the loss of a degradation motif located in the last 65 amino acids of PPM1D18,22. Besides somatic mutations, age-related truncating mutations in PPM1D occur in a fraction of hematopoietic stem cells (HSCs) leading to clonal hematopoiesis22,23. The importance of these mutations is highlighted in mutation carriers receiving chemotherapy, because HSCs carrying the truncated PPM1D show better survival and potentially may allow development of secondary cancers including acute myeloid leukemia (AML) and myelodysplastic syndrome23,24.
Most of the supporting evidence for oncogenic properties of PPM1D comes from cell-based assays or from the knock-out mouse model, however, contribution of the truncated PPM1D to tumor growth has not been addressed in vivo so far. Here we generated a mouse model mimicking the truncating mutation in PPM1D identified in human cancers. Subsequently, we studied the impact of truncated Ppm1d on cell response to DNA damage, as well as its ability to potentiate colon carcinoma growth in vivo. We show that truncated Ppm1d can suppress p53-mediated response in ISCs. As a result, ISCs carrying the mutated Ppm1dT allele survive in the presence of genotoxic stress better than the wild-type ISCs. In addition, Ppm1dT/+ mice showed accelerated growth of Apcmin-driven adenocarcinoma in the colon. Tissue organoids derived from tumors expressing truncated PPM1D were resistant to 5-fluorouracil (5-FU), whereas they responded well to combined treatment with 5-FU and a small-molecule inhibitor of PPM1D. Finally, we identified recurrent somatic truncating PPM1D mutations in a fraction of human colon adenocarcinomas that were associated with defects in mismatch DNA repair pathway (MMR), while retaining wild type (wt) p53. In summary, we provide the first in vivo evidence that truncation of PPM1D contributes to tumorigenesis and may affect response of tumor cells to chemotherapy.
Materials and methods
Ethical approval
All animal models and experiments of this study were ethically reviewed and approved by the Institute of Molecular Genetics (c.j. 1/2016). All tumor samples were provided from subjects that gave their written informed consent approved by the local ethical committees and the research complies with the Declaration of Helsinki. The project was approved by the Regional Committee for Medical and Health Research Ethics, South Eastern Norway (REC number 1.2005.1629; 2010/1805) and the Norwegian Data Inspectorate.
Patient samples
A total of 947 primary CRC from three series were analyzed for gene mutations and MSI. Fresh-frozen tumor specimens were consecutively collected from patients (n = 364) treated for primary CRC (stage I–IV) at Oslo University Hospital in the period December 2005–June 2011. Czech cohort consisted of 243 non-selected sporadic CRC patients. Tumor tissue and adjacent non-malignant tissue were resected during primary surgery and collected to the dry ice. Swedish cohort included 340 primary CRC tissue and paired distant normal mucosa from patients diagnosed at the University Hospital in Linköping and Vrinnevi Hospital in Norrköping. Representative tumor tissues, evaluated by pathologist, were stored for subsequent analyses at dry ice.
Gene mutation analyses
Variants affecting PPM1D exon 6 were identified as described previously18. Briefly, DNA from non-cancer mucosa and colorectal tumor tissues were isolated by a routine procedure and was PCR amplified in two overlapping amplicons covering PPM1D exon 6 and directly sequenced. Paired non-cancer and tumor samples with identified PPM1D variants were subjected for analysis by next generation sequencing (NGS) using CZECANCA panel targeting 219 cancer-predisposition and candidate genes and bioinformatic analysis was performed as described25. All de novo indels identified in the tumor samples were visually inspected in IGV software. Recurrent TP53, KRAS, and BRAF-V600E mutations were identified by a routine pathological assessment in tumor samples and retrieved from patients’ records when available or analyzed from tumor DNA as described previously26,27,28,29. Microsatellite instability (MSI) status of the tumors was determined using the consensus markers provided by the National Cancer Institute (Bethesda marker panel), as previously described27.
Cell lines
hTERT-RPE1 (RPE) cells were obtained from ATCC, immortalized human colon cells from Applied Biological Materials (Cat. no. T0570) and HCT116 cells from Dr. Medema (NKI, Amsterdam). For Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-mediated editing, RPE cells or human colon cells were transfected with pSpCas9-(BB)-2A-Puro plasmid (Addgene ID48139) carrying targeting sequences GAAGGCATTGCTACGAACCAGGG (CR1) or ATAGCTCGAGAGAATGTCCAAGG (CR2) located in the exon 6 of PPM1D and single cell clones were expanded30. Alternatively, RPE cells were transfected with a combination of pZGB-3L-EGFP and pZGB-3R-mCherry plasmids containing coding sequence for FokI nuclease and transcription activator-like effector nuclease (TALEN) repeats targeting TGAAGAAAATTGCGCTA and TCAAAGAATC-ATGTATC regions in exon6 of human PPM1D (ZgenBio Biotech, Taiwan). Cells positive for mCherry and EGFP were sorted by Influx instrument (BD Biosciences) and single cell clones were expanded. Correct editing in the targeted region was confirmed by sequencing of genomic DNA and the stabilization of PPM1D protein by immunoblotting. RPE-TP53-KO cells with knocked-out TP53 were generated as described31. Relative cell proliferation after various treatments was determined by resazurin assay as described previously31.
Cell cycle checkpoint
Cells were pulsed with 5-ethynyl-2′-deoxyuridine (EdU, Jena Bioscience) and subsequently exposed to ionizing radiation generated by Precision X-RAD 225XL (dose 3, 6, or 10 Gy) and were grown for further 20 h in the presence of nocodazole (250 ng/mL). Cells were stained with anti-pSer10-histone H3 (mitotic marker, Santa Cruz) and 4′,6-diamidino-2-phenylindole (DAPI as a marker of DNA content) and EdU was labeled with AlexaFluor 647 using Click-iT reaction (Thermo Scientific). Cells were analyzed by flow cytometry using LSRII (BD Biosciences) and FlowJo software (FlowJo). EdU-positive cells were assayed for progression through the G2 phase to mitosis (4n DNA content, pH3+). EdU-negative cells with 2n content were used for quantification of cells arrested in G1 checkpoint.
Antibodies and reagents
Following antibodies were used: WIP1 (clone D4F7) Rabbit mAb (Cell Signaling, #11901) for detection of mouse PPM1D, WIP1 antibody (clone F-10, Santa Cruz sc-376257) for detection of human PPM1D, Importin-β (Santa Cruz, sc-137016), p21 for immunoblotting (Santa Cruz, sc-397), p21 (BD pharmingen, #556431) for IHC, p53 (clone D01, Santa Cruz, sc-126), phospho-S15-p53 (Cell Signaling, #9284), RPA2 (Abcam), phospho-S824-KAP1 (Genetex, GTX63711), KAP1 (Genetex, GTX62973), 14-3-3 (Santa Cruz, sc-133233), γH2AX (Millipore, 05-636 and Cell Signaling, #9718), TFIIH (Santa Cruz, sc-293), and Ki-67 (Genetex, GTX16667).
Animals
Mice were maintained in the animal facility of the Institute of Molecular Genetics on a standard diet and 12 h light–dark cycle with free access to food and water. Ppm1d mutant mice were generated by TALEN-mediated genome editing32. The following TALEN-repeat domain sequences were used to target exon 6 of Ppm1d gene: left TALEN: NI NI NG NN HD HD NG NG HD NG HD NI NN NI NN NI NI NN, right TALEN: NG HD HD HD NG HD NG NI NN HD NG NI NG HD NG HD NI NN HD. RNA precursors of TALENs were microinjected into male nucleoli of zygotes isolated from C57BL/6NCrl mice as described33. These zygotes were subsequently implanted into pseudopregnant females. Tail biopsies from newborn pups were tested for the presence of TALEN-mediated frameshift mutations using Sanger sequencing. Founder mouse carrying a c.1324_1339del16 frameshift-inducing mutation (resulting in p.F442Lfs*3) within exon 6 of Ppm1d gene was further bred to establish Ppm1d mutant line (referred to as Ppm1dT/+). Ppm1dT/+ mice were further crossed with previously described strains ApcMin/J, Lgr5tm1(cre/ERT2)Cle (Lgr5-EGFP-IRES-CreERT2 mice) and Trp53tm1Tyj (p53 knock-out mice)34,35,36. Animals were sacrificed at indicated times or when tumor burden was apparent or when moribund. Where indicated, mice or cells were irradiated with Precision X-RAD 225XL (dose 3 or 4 Gy as indicated; Cu filter 0.5 mm). For immunoblotting, mouse tissues were lyzed in 250 mM Tris–HCl (pH 6.8), 40% glycerol and 8% SDS buffer.
Genotyping
DNA was isolated from the tails by incubating overnight at 56 °C in 200 μL of a lysis solution containing [50 mM Tris pH 8.0, 100 mM EDTA, and 0.5% SDS] supplemented with proteinase K (100 μg/mL), precipitated by 0.5 M NaCl and isopropanol and resuspended in water.
Genotypes were determined by PCR. PCR fragment was digested by AvaII restriction enzyme, to distinguish between wt Ppm1d+/+ (cut) or Ppm1dT/+ (uncut) alleles. Following PCR primers were used: Ppm1d: CTAAGGACCATATACCTGCCCTTGTTCGCAG, and GATAGTATTT-GTTGAATTGGTTGGAATGAGGCC; Apcmin: common primer GCCATCCCTTCACGTTAG and TTCCACTTTGGCATAAGGC specific for wt allele or TTCTGAGAAAGACAGAAGTTA specific for mutant allele; Tp53: common primer GAAACAGGCTAACCTAACCTACCACGC and ATCCCGAGTATCTGGAAGACAGGC specific for wt allele or TTTGAATGGAAGGATTGGAGCTACGG specific for mutant allele; Lgr5: common primer CTGCTCTCTGCTCCCAGTCT and ATACCCCATCCCTTTTGAGC specific for wt allele or GAACTTCAGGGTCAGCTTGC specific for mutant allele.
Immunohistochemistry
The colon was harvested and placed in a 4% formaldehyde diluted in PBS overnight and then transferred into 70% ethanol. Paraffin-embedded colon sections were cut 4 μm thick and deparaffinized, cleared, and rehydrated in graded ethanol concentrations. Antigen retrieval was performed in a steam bath in Na–citrate buffer (10 mM, pH 6.0) for 30 min. Sections were pretreated with peroxidase blocking buffer (3% H2O2 in methanol) for 20 min at room temperature (RT). Blocking of non-specific binding was performed using 5% BSA for 1 h at RT. All antibodies were incubated overnight at 4 °C. Tissue sections were stained with p21 (BD Pharmingen #556431), Ki-67 (GeneTex #GTX16667), γH2A.X (Cell Signaling #9718). Secondary anti-rabbit-Alexa-488/594 or Biotin-XX Goat anti-Rabbit IgG biotin-conjugated (Thermo Fisher Scientific) were applied for 1 h at RT. TUNEL assay was performed using the In Situ Cell Death Detection kit (Invitrogen, #10618). To examine the morphological changes in the colon, standard haematoxylin and eosin (H&E) staining protocol was followed.
Isolation of colon crypts
Isolation of colon crypts and the dissociation of cells for flow cytometry analysis were performed as previously described37. In brief, colon crypts were isolated from 8 weeks old mice. Luminal contents were flushed out with cold PBS, tissue was split open longitudinally and incubated in 5 mM EDTA (pH 8) in PBS at 4 °C for 90 min. To isolate single cells from colon crypts, the pellet was further incubated with Dispase (Thermo Scientific; stock solution 100 mg/mL diluted 1:300 in serum-free DMEM) using rigorous shaking on rotating platform (800 RPM, 3 × 5 min, 37 °C). Large pieces of colon were then allowed to settle by gravity for 1 min, leaving the isolated crypts in suspension. These were collected and retained. Supernatant was diluted in DMEM containing 10% FBS (Gibco), and passed through a 70-μm strainer (Corning) to obtain a single-cell suspension. The step was repeated three times until the supernatant appeared clear. The sample was centrifuged for 5 min at 300 × g and cells were resuspended in PBS.
Flow cytometry and cell sorting
Sorting of ISCs and differentiated cells from small intestine or colon was performed as we described previously38,39. The single-cell suspension of colon crypts from Lgr5-EGFP-IRES-CreERT2 mice was stained with phycoerythrin-conjugated anti-CD24 antibody (Thermo Fisher Scientific, #12-0242-81) for 30 min at 4 °C. Sorting was performed using an Influx cell sorter (BD Biosciences) and cells were gated by forward scatter (FSC), side scatter (SSC), and negative staining for Hoechst 33258. The crypt base colon stem cells were sorted according to the cell size, CD24 stem cell marker and GFP signal (CD24+/GFP+) and compared to differentiated cells (CD24−/GFP−). Alternatively, single-cell suspension from small intestinal crypts was stained with antibody against Ulex europaeus agglutinin (UEA), marker of Paneth cells, and ISCs were sorted according to GFP+/UEA− signal.
Real-time quantitative reverse transcriptase PCR (qRT-PCR)
Colon mucosa was homogenized using TissueLyser LT (Qiagen) and RNA was extracted with the RNAeasy Mini Kit (Qiagen) according to the manufacturer’s instructions and cDNA was synthesized using the 0.5 μg RNA, random hexamer, and RevertAid H Minus Reverse Transcriptase (Thermo Scientific). Alternatively, RNA was isolated from ISCs or differentiated cells (5 × 103 cells) sorted by flow cytometry using RNeasy Micro Kit (Qiagen) and cDNA was generated using MAXIMA Reverse Transcriptase (Thermo Scientific). RT-qPCR was performed using LightCycler 480 SYBR Green I Master mix, Light Cycler LC480 (Roche, Basel, Switzerland) and following cycle conditions: initial denaturation 95 °C for 7 min, followed by 45 cycles of denaturation 95 °C for 15 s, annealing 60 °C for 15 s and extension 72 °C for 15 s. Melting curve analysis was used to confirm the specificity of amplification, and cycle threshold (Ct) values were determined using LigtCycler480 software. All qRT-PCR experiments were performed in triplicates and data are presented as the ratio of the tested mRNA to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. Following primers were used for qRT-PCR: Ppm1d: AGCCAGGAGACCTGTGTGAT and GGCATTACTGCGAACAAGGG; CDKN1A: TGAGGAGGAGCATGAATGGAGACA and AACAGGTCGGACATCACCAGGATT; PUMA: CCTGGAGGGTCATGTACAATCT and TGCTACATGGTGCAGAAAAAGT; GAPDH: TGGCAAAGTGGAGATTGTTGCC and AAGATGGTGATGGGCTTCCCG; LGR5: CAAGCCATGACCTTGGCCCTG and TTTCCCAGGGAGTG-GATTCTATT.
Organoid culture
Organoid cultures were established from freshly isolated colon tumors from 16 weeks old mice as described previously37,40. The organoids were embedded in Matrigel (BD Biosciences) and cultured with Advanced DMEM/F12 supplemented with penicillin/streptomycin, 10 mM HEPES, 2 mM GlutaMAX, N-2 supplement, B27 supplement (all Thermo Scientific), N-acetylcysteine (1 mM; Sigma-Aldrich), mRspo1 (500 ng/mL; Peprotech), mNoggin (100 ng/mL; Peprotech), and primocin (100 μg/mL, Invivogen). The following niche factors were added to the basal culture medium depending on the niche requirements of CRC organoid lines: gastrin I (10 nM; Sigma Aldrich #G9145), mouse recombinant EGF (50 ng/mL; Invitrogen Biosource #PMG8043), TGFβ type I receptor inhibitor (500 nM; Sigma Aldrich #A83-01) and p38 inhibitor (3 µM; Sigma Aldrich #SB202190). Tumor organoid proliferation was measured using resazurin proliferation assay. Organoids were seeded and embedded in Matrigel in 96-well plates. Following day, organoids were treated with 5-FU (20 µM; Sigma) and with DMSO or GSK2830371 (Medchemexpress). After 3 days, a mixture of resazurin (30 μg/mL) and complete organoid medium was added to each well, incubated for 2 h and fluorescence signal (Ex/Em = 540/590 nm) was measured by Envision plate reader (PerkinElmer). Samples were measured in hexaplicates and three independent experiments were performed.
Tumor size determination
The small intestine and colon of 16-week-old mice of indicated genotypes were resected longitudinally, washed with cold PBS and examined for polyp number count and tumor size. Length, width, and height of the tumor were measured by vernier caliper and tumor volume was calculated using formula (L*W*H)/2. The investigator was blinded to the group allocation during the experiment.
Statistical analysis
Each experiment was repeated at least three times and statistical significance was evaluated using Graph Pad Prism 5.04 software. Unless stated otherwise, two-sided unpaired t-test was used and p values < 0.05 are considered statistically significant. Kaplan–Meier survival plot was evaluated using log rank test. Error bars indicate standard deviation.
Results
Truncation of PPM1D impairs the checkpoint and p53 response in human cells
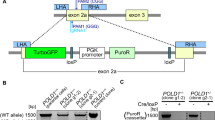
We have previously shown that heterozygous truncating mutations in exon 6 of the PPM1D impair activation of the p53 pathway in several human cancer cell lines, including U2OS and HCT116 cells18. Since cancer cells are genetically instable, we tested whether PPM1D truncation would cause similar defects in non-transformed cells. To this end, we introduced truncating mutations in exon 6 of the PPM1D gene in hTERT-RPE1 (RPE) cells using CRISPR/Cas9 technology and evaluated cell response to IR (Fig. 1, Supplementary Fig. S1a, b). Truncated PPM1D was expressed at considerably higher level than the full-length protein which is in agreement with the proposed role of the C-terminal degron motif in PPM1D turn over18,22. As expected, cells carrying the stabilizing PPM1D mutation showed lower level of KAP1-pS824, p53-pS15, and γH2AX phosphorylation, three established PPM1D targets (Fig. 1a)9,41. In addition, these cells failed to activate p53 and did not arrest in the G1 checkpoint after exposure to IR (Fig. 1b). Cells with truncated PPM1D also entered prematurely to mitosis after exposure to a low dose of IR which was consistent with the established role of p53 in maintenance of the G2 checkpoint42 (Fig. 1c). However, high dose of IR allowed full activation of the G2 checkpoint suggesting that the negative impact of the PPM1D activity on the p53 pathway can be compensated by high activity of upstream ATM/ATR kinases. We observed similar effects of the truncated PPM1D on DDR in independent RPE clones where genome editing was performed by TALEN technology (Supplementary Fig. S1c–e), and in immortalized human colon cells (Supplementary Fig. S1f, g), suggesting that mechanisms controlling PPM1D stability and its function in negative regulation of the p53 pathway do not depend on specific genetic or cellular background. Next, we performed colony formation assays to study the long-term effects of PPM1D truncation on cell proliferation under genotoxic stress. Whereas proliferation of the wt RPE cells was efficiently suppressed by a DNA damage-inducing drug etoposide, RPE cells producing truncated PPM1D formed comparable amount of colonies as RPE cells with inactivated p53 (Fig. 1d).

a Parental RPE and RPE-PPM1D-cr1.1 and RPE-PPM1D-cr2.1 cells were exposed to 3 or 10 Gy of IR, collected at indicated times and analyzed by immunoblotting. Arrow indicates position of the full length PPM1D. b Parental RPE and RPE-PPM1D-cr1.1 and RPE-PPM1D-cr2.1 cells were pulse labeled by EdU and 20 h after exposure to 3 or 10 Gy of IR were analyzed by flow cytometry. Plotted are EdU negative cells with 2n DNA content that correspond to cells arrested in G1. Bars indicate SD (n = 3). c Parental RPE and RPE-PPM1D-cr1.1 and RPE-PPM1D-cr2.1 cells were pulse labeled by EdU and 20 h after exposure to 3 or 10 Gy of IR were analyzed by flow cytometry. Plotted are pS10-histone H3-positive cells from EdU-positive cells corresponding to cells that progressed from S phase to mitosis. Bars indicate SD (n = 3). d Parental RPE and RPE-PPM1D-cr1.2, RPE-PPM1D-cr2.2 and RPE-PPM1D-tal1 cells were seeded in six-well plate and treated with etoposide for 24 h. Subsequently fresh media was added and colony formation was assayed after 14 days
Truncation of PPM1D impairs response to genotoxic stress in the mouse colon
Next, we aimed to determine the impact of truncated PPM1D in the tissue context of and to evaluate its potential role in cancer growth. To this end, we employed TALEN technology and generated a mouse strain producing the C-terminally truncated PPM1D (Fig. 2a, b). Similarly to human cells, heterozygote Ppm1dT/+ mice showed higher level of truncated Ppm1d protein compared to the full-length Ppm1d in all tested tissues including small intestine, colon, liver, and spleen (Fig. 2c). Expression of Ppm1d was previously detected by in situ RNA hybridization at the bottom of the crypts in small intestine12. This compartment is composed of terminally differentiated Paneth cells and self-renewing ISCs; the latter cells specifically express leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5)34,43,44. We used Lgr5-EGFP-IRES-CreERT2 mice carrying either wt or truncated Ppm1d to isolate different subpopulations of cells from intestinal epithelium and analyzed PPM1D expression. Using real-time qRT-PCR, we confirmed that Ppm1d mRNA was highly expressed in the small intestinal ISCs (EGFP+/UEA−) but not in Paneth cells (EGFP−/UEA+) (Fig. 2d). Similarly, Ppm1d was highly expressed in colon ISCs double positive for Lgr5-EGFP and a surface antigen CD24 (Fig. 2e). Next, we performed histological analysis of the wt and Ppm1dT/+ mice and found that truncation of PPM1D did not impair the general organization of the small intestinal and colon epithelium (Fig. 2f). To study the role of PPM1D in response to genotoxic stress, we exposed mice carrying the Ppm1dT allele to IR and analyzed expression of candidate genes in the isolated colon mucosa. As expected, expression of Cdkn1a (coding for the cell cycle inhibitor p21) was highly induced in wt animals after exposure to a low dose of IR (Fig. 3a). In contrast, expression of Cdkn1a was significantly decreased in Ppm1dT/+ mice exposed to IR (Fig. 3a). Interestingly, the suppression of Cdkn1a in Ppm1dT/+ was similar to the level observed in Tp53+/− mice suggesting that truncated PPM1D can partially inhibit the p53 pathway in the mouse colon (Fig. 3a). In addition, truncated PPM1D-impaired expression of PUMA, an established p53 target and the main pro-apoptotic gene in ISCs (Fig. 3b)45,46. Furthermore, expression analysis performed in LGR5+ cells isolated from mice exposed or not to IR revealed lower expression of Cdkn1a and two pro-apoptotic genes Trp53inp1 and PHLDA3 in Ppm1dT/+ confirming that truncated Ppm1d impairs the p53-dependent response of colon ISCs to genotoxic stress (Fig. 3c, Supplementary Fig. S2)47,48. In agreement with the expression analysis, we observed lower amounts of p21-positive cells in the colon crypts of Ppm1dT/+ mice using immunohistochemistry (Fig. 3d). Similarly, we detected lower amounts of apoptotic cells in the colon crypts of Ppm1dT/+ mice after exposure to IR compared to wt mice (Fig. 3e). Conversely, we found higher levels of the proliferation marker Ki-67 in the Ppm1dT/+ colon epithelium when compared to wt mice (Fig. 3f), which was consistent with the role of truncated PPM1D in blocking p53-dependent suppression of the cell proliferation and survival.

a Schematic representation of TALEN-mediated editing of the Ppm1d locus. DNA and protein sequences of the wild type Ppm1d+ and Ppm1dT alleles are shown. Bases lost by editing are indicated in red. b Genotyping of F2 generation. Exon 6 of Ppm1d was PCR amplified from Ppm1d+/+, Ppm1dT/+, and Ppm1dT/T mice and subjected to AvaII digestion. c Protein levels of wt and truncated PPM1D in intestine, colon, liver, and spleen were analyzed by western blotting. Empty and full arrowhead indicate positions of wt and truncated PPM1D, respectively. Importin was used as a loading control. d Expression of Ppm1d was determined by RT-qPCR in ISCs (GFP+UEA−), Paneth cells (GFP−UEA+) and other differentiated cells isolated from small intestine of Lgr5-EGFP-IRES-CreERT2 mice and was normalized to the GAPDH expression levels. e Expression of Ppm1d was determined by RT-qPCR in colon ISCs (GFP+CD24+) and differentiated colon cells (GFP−CD24−) isolated from Lgr5-EGFP-IRES-CreERT2-Ppm1d+/+ or Lgr5-EGFP-IRES-CreERT2-Ppm1dT/+ and was normalized to GAPDH (n = 3). f Representative H&E-stained sections of the colon from 8 weeks old wt Ppm1d+/+, Ppm1dT/+, and Ppm1dT/T mice

a Expression of Cdkn1a in colon mucosa 6 and 12 h after exposure of mice of indicated genotypes to 3 Gy of IR was measured by RT-qPCR and normalized to GAPDH (n ≥ 3). b) Expression of PUMA in the colon mucosa 48 h after exposure to 3 Gy of IR was measured by RT-qPCR and normalized to GAPDH (n ≥ 3). c LGR5-EGFP-positive ICS cells were sorted from colon of wt Ppm1d+/+, and Ppm1dT/+ mice exposed or not to 3 Gy of IR and expression of Cdkn1a was measured by RT-qPCR and normalized to GAPDH (n = 3). d Levels of p21 were determined in the mouse colon 48 h after exposure to 3 Gy of IR by immuno-histochemistry. The percentage of p21-positive cells per crypt was scored (n ≥ 4). At least 90 crypts were counted for each group. Representative images are shown. Scale bar 25 µm. e Apoptotic cells were detected in colon epithelia of mice of indicated genotypes 48 h after exposure to IR (3 Gy) by TUNEL apoptosis assay (n = 5). At least 90 crypts were counted for each group. Representative images are shown. Scale bar 50 µm. f Levels of a proliferation marker Ki-67 were determined in the mouse colon 48 h after exposure to 3 Gy of IR by immuno-histochemistry (n ≥ 4). At least 90 crypts were counted for each group. Representative images are shown. Scale bar 50 µm
Truncation of PPM1D promotes growth of colon tumors in Apcmin mice
Loss of Ppm1d was previously shown to delay development of various solid tumors in mice including mammary and intestinal adenocarcinoma12,49. As the truncating PPM1D mutation was originally identified in HCT116 cells derived from colon adenocarcinoma18, we aimed to test the contribution of the truncated Ppm1d to tumor development in mouse intestine. Stem cell proliferation in the gastrointestinal tract is controlled by secreted glycoproteins of the Wnt family that trigger β-catenin/T-cell factor (TCF)-dependent expression of a specific set of target genes50. Constitutive activation of the Wnt pathway occurs in the majority of colorectal cancer (CRC) and is caused mainly by mutations in the genes encoding tumor suppressor adenomatous polyposis coli (APC) (~80% of cases), β-catenin (~4.7%), the nuclear mediator of Wnt signaling TCF4 (~4.1%) or ubiquitin ligase F-box/WD repeat-containing protein 7 (FBXW7) (~10.4%)51. Inactivating mutations in APC promote gradual development of dysplasia, adenoma, and adenocarcinoma in human cancers and corresponding mouse models52. Here, we crossed Ppm1dT/+ mice with Apcmin mice, an established model of gastrointestinal cancer in which the mice carry one non-functional allele of Apc and the second wt allele is randomly inactivated during the life span of the animals53. Typically, Apcmin mice develop multiple polyps in intestine and die approximately at 6 months of age. In contrast to humans, Apcmin promotes mostly small intestinal tumors, whereas tumors in the colon are less frequent53. As expected, multiple polyps were present in the small intestine in Apcmin mice and the number of polyps significantly increased in ApcminPpm1dT/+ mice (Fig. 4a). In addition, we observed single tumors in colon in about 34% of Apcmin mice at 16 weeks of age (Fig. 4b, c). Strikingly, formation of the colon tumors was enhanced to 68% in ApcminPpm1dT/+ mice and typically these mice developed multiple (usually 2–3 tumors) colon tumors at 16 weeks of age (Fig. 4b, c). Although the mean volume of the tumors in the colon was comparable in Apcmin and ApcminPpm1dT/+ mice, we observed several very large tumors in a fraction of ApcminPpm1dT/+ mice (Fig. 5d, e). The median survival of ApcminPpm1dT/+ mice was significantly reduced when compared to Apcmin (28 vs. 30 weeks, log rank test, p = 0.019), probably reflecting a higher tumor burden in the whole intestine observed in these mice (Fig. 4f). In addition, survival of ApcminPpm1dT/+ and ApcminTp53+/− was comparable (p = 0.717, log rank test), indicating that the impact of truncated PPM1D on promoting tumor growth was mediated by the p53 pathway. Finally, we observed significantly reduced survival of Ppm1dT/+ heterozygote compared to Ppm1d+/+ mice (p = 0.014, log rank test) (Fig. 4f). Within the period of 50 weeks of age we did not observe any tumor growth in the colon of Ppm1dT/+ mice suggesting that truncated PPM1D does not represent the tumor-initiating mutation leading to ISCs transformation. This is consistent with previous observation when overexpression of Ppm1d in mouse embryonic fibroblasts failed to stimulate cell growth in the soft agar but increased transformation efficiency of the RAS oncogene15. In summary, our data suggest that truncated PPM1D promotes colon tumor growth in cooperation with other driver mutations including loss of the tumor suppressor APC.

a Wild-type Ppm1d+/+, Ppm1dT/+, Apcmin, and ApcminPpm1dT/+ mice were sacrificed at 16 weeks and the number of intestinal polyps was scored (p = 0.044). Numbers indicate numbers of animals of each genotype. b Wild-type Ppm1d+/+, Ppm1dT/+, Apcmin, and ApcminPpm1dT/+ mice were sacrificed at 16 weeks and tumor formation in colon was scored. Proportion of animals developing tumor(s) is shown, N indicates number of animals of each genotype. Statistical significance was determined by two tailed Fisher’s exact test (***p < 0.001, **p < 0.01, *p < 0.05). c Proportion of animals with multiple tumors in mice from b was scored. N indicates number of animals that developed at least one tumor. Statistical significance was determined by two tailed Fisher’s exact test (***p < 0.001, **p < 0.01, *p < 0.05). d Volume of the colon tumors developed in mice with indicated genotypes. In case of multiplicity, the largest tumor was quantified. Line indicates mean. e Representative sections of colon tumors from 16 weeks old Apcmin, ApcminPpm1dT/+ mice stained for Ki-67. Scale bars 50 and 25 μm. f Kaplan–Meier survival plot of mice of indicated genotypes over a period of 50 weeks. Statistical significance was determined by log rank test as follows Apcmin vs. ApcminPpm1dT/+ p = 0.019; Apcmin vs. ApcminTp53+/− p = 0.008; ApcminPpm1dT/+ vs. ApcminTp53+/− p = 0.717; Ppm1d+/+ vs. Ppm1d T/+ p = 0.014). Numbers of animals per group are shown

a Parental RPE, RPE-PPM1D-cr1.1 or RPE-PPM1D-cr2.1 cells were mock treated, exposed to camptothecin (5 nM) or to IR (2 Gy). Relative cell proliferation was determined after 7 days by resazurin assay (n = 3). b Parental RPE, RPE-PPM1D-cr1.1 or RPE-PPM1D-cr2.1 cells were grown in indicated doses of GSK2830371 (PPM1Dinh) for 7 days and relative cell proliferation was determined by resazurin assay. c Parental HCT116 cells or HCT116-PPM1D-KO cells were treated with indicated combinations of DMSO, GSK2830371 (PPM1Dinh; 0.5 µM) and 5-FU (1 µM) and relative cell proliferation was determined after 7 days by resazurin assay (n = 3). d Organoids derived from colon tumors of Apcmin or ApcminPpm1dT/+ mice were grown in the presence of 5-FU for 3 days and imaged by light microscopy. Representative image is shown. Scale bar 250 µm. e Tumor organoids derived from Apcmin or ApcminPpm1dT/+ mice (n > 3) were grown in the presence of 5-FU (20 µM) and GSK2830371 (PPM1Dinh; 10 µM) and survival was determined after 3 days
Truncation of PPM1D impairs sensitivity to chemotherapy
As we observed altered response to genotoxic stress in cells carrying truncated PPM1D, we wished to address if truncated PPM1D may affect sensitivity of cells to chemotherapeutic agents. We found that RPE with truncated PPM1D proliferated in the presence of a topoisomerase inhibitor camptothecin or after exposure to the low level of IR, whereas parental RPE cells were sensitive to both treatments (Fig. 5a). Conversely, RPE with truncated PPM1D were more sensitive to GSK2830371, a specific small-molecule inhibitor of PPM1D, compared to parental RPE cells (Fig. 5b)31,54. These data suggested that stabilization of PPM1D can impair sensitivity of cells to DNA-damaging agents, while making them more sensitive to inhibition of PPM1D activity. To test the sensitivity in cancer cells, we used HCT116 cells that intrinsically carry PPM1D truncation, and exposed them to 5-FU, a chemotherapeutic agent commonly used in CRC treatment. We found that PPM1D inhibitor improved sensitivity of HCT116 cells to 5-FU (Fig. 5c). Similarly, genetic inactivation of PPM1D by the CRISPR/Cas9 system also sensitized HCT116 cells to 5-FU confirming the specificity of GSK2830371 (Fig. 5c). Additionally, we established organoid cultures from Apcmin and ApcminPpm1dT/+ colon tumors and compared their growth in the presence 5-FU. We found that organoids derived from Apcmin tumors were sensitive to 5-FU, whereas ApcminPpm1dT/+ organoids continued proliferation despite the presence of 5-FU (Fig. 5d, e). To further validate the impact of Ppm1dT on resistance to 5-FU, we treated organoids with GSK2830371 and found that inhibition of PPM1D restored the sensitivity of ApcminPpm1dT/+ organoids to 5-FU (Fig. 5e). We concluded that cancer cells carrying truncating mutations in the PPM1D gene are resistant to 5-FU treatment, whereas inhibition of PPM1D can restore the sensitivity to 5-FU.
PPM1D is truncated in a fraction of colon adenocarcinomas
Finally, we wished to address potential contribution of PPM1D truncation to CRC development in humans. Search for the cancer cell lines harboring the truncating mutations in exon 6 of the PPM1D in COSMIC database (https://cancer.sanger.ac.uk/cosmic; v87) revealed that besides HCT116 cells with c.1349delT (p.L450*), similar mutations are present in SNU175 (c.1528_1529insA; p.N512Kfs*16) and CL-34 (c.1714C>T; p.R572*) cells derived from colon adenocarcinoma55. Next, we performed PPM1D mutation analysis in 364 unselected CRC samples from Norway. We found two recurrent mutations: single nucleotide deletions c.1349delT (p.L450*), and c.1535delA (p.N512Ifs*2) in five and six patients, respectively, single nucleotide duplication c.1535dupA (p.N512Kfs*16) in another patient and several individual missense variants throughout the exon 6 (Table 1, Supplementary Table S1). Interestingly, all frameshift mutations reside in two short homopolymer “hot spot” regions in exon 6, namely c.1344_1349TTTTTT and c.1529_1535AAAAAAA. Further analysis revealed that only two of 12 PPM1D-mutated samples also carried a mutation in TP53 indicating that most of these tumors developed in the presence of wt p53 (Table 1). Out of the 10 PPM1D mutated CRC samples with wt TP53, all showed high level of MSI and seven carried activating BRAF mutations strongly suggesting that PPM1D mutations clustered into the consensus molecular subtype 1 (CMS1) of CRC characterized by presence of MSI and BRAF mutation56,57. Identified PPM1D indels resided in short homopolymer sequences, typically altered in MMR-deficient tumors58,59,60,61,62. In addition, we identified the same frameshift mutations in the hot spot region in a smaller cohort of 243 Czech colon cancer patients. Both tumors samples with truncated PPM1D carried somatic BRAF-V600E mutations, wt TP53 and were classified as MSI (Table 1). Subsequently, tumors and matched non-tumor tissues were subjected to panel sequencing that revealed de novo somatic frameshift mutations in homopolymer regions of several genes including MSH3 and MLH3, confirming a defect in MMR in the tumor tissue (Supplementary Table S2). Next, we screened also a cohort of 340 unselected Swedish CRC patients revealing four samples with PPM1D truncating mutation, which included recurrent p.L450* mutation, a deletion affecting c.1632_1636CCCCC homopolymer (c.1636delC; p.L546*), one missense mutation (c.1358C>A; p.S453*) and one insertion (c.1476dupT; p.S493*) outside the homopolymeric regions (Table 1). Finally, we searched the COSMIC database and found that PPM1D truncations in exon 6 harbor 89% of all PPM1D mutations (166/186) identified in various tumor types. The most frequent mutations occurred in samples of endometrial cancer (10/936; 1.07%), hematological tumors (46/4816; 0.96%), and CRC (29/3490; 0.83%). The distribution of exon 6 alterations further confirmed that PPM1D truncations in CRC, but not in other tumors, clustered into the hot spots identified in the present samples (Supplementary Fig. S3). Moreover, 23/29 (79%) mutations referred in the CRC samples in COSMIC (and 16/19; 84% in the present series) appeared in short homopolymers, in contrast with 18/64 (28%) and 2/10 (20%) of such mutations in samples from hematopoietic and endometrial tumors, respectively. Altogether, we conclude that recurrent somatic truncating PPM1D mutations occur dominantly in a fraction of human CRCs that remain p53-proficient and are associated with MMR pathway defects. Truncating PPM1D mutations in non-CRC tumors, including hematological malignancies, may arise by different mechanisms than MMR defects.
Discussion
By exploiting a newly generated mouse model we demonstrate that truncated Ppm1d can partially suppress the p53 pathway in ISCs in the small intestinal and colon epithelium. As a result, stem cells carrying the truncated form of Ppm1d survive in the presence of genotoxic stress whereas wt stem cells are more prone to cell death. Interestingly, Ppm1dT/+ and Ppm1dT/T mutants showed comparable effects on suppressing p53 functions suggesting that alteration of only one Ppm1d allele is sufficient to induce the gain-of-function phenotype. We noted that suppression of the p53 response in Ppm1dT/+ mice is partial as more robust decrease of Cdkn1a and PUMA expression was observed in Tp53 knock-out mice.
Although we did not observe increased spontaneous tumor formation in Ppm1dT/+ mice, we noted moderately decreased overall survival towards the end of this study (50 weeks) warranting a future follow-up of these mice. Whereas truncated PPM1D may be insufficient to fully transform ISCs and drive tumorigenesis of otherwise normal cells, in genetically sensitized background it promoted growth of colon tumors. Consistent with a previously reported suppression of the polyp formation in the small intestine in Ppm1d knock-out mice, we observed that truncation of Ppm1d significantly increased the number of intestinal polyps in ApcminPpm1dT/+ mice12. More strikingly, we observed increased frequency of the APC-driven colon tumors in Ppm1dT/+ mice and their reduced survival. The phenotype is more closely resembling the tumorigenic process observed in human patients. The reason why the increased stability of PPM1D in the mouse model shifts the tumor burden from the small intestine to the colon remains unclear. We suggest the tumor-promoting role of truncated PPM1D may be related to some mutational signature that differs between small intestinal and colon tissue.
We have identified various types of somatic frameshift and/or nonsense mutations in the last exon of PPM1D in the colon tumors in cohorts of CRC patients from three countries and similar mutations were also found in publicly available databases. Out of these, most common are the frameshift mutations N512Ifs*2 and L450stop that are both caused by a single nucleotide deletion in the homopolymeric region in exon 6 of PPM1D. Tumors carrying these two mutations were typically microsatellite instable, p53-proficient, carried BRAF mutations and were located in the right colon. In addition, NGS of two tumors with PPM1D mutations revealed somatic defects in MSH genes. In summary, we conclude that the truncating PPM1D mutations arise primarily in MMR-deficient CRC. It will be interesting to determine the contribution of truncated PPM1D to tumor development using the BRAF-V600E mouse model.
Besides the two recurrent frameshift mutations, we identified several individual nonsense mutations outside the hotspots. Similar nonsense PPM1D mutations were recently found in a fraction of HSCs and their frequency typically increases with advancing age22. Clonal hematopoiesis may represent a serious threat when resistant cell clones are selected during chemotherapy and the clones promote development of secondary malignancies including therapy-related AML and myelodysplastic syndrome24. With new technological advances in sequencing of individual stem cells, it is now becoming clear that similar age-related clonal differences in the stem cell populations may occur also in other tissues including the small intestine, colon, and liver63. We hypothesize that some of the PPM1D truncating mutations occur randomly in a fraction of colon stem cells, providing a selective advantage compared to healthy stem cells and potentially contributing to cancer development.
Using cell competition assays, hematopoietic cells carrying truncated PPM1D were recently shown to outcompete wt cells when transplanted into lethally irradiated mice treated with cytarabine, cisplatin, or etoposide22,24. Here, we tested the sensitivity of organoid cultures derived from Apcmin or ApcminPpm1dT/+ colon tumors to 5-FU, used as a standard chemotherapy for CRC. We found that ApcminPpm1dT/+ organoids were less sensitive to 5-FU compared to Apcmin organoids. Importantly, inhibition of PPM1D by a small molecule inhibitor increased sensitivity of organoids carrying truncated PPM1D to 5-FU, likely reflecting increased activity of p53, which is an important factor determining the sensitivity to 5-FU64. Allosteric inhibitor GSK2830371 potently suppresses the PPM1D activity; however, its low stability prevented us from testing its efficiency directly in mice. Recently it has been suggested that combining chemotherapy with inhibition of PPM1D could prevent development of secondary therapy-related tumors22. Based on our results we propose that inhibition of PPM1D may represent a promising strategy for improving efficacy of 5-FU in colon cancer treatment, especially in patients with MMR-deficient tumors located in caecum and ascending colon.
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Lukas, J., Lukas, C. & Bartek, J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair 3, 997–1007 (2004).
Bieging, K. T., Mello, S. S. & Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359 (2014).
Bartkova, J. et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870 (2005).
Bartkova, J. et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006).
Lord, C. J. & Ashworth, A. The DNA damage response and cancer therapy. Nature 481, 287–294 (2012).
Lindqvist, A. et al. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J. 28, 3196–3206 (2009).
Jaiswal, H. et al. ATM/Wip1 activities at chromatin control Plk1 re–activation to determine G2 checkpoint duration. EMBO J. 36, 2161–2176 (2017).
Shaltiel, I. A. et al. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. PNAS 111, 7313–7318 (2014).
Demidov, O. N. et al. The role of the MKK6//p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene 26, 2502–2506 (2006).
Demidov, O. N. et al. Wip1 phosphatase regulates p53-dependent apoptosis of stem cells and tumorigenesis in the mouse intestine. Cell Stem Cell 1, 180–190 (2007).
Demidov, O. N. et al. Role of Gadd45a in Wip1-dependent regulation of intestinal tumorigenesis. Cell Death Differ. 19, 1761–1768 (2012).
Shreeram, S. et al. Regulation of ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. J. Exp. Med. 203, 2793–2799 (2006).
Bulavin, D. V. et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 31, 210–215 (2002).
Le Guezennec, X. & Bulavin, D. V. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 35, 109–114 (2010).
Li, J. et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat. Genet. 31, 133–134 (2002).
Kleiblova, P. et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J. Cell Biol. 201, 511–521 (2013).
Ruark, E. et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 493, 406–410 (2013).
Agrawal, N. et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 (2014).
Zhang, L. et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat. Genet. 46, 726–730 (2014).
Kahn, J. D. et al. PPM1D truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 132, 1095 (2018).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472 (2014).
Hsu, J. I. et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell 23, 700–713.e706 (2018).
Soukupova, J. et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS ONE 13, e0195761 (2018).
Brozova, M. et al. Establishment, growth and in vivo differentiation of a new clonal human cell line, EM-G3, derived from breast cancer progenitors. Breast Cancer Res. Treat. 103, 247–257 (2007).
Vedeld, H. M. et al. CpG island methylator phenotype identifies high risk patients among microsatellite stable BRAF mutated colorectal cancers. Int. J. Cancer 141, 967–976 (2017).
Smeby, J. et al. CMS-dependent prognostic impact of KRAS and BRAFV600E mutations in primary colorectal cancer. Ann. Oncol. 29, 1227–1234 (2018).
Berg, M. et al. DNA sequence profiles of the colorectal cancer critical gene set KRAS-BRAF-PIK3CA-PTEN-TP53 related to age at disease onset. PLoS ONE 5, e13978 (2010).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281 (2013).
Pechackova, S. et al. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 7, 14458–14475 (2016).
Cermak, T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82–e82 (2011).
Kasparek, P. et al. Efficient gene targeting of the Rosa26 locus in mouse zygotes using TALE nucleases. FEBS Lett. 588, 3982–3988 (2014).
Barker, N. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003 (2007).
Su, L. et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670 (1992).
Jacks, T. et al. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7 (1994).
Sato, T. et al. Single Lgr5 stem cells build crypt–villus structures in vitro without a mesenchymal niche. Nature 459, 262 (2009).
Fafilek, B. et al. Troy, a tumor necrosis factor receptor family member, interacts with Lgr5 to inhibit Wnt signaling in intestinal stem cells. Gastroenterology 144, 381–391 (2013).
Stancikova, J. et al. NKD1 marks intestinal and liver tumors linked to aberrant Wnt signaling. Cell. Signal. 27, 245–256 (2015).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Macurek, L. et al. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 29, 2281–2291 (2010).
Bunz, F. et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 (1998).
Sato, T. et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418 (2011).
Snippert, H. J. et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144 (2010).
Nakano, K. & Vousden, K. H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7, 683–694 (2001).
Qiu, W. et al. PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell 2, 576–583 (2008).
Okamura, S. et al. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol. Cell 8, 85–94 (2001).
Kawase, T. et al. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell 136, 535–550 (2009).
Bulavin, D. V. et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19Arf pathway. Nat. Genet. 36, 343–350 (2004).
Krausova, M. & Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell. Signal. 26, 570–579 (2014).
Zhan, T., Rindtorff, N. & Boutros, M. Wnt signaling in cancer. Oncogene 36, 1461–1473 (2017).
Fearon, E. R. & Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990).
Moser, A., Pitot, H. & Dove, W. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247, 322–324 (1990).
Gilmartin, A. G. et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 10, 181–187 (2014).
Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350 (2015).
Sveen, A. et al. Colorectal cancer consensus molecular subtypes translated to preclinical models uncover potentially targetable cancer cell dependencies. Clin. Cancer Res. 24, 794–806 (2018).
Zhao, H. et al. Mismatch repair deficiency endows tumors with a unique mutation signature and sensitivity to DNA double-strand breaks. eLife 3, e02725 (2014).
Markowitz, S. et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268, 1336–1338 (1995).
Rampino, N. et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 275, 967–969 (1997).
Thorstensen, L. et al. WNT1 inducible signaling pathway protein 3, WISP-3, a novel target gene in colorectal carcinomas with microsatellite instability. Gastroenterology 121, 1275–1280 (2001).
Duval, A. et al. Variable mutation frequencies in coding repeats of TCF-4 and other target genes in colon, gastric and endometrial carcinoma showing microsatellite instability. Oncogene 18, 6806 (1999).
Blokzijl, F. et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260 (2016).
Longley, D. B., Harkin, D. P. & Johnston, P. G. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330 (2003).
Acknowledgements
Authors are grateful to Rene H. Medema for commenting on the manuscript, to Katerina Krejcikova, Anna Makarova, Zdeněk Cimburek, and Miroslav Stoyanov for technical assistance. This work was supported by the Czech Science Foundation (16-19437S), Academy of Sciences of the Czech Republic (RVO 68378050), and EEA Czech-Norwegian Research Programme—Norwegian Financial Mechanism 2009–2014 (PHOSCAN, 7F14061). Animal work was partially supported by Czech Centre for Phenogenomics (project no. LM2015040), OP RDI CZ.1.05/2.1.0019.0395 (project: Higher quality and capacity for transgenic models) and Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University (CZ.1.05/1.1.00/02.0109). A.S.M. is a Ph.D. student of Developmental and Cell Biology and received a stipend from Charles University. K.B. was partially supported by Worldwide Cancer Research foundation (14-1176), P.V. by the Czech Science Foundation (18-09709S), V.L. by National Sustainability Program I (NPU I, LO1503), X.-F.S. by Swedish Cancer Foundation, Swedish Research Council, and the Health Research Council in South-East Sweden, and R.A.L. by the Research Council of Norway (179571 and 250993), the Norwegian Cancer Society (182759-2016) and the South-Eastern Regional Health Authorities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by U. Moll
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Burocziova, M., Burdova, K., Martinikova, A.S. et al. Truncated PPM1D impairs stem cell response to genotoxic stress and promotes growth of APC-deficient tumors in the mouse colon. Cell Death Dis 10, 818 (2019). https://doi.org/10.1038/s41419-019-2057-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-2057-4
This article is cited by
-
PPM1D activity promotes cellular transformation by preventing senescence and cell death
Oncogene (2024)
-
Ppm1d truncating mutations promote the development of genotoxic stress-induced AML
Leukemia (2023)
-
A stop-gain mutation in GXYLT1 promotes metastasis of colorectal cancer via the MAPK pathway
Cell Death & Disease (2022)
-
PPM1D suppresses p53-dependent transactivation and cell death by inhibiting the Integrated Stress Response
Nature Communications (2022)
