
Abstract
Osteosarcoma (OS) is a primary malignant bone tumour. However, the genetic basis for the pathogenesis of OS remains elusive. In this study, we uncovered the role of the histone methyltransferase NSD2 in regulating tumourigenesis and chemosensitivity in OS. We show that NSD2 knockdown leads to increased apoptosis in OS cells in vitro and in vivo. Additionally, NSD2 knockdown significantly enhances the efficacy of cisplatin against OS cells and accordingly inhibits properties associated with cancer stem cells (CSCs). Furthermore, RNA sequencing (RNAseq) and Gene Ontology (GO) analysis revealed that NSD2 promotes transcription of genes associated with negative regulation of apoptotic signalling pathways and CSC properties. The results of chromatin immunoprecipitation quantitative polymerase chain reaction (ChIP-qPCR) assays indicated that NSD2 knockdown leads to decreased H3K36me2 modification at BCL2 and SOX2 loci, thus inhibiting the transcription of these two genes that are closely correlated with apoptosis, CSC properties and chemosensitivity in OS cells. Pathway analysis demonstrated that the ERK and AKT pathways mediate the regulation of OS progression and chemosensitivity by NSD2. Overall, our study is the first to uncover the function of NSD2 in OS chemosensitivity. NSD2 regulates the expression of the apoptosis regulatory proteins BCL2 and SOX2 through the ERK and AKT pathways. Our results suggest that NSD2 is a new target for combined chemotherapy and is a prognostic factor for OS.
Similar content being viewed by others


Introduction
Osteosarcoma (OS) is one of the most common malignant bone tumours and arises primarily in children and adolescents1. In adults aged >65 years, OS develops as a secondary malignancy related to Paget’s disease of bone2. To date, chemotherapy has been frequently included in conventional treatment of OS along with surgical resection. Since the 1970s, the introduction of adjuvant chemotherapies to OS treatment has impressively improved the 5-year survival rate. In addition, in patients with localized disease, the strongest predictor of overall survival is the patient’s response to preoperative combination chemotherapy3. Unfortunately, the 5-year survival rate has remained at approximately 20% over the past 20 years without changing4,5, which is partly due to chemoresistance. Cisplatin is a National Comprehensive Cancer Network first-line chemotherapy medication commonly used for clinical treatment of OS and a variety of other tumours3. In OS patients, cisplatin shows a response rate of approximately 30%, indicating that a significant proportion of OS patients are intrinsically resistant to cisplatin. Therefore, there is an urgent need to develop new strategies to further improve the long-term survival rates of OS patients.
Epigenetic perturbations caused by histone methyltransferases or demethylases are recognized as important contributing factors to a variety of tumours6, and epigenetic markers have frequently been found to be mutated or dysregulated in multiple cancers7. NSD2, which belongs to the NSD family of histone lysine methyltransferases (HMTases)8, is a histone methyltransferase that mediates dimethylation of histone 3 lysine 36 (H3K36me2); H3K36 methylation is typically a permissive marker associated with transcriptional activation9,10,11. NSD2 was first reported to function as an oncogenic gene in multiple myeloma12,13,14. In addition, NSD2 is highly expressed in many solid tumours, such as those in prostate cancer, breast cancer and head and neck cancer12,13,14. However, whether NSD2 mediates chemosensitivity in OS has not been reported.
In this study, the results indicated that NSD2 is upregulated in OS tissues compared with normal tissues and that NSD2 knockdown can enhance OS apoptosis and sensitize OS to cisplatin by directly decreasing H3K36me2 levels at BCL2 and SOX2 gene loci, as measured by chromatin immunoprecipitation (ChIP) analysis. Pathway analysis illustrated that the extracellular signal–regulated kinase (ERK) and AKT signalling pathways are reversed when NSD2 is knocked down. Together, our results suggest that NSD2 is a novel target for overcoming chemoresistance in OS.
Results
NSD2 expression is upregulated in OS and in cisplatin-resistant patients
To assess the role and clinical relevance of NSD2 in OS, we first assessed NSD2 expression in OS and matched normal peritumoural specimens. Real-time quantitative polymerase chain reaction (RT-qPCR) analysis showed that NSD2 mRNA expression was significantly higher in OS tissues than in matched normal tissues in 20 OS patient biopsies (Fig. 1a). The clinical information of the 20 OS patients is summarized in Table S1. Furthermore, NSD2 expression was higher in cisplatin-resistant OS biopsies than in cisplatin-sensitive ones, as measured by immunohistochemistry (IHC) and RT-qPCR (Fig. 1b, c). Patients with lower NSD2 expression presented better prognoses (Fig. 1d); the relative clinical information is listed in Table S2. Together, these findings suggest that NSD2 is upregulated in OS patients, especially in cisplatin-resistant OS patients, and that lower NSD2 expression predicts better prognoses in OS patients.

a NSD2 mRNA levels in 20 tumoural and matched peritumoural tissues. NSD2 expression in cisplatin-sensitive and cisplatin-resistant specimens was measured by immunohistochemistry (b) and real-time quantitative polymerase chain reaction (c). d Kaplan–Meier plot of survival time in patients with low and high NSD2 expression. IOD integrated optical density
NSD2 knockdown promotes OS apoptosis in vitro
To further test the role of NSD2 in OS, we knocked down NSD2 (NSD2-KD) with a lentiviral system. We generated NSD2-depleted 143B and HOS cells using packaged lentiviruses including two short hairpin RNA (shRNA) sequences targeting NSD2 (the sequences are listed in Table S4) that displayed similar knockdown efficiencies (Fig. 2a). Although previous research on myeloma has indicated that NSD2 can affect the enhancer of zeste homologue 2 (EZH2, a methyltransferase), increase histone 3 lysine 27 trimethylation (H3K27me3) levels and ultimately lead to repression of gene expression15,16, the results in our study indicated that H3K27me3 levels were not changed significantly by knockdown of NSD2 (Fig. S1) which is in accordance with the results of Kuo et al.9. Moreover, the results also indicated that NSD2 knockdown reduced the growth of OS cells, as measured by Cell Counting Kit-8 (CCK-8) and colony-formation assays (Fig. 2b, c). Notably, the outcomes of flow cytometric analysis indicated that there were no significant differences in cell cycle progression between control and NSD2-KD OS cells (Fig. S2). In addition, the percentage of cells that underwent apoptosis was higher among NSD2-KD OS cells than among parental cells (Fig. 2d). Taken together, our results indicate that NSD2 promotes tumourigenesis and inhibits apoptosis in OS.

a Real-time quantitative polymerase chain reaction and western blot analysis of the indicated proteins in control and NSD2-KD cells. In vitro growth of control and NSD2-KD 143B and HOS cells was assessed by Cell Counting Kit-8 assay (b) and colony-formation assay (c). d Flow cytometric analysis of apoptosis in control and NSD2-KD OS cells. *P < 0.05; **P < 0.01 compared with the control group
NSD2 knockdown inhibits OS genesis in vivo
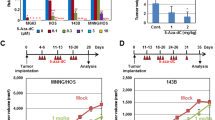
Next, we used a xenograft mouse model to determine the role of NSD2 in OS tumourigenesis. Three groups of 6–8-week-old athymic nude mice were subcutaneously injected with control or NSD2-KD 143B cells. Tumour volume was monitored every 7 days, and tumour weight was measured on day 21 when the mice were sacrificed. The data indicated that there were dramatic reductions in the size and weight of OS tumours in the NSD2-KD groups compared with the control group (Fig. 3a–c). Then we assessed a range of markers related to tumour cell apoptosis and proliferation via IHC, the results of which indicated that the numbers of cleaved caspase 3-positive and transferase-mediated deoxyuridine triphosphate-biotin nick end labelling (TUNEL)-positive tumour cells were significantly higher in the two NSD2-KD groups than in the control group, although there were no significant differences in Ki67 expression among the groups (Fig. 3d). Taken together, these data support and verify the observations made in vitro and demonstrate that NSD2 knockdown suppresses OS tumourigenesis and enhances apoptosis in vivo.

a Macroscopic image of tumour size. Diagrams of tumour volume (b) and tumour weight (c) for mice sacrificed at the indicated time points are shown. d Haematoxylin–eosin staining and transferase-mediated deoxyuridine triphosphate-biotin nick end labelling, cleaved caspase 3 and Ki67 staining of representative tumours in the control group and two NSD2-KD groups. **P < 0.01. Scale bar = 50 μm
NSD2 deficiency enhances cisplatin efficacy
Owing to the significant apoptotic enhancement that occurred after NSD2 knockdown in OS cells, we further estimated the synergistic effect of NSD2 on OS chemosensitivity to cisplatin. The results of CCK-8 and colony-formation assays suggested that NSD2 knockdown significantly sensitized OS cells to cisplatin (Fig. 4a, b), and flow cytometric analysis demonstrated that NSD2 deficiency could magnify the cytotoxic effects of cisplatin (Fig. 4c). The percentage of apoptotic cells was approximately two-fold greater among NSD2-KD cells than among control cells after cisplatin (1 μM) treatment. Thus these results indicate that NSD2 knockdown can promote cisplatin efficacy in vitro.

In vitro growth of control and NSD2-KD 143B and HOS cells with or without cisplatin as assessed by Cell Counting Kit-8 assay (a) and colony-formation assay (b). c Apoptosis in control and NSD2-KD osteosarcoma cells with the indicated treatments was assessed by staining by Annexin V and 7-aminoactinomycin D. *P < 0.05 and **P < 0.01 compared with the control group
NSD2 regulates cancer stem cell (CSC) properties in OS
Accumulating evidence indicates that a specific subset of cells in tumour cell lines or in tumour masses can be classified as CSCs17. CSCs are closely correlated with chemoresistance and tumourigenesis in many solid tumours18,19. Thus we explored whether NSD2 influences OS CSC properties (stemness). The results of sphere-formation experiments revealed that NSD2-KD decreased the number and size of tumourspheres in OS cells (Fig. 5a), indicating that NSD2 knockdown was associated with reduced CSC properties. Likewise, RT-qPCR also indicated that CD117, CD133 and SOX2 were repressed in the absence of NSD2 (Fig. 5b). Collectively, these findings highlight that NSD2 promotes CSC properties in OS.

a Tumoursphere formation in control and NSD2-KD 143B and HOS cells. b CD117, CD133 and SOX2 mRNA expression in parental and NSD2-KD OS cells. *P < 0.05; **P < 0.01 compared with the control group
NSD2 regulates apoptosis- and CSC-related genes
To gain insight into the mechanism by which NSD2 knockdown promotes apoptosis in OS cells and sensitizes the cells to cisplatin, we performed RNA sequencing (RNAseq) using parental and NSD2-KD 143B cells. Gene Ontology (GO) analysis indicated that a number of GO terms associated with cell apoptosis and CSCs, such as “intrinsic apoptotic signalling pathway in response to DNA damage”, “positive regulation of apoptotic signalling pathway”, “response to drug” and “stem cell differentiation”, were enriched for the genes that were differentially expressed in NSD2-KD cells compared with control cells in the presence or absence of cisplatin (Fig. 6a). Based on the results of GO analysis, we then analysed the expression of several genes with RT-qPCR. The results indicated that BCL2 and Sex-determining region Y-box 2 (SOX2) were downregulated but that BAD was upregulated when NSD2 was knocked down in OS cells (Fig. 6b). Western blot analysis further verified that with the knockdown of NSD2, BCL2 and SOX2 were downregulated (Fig. 6c). BCL2 and BAD belong to the BCL2 family, which governs the intrinsic apoptosis pathway20,21,22,23,24. In general, BCL2 plays an antiapoptotic role, while BAD plays a proapoptotic role. SOX2 belongs to the highly conserved HMG box family of SOX transcription factors and plays essential roles during embryogenesis as a master regulator in pluripotent embryonic stem cells25. Several recent studies have shown that exogenous upregulation of SOX2 can promote resistance to chemotherapeutics26,27,28,29,30,31,32,33,34,35. In addition, higher SOX2 levels have been reported to correlate with poorer prognosis in cancers, such as breast, colorectal, oesophageal, ovarian, prostate, and lung cancer as well as in nasopharyngeal and sinonasal carcinoma32,33,35,36,37,38,39,40,41,42. Taken together, these results indicate that NSD2 deficiency downregulates the expression of the antiapoptotic gene BCL2 and the CSC-associated gene SOX2 but upregulates the expression of the proapoptotic gene BAD.

a Gene Ontology analysis of differentially expressed genes in NSD2-KD cells compared with control cells without cisplatin (left panel) or with cisplatin (right panel). b Real-time quantitative polymerase chain reaction confirmation of NSD2, BCL2, BAD and SOX2 expression in control and NSD2-KD 143B cells (upper panel) and in HOS cells (lower panel). c Western blot analysis of SOX2, BCL2 and NSD2 expression in parental and NSD2-KD OS cells. *P < 0.05 and **P < 0.01 compared with the control group; #P < 0.05 and ##P < 0.01 compared with the cisplatin-treated group
NSD2 mediates H3K36me2 modification at BCL2 and SOX2 loci and regulates the ERK and AKT signalling pathways
NSD2 is a histone methyltransferase that catalyses H3K36me2, and H3K36 methylation is a histone marker that generally promotes gene transcription. Thus we further investigated whether NSD2 regulates BCL2 and SOX2 expression directly by modifying H3K36me2 at their gene loci. The results of ChIP-qPCR revealed that NSD2 knockdown reduced H3K36me2 modification and NSD2 enrichment on the BCL2 and SOX2 gene promoters (Fig. 7a and Fig. S3), indicating that knockdown of NSD2 may increase chromatin condensation at BCL2 and SOX2 gene loci and thereby blunt the transcription of these two genes in OS cells. Moreover, pathway analysis demonstrated that 45, 69 and 61 pathways were enriched between the control (Ctrl) and cisplatin-treated (Cis) groups (circle a), the Ctrl and NSD2-KD (KD) groups (circle b) and the control with cisplatin (Ctrl+Cis) and NSD2-KD with cisplatin (KD+Cis) groups (circle c), respectively (Fig. 7b). Notably, 14 pathways were shared among circles a, b and c, including the phosphoinositide-3 kinase–AKT pathway and the mitogen-activated protein kinase pathway (Fig. 7c). Western blot analysis illustrated that phosphorylated ERK and phosphorylated AKT were downregulated upon NSD2 knockdown, while the levels of ERK and AKT were not significantly altered (Fig. 7d). Taken together, these results demonstrate that NSD2 directly regulates BCL2 and SOX2 expression by mediating H3K36me2 modification at their gene loci and also regulates the ERK and AKT signalling pathways (Fig. 7e).

a Chromatin immunoprecipitation–quantitative polymerase chain reaction analysis of H3K36me2 enrichment in the BCL2 and SOX2 gene promoters in control and NSD2-KD osteosarcoma (OS) cells. b Venn diagram of the enriched pathways between the indicated groups. c Fourteen pathways shared by circles a, b and c in b. d Western blot analysis of GAPDH, AKT, pAKT, ERK and pERK expression in control and NSD2-KD OS cells with or without cisplatin. e Schematic diagram of the mechanism by which NSD2 mediates OS genesis and chemosensitivity. NSD2 can directly increase H3K36me2 levels at BCL2 and SOX2 loci and activate ERK and AKT signalling pathways to facilitate OS genesis and chemoresistance. *P < 0.05; **P < 0.01
Discussion
OS is a highly malignant bone tumour with poor prognosis. The conventional therapeutic treatments for OS rely on surgical resection of the tumour bulk combined with chemotherapy and/or radiotherapy and significantly improve the 5-year survival rate of OS patients up to approximately 60–70%. However, the frequency of recurrence and chemotherapy resistance is high, and these issues are the leading contributors to decreased survival time in patients43,44. Thus new strategies need to be introduced into treatment regimens.
Since it is widely acknowledged that epigenetic modulation plays an important role in cancer progression and resistance to chemotherapy, introduction of agents targeting epigenetic changes could be a new anticancer therapeutic strategy to treat the chemoresistance problem in OS45. In our study, we first found that NSD2 is upregulated in OS patients, especially in those who are resistant to cisplatin treatment. Additionally, we found that patients with lower NSD2 expression have better prognoses. Then we conducted several experiments in vitro and in vivo, and the results demonstrated that knockdown of NSD2 enhances OS cell apoptosis and has synergistic effects with cisplatin against OS. Lu et al.46 likewise reported that NSD2 knockdown could inhibit OS progression, but these researchers did not further investigate the effects of NSD2 on chemosensitivity in OS. In our study, NSD2 knockdown also repressed the expression of genes associated with CSC properties, such as CD133, CD117 and SOX2. CD133 (also known as prominin-1) is a cell surface transmembrane glycoprotein that has been used to identify and isolate putative CSCs in several types of solid tumours40, and CD117 (KIT) is also a well-known cell surface marker for stem cells47. SOX2 has been shown to maintain OS CSCs48. Besides, Tang et al.49 reported that knockdown of SOX2 inhibited OS cells invasion and migration and Maurizi et al.50 also reported that SOX2 was required for OS development and proliferation. Mechanistically, GO analysis revealed several apoptotic and stem cell-related terms that were enriched for differentially expressed genes between the NSD2-KD group and the control group with or without cisplatin treatment. Upon NSD2 knockdown in OS, BCL2 and SOX2 were downregulated, but BAD was upregulated. Pathway analysis further clarified that the ERK and AKT signalling pathways might mediate the process by which NSD2 knockdown enhances OS apoptosis and strengthens cisplatin efficacy.
However, although we revealed direct regulation of BCL2 and SOX2 expression by NSD2, the core subunits or relevant co-factors were not elucidated. Therefore, the underlying mechanisms by which NSD2 recognizes specific genes and modulates their epigenetic modifications remain undefined. In addition, histone methyltransferases can also be involved in methylation of non-histone proteins. For example, the H3K36me3 methyltransferase SETD2 can directly bind to signal transducer and activator of transcription factor 1 (STAT1) and promote its methylation on lysine 525 (K525), leading to STAT1 phosphorylation and activation51; however, whether NSD2 mediates modification of certain non-histone proteins to regulate tumourigenesis or chemosensitivity warrants further investigation.
In summary, our evidence of the involvement of NSD2 in OS provides a new perspective regarding possible epigenetic regulation in tumourigenesis and apoptosis as well as in chemosensitivity. The results of this study suggest that NSD2 could be a new therapeutic target in combined chemotherapy to enhance OS apoptosis and the response of OS to chemotherapy. In addition, NSD2 could be a prognostic factor for OS. Thus this study has elucidated new strategies for clinical OS treatment regimens.
Materials and methods
Patients and tissue sample collection
The patients with histologically confirmed OS involved in this study were hospitalized at Shanghai Ninth People’s Hospital. All participants signed informed consent documents, and all experiments were conducted according to the guidelines of the ethics committee of Shanghai Ninth People’s Hospital. The clinical information of the patients is summarized in Tables S1 and S2. Based on analysis of cisplatin efficacy, patient biopsies were divided into a cisplatin-sensitive group (inhibition rate 50–100%) and a cisplatin-resistant group (inhibition rate 0–50%). There were no significant differences regarding age or sex between the two groups (P > 0.05).
Immunohistochemistry
IHC was performed on 5-μm-thick formalin-fixed paraffin-embedded slices. After deparaffinization, rehydration, antigen retrieval and endogenous peroxidase inhibition, the samples were incubated with anti-NSD2 (Abcam), anti-TUNEL (Roche) and anti-cleaved caspase 3 (Servicebio) antibodies at 4 °C for 8–10 h. After the secondary antibody was applied, diaminobenzidine (DAKO) solution was used as a chromogen. Furthermore, haematoxylin (Sigma) staining was performed to identify nuclei. Images were acquired using a microscope (Leica DM 4000B).
Cell lines and cell culture
The human OS cell lines 143B and HOS were purchased from the American Type Culture Collection. The cells were grown in Dulbecco’s modified Eagle’s medium (HyClone) with 10% foetal bovine serum (Gibco, Australia) and antibiotics (100 U/ml penicillin, 100 µg/ml Streptomycin) in a 37 °C humidified atmosphere with 5% CO2.
Cell viability assay
A cell viability assay was performed as previously described52. In brief, cells were added into 96-well plates at a density of 5 × 103 cells/well and incubated with or without reagents for 72 h. Then a 1:10 dilution of CCK-8 reagent in culture medium was added into each well, and the cells were cultured for 2 h while protected from light. Then the optical density was read at 450 nm, and data were collected.
Real-time quantitative polymerase chain reaction
RNA was isolated using TRIzol Reagent (Invitrogen), and 1 μg of each sample was subjected to RT-qPCR using a TaKaRa RT-PCR kit (Takara, Shiga, Japan) following the manufacturer’s protocols. Relative quantification (RQ) was derived from the cycle threshold (Ct) using the equation RQ = 2−ΔΔCt. The sequences of the primers are listed in Table S3.
Western blot analysis
Protein was extracted under the indicated conditions, and the protein lysate was separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis. The proteins were then transferred to polyvinylidene fluoride membranes. After being blocked in 10% non-fat milk, the membranes were incubated with specific primary and secondary antibodies. The antibodies used were anti-SOX2 (Active Motif), anti-NSD2 (Cell Signaling Technology, CST), anti-H3K36me2 (Active Motif), anti-H3K27me3 (CST), anti-BCL2 (CST), anti-GAPDH (CST), anti-H3 (Abways), anti-ERK (CST), anti-pERK (CST), anti-AKT (CST) and anti-pAKT (CST).
Colony-formation assay
A colony-formation assay was conducted as previously described52. Briefly, 1000 cells were added into 6-well plates, and after cell attachment, different treatments were added. On day 7, the colonies were fixed, stained with 0.1% crystal violet and counted.
Flow cytometric analysis
The percentage of cells undergoing apoptosis was evaluated by flow cytometry (Beckman Gallios). After incubation with cisplatin for 48 h, the cells were harvested, washed and resuspended in 100 μl of binding buffer with 1 µl of 7-aminoactinomycin D (7-AAD; BD Biosciences) and 5 µl of annexin V (BD Biosciences). To assess cell proliferation, the cells with the indicated treatments for 24 h and subsequent experiments were performed using an EdU Cell Proliferation Kit (Beyotime) as instructed. Briefly, the cells were incubated with 10 mM EdU for 2 h, washed, immobilized and permeabilized. Then the cells were treated with Click Additive Solution for 30 min and stained with 7-AAD.
Tumoursphere formation
Single-cell suspensions were added to ultralow-attachment plates in defined medium53 at a density of 1000 cells/well. Half of the volume of the medium was changed every second day. Images were obtained on day 7 under an inverted microscope. Only spheres >100 μm were quantitated.
Short hairpin RNAs
Two NSD2-specific shRNAs (sequences listed in Table S4) were designed, and a scramble nonsense sequence was used as a negative control. Lentivirus was packaged using a three-plasmid system including pLVX-shRNA1 with targeted or nonsense sequences, psPAX2 and pMD2G. Stable cell lines were screened with 2 μg/ml puromycin for 2 weeks.
Mice
Mice were bred and maintained in specific pathogen-free conditions at the animal experimental centre of Shanghai Ninth People’s Hospital. All animal experiments were approved by the ethics committee of Shanghai Ninth People’s Hospital. Twenty-four BALB/c athymic nude mice (6–8-week old) were subcutaneously injected with 1 × 106 of the indicated OS cells. Tumour volume was evaluated every 7 days as 1/2 (a ×b × b), where a is the major tumour axis length and b is the minor tumour axis length. Tumour weight was measured after the mice were sacrificed on day 21.
RNAseq and GO analysis
RNA samples were obtained from parental and NSD2-KD1 143B cells that were untreated or treated with cisplatin (1 μM) for 24 h. After the proper pretreatment, the samples were sent to Oebiotech Co. Ltd. for sequencing. Gene expression levels were analysed with the htseq-count54 and Cufflinks55 programs. All differentially expressed gene lists (q value < 0.05) generated by DESeq56 were subsequently analysed for enrichment of biological terms with the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics platform.
ChIP-qPCR analysis
Control and NSD2-KD1 143B cells (2 × 107 cells) were crosslinked, lysed and sheared using a UCD-300 (Bioruptor) to ~200–700 base pairs in length. ChIP was then performed using an EZ ChIP Kit (Millipore; 17-371) according to the manufacturer’s instructions. ChIP-enriched DNA was then quantified by RT-qPCR. The ChIP antibodies used were anti-H3K36me2 (Active Motif) and anti-NSD2 (Abcam). The primer sequences are listed in Table S3.
Statistical analysis
All experiments were repeated at least three times as indicated. The mean, standard error of the mean (SEM) and p values based on two-tailed t tests were calculated with Excel (Microsoft). Differences were considered significant at P < 0.05.
References
Kansara, M., Teng, M. W., Smyth, M. J. & Thomas, D. M. Translational biology of osteosarcoma. Nat. Rev. Cancer 14, 722–735 (2014).
Jaffe, N., Bruland, O. S. & Bielack, S. (eds) Pediatric and Adolescent Osteosarcoma (Springer, Boston, MA, 2010). https://doi.org/10.1007/978-1-4419-0284-9.
Collins, M. et al. Benefits and adverse events in younger versus older patients receiving neoadjuvant chemotherapy for osteosarcoma: findings from a meta-analysis. J. Clin. Oncol. 31, 2303–2312 (2013).
Meyers, P. A. et al. Addition of pamidronate to chemotherapy for the treatment of osteosarcoma. Cancer 117, 1736–1744 (2011).
Link, M. P. et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N. Engl. J. Med. 314, 1600–1606 (1986).
Li, N. et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Invest. 127, 1284–1302 (2017).
Janczar, S. et al. The role of histone protein modifications and mutations in histone modifiers in pediatric B-cell progenitor acute lymphoblastic leukemia. Cancers (Basel) 9, 1–14 (2017).
Bennett, R. L., Swaroop, A., Troche, C. & Licht, J. D. The role of NSD family histone lysine methyltransferases in cancer. Cold Spring Harb. Perspect. Med. 7, a026708 (2018).
Kuo, A. J. et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 (2011).
Li, Y. et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295 (2009).
Asangani, I. A. et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol. Cell 49, 80–93 (2013).
Chesi, M. et al. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 92, 3025–3034 (1998).
Santra, M., Zhan, F., Tian, E., Barlogie, B. & Shaughnessy, J. A subset of multiple myeloma harboring the t(4;14)(p16; q32) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. Blood 101, 2374–2376 (2003).
Keats, J. J. et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16; q32)-positive multiple myeloma patients. Blood 105, 4060–4069 (2005).
Zheng, Y. et al. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. 109, 13549–13554 (2012).
Popovic, R. et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. https://doi.org/10.1371/journal.pgen.1004566 (2014).
Patrawala, L. et al. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 65, 6207–6219 (2005).
Britton, K. M. et al. Breast cancer, side population cells and ABCG2 expression. Cancer Lett. 323, 97–105 (2012).
Brown, H. K., Tellez-Gabriel, M. & Heymann, D. Cancer stem cells in osteosarcoma. Cancer Lett. 386, 189–195 (2017).
Hata, A. N., Engelman, J. A. & Faber, A. C. The BCL2 family: Key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 5, 475–487 (2015).
Safe, S. Targeting apoptosis pathways in cancer - Letter. Cancer Prev. Res. 8, 338 (2015).
Brown, J. M. & Attardi, L. D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 5, 231–237 (2005).
Reed, J. C. Apoptosis-based therapies. Nat. Rev. Drug Discov. 1, 111–121 (2002).
Gougeon, M. L. Apoptosis as an HIV strategy to escape immune attack. Nat. Rev. Immunol. 3, 392–404 (2003).
Rizzino, A. Concise review: the Sox2-Oct4 connection: critical players in a much larger interdependent network integrated at multiple levels. Stem Cells 31, 1033–1039 (2013).
Wuebben, E. L. et al. SOX2 functions as a molecular rheostat to control the growth, tumorigenicity and drug responses of pancreatic ductal adenocarcinoma cells. Oncotarget 7, 34890–34906 (2016).
Li, D. et al. Sox2 is involved in paclitaxel resistance of the prostate cancer cell line PC-3 via the PI3K/Akt pathway. Mol. Med. Rep. 10, 3169–3176 (2014).
Ma, L., Lai, D., Liu, T., Cheng, W. & Guo, L. Cancer stem-like cells can be isolated with drug selection in human ovarian cancer cell line SKOV3. Acta Biochim. Biophys. Sin. (Shanghai) 42, 593–602 (2010).
Hägerstrand, D. et al. Identification of a SOX2-dependent subset of tumor- and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro Oncol. 13, 1178–1191 (2011).
Bareiss, P. M. et al. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer Res. 73, 5544–5555 (2013).
Singh, S. K. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828 (2003).
Piva, M. et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 6, 66–79 (2014).
Chou, Y.-T. et al. The emerging role of SOX2 in cell proliferation and survival and its crosstalk with oncogenic signaling in lung cancer. Stem Cells 31, 2607–2619 (2013).
Tian, Y. et al. SOX2 oncogenes amplified and operate to activate AKT signaling in gastric cancer and predict immunotherapy responsiveness. J. Cancer Res. Clin. Oncol. 140, 1117–1124 (2014).
Jia, X. et al. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. J. Mol. Cell Biol. 3, 230–238 (2011).
Wuebben, E. L. & Rizzino, A. The dark side of SOX2: cancer - a comprehensive overview. Oncotarget 8, 44917–44943 (2017).
Sholl, L. M., Barletta, J. A., Yeap, B. Y., Chirieac, L. R. & Hornick, J. L. Sox2 protein expression is an independent poor prognostic indicator in stage I lung adenocarcinoma. Am. J. Surg. Pathol. 34, 1193–1198 (2010).
Wang, X. et al. Prognostic significance of SOX2 expression in nasopharyngeal carcinoma. Cancer Invest. 30, 79–85 (2012).
Li, W. et al. SOX2 as prognostic factor in head and neck cancer: a systematic review and meta-analysis. Acta Otolaryngol. 134, 1101–1108 (2014).
Saigusa, S. et al. Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann. Surg. Oncol. 16, 3488–3498 (2009).
Wang, Q. et al. Oct3/4 and Sox2 are significantly associated with an unfavorable clinical outcome in human esophageal squamous cell carcinoma. Anticancer Res. 29, 1233–1241 (2009).
Zhang, J., Chang, D. Y., Mercado-Uribe, I. & Liu, J. Sex-determining region Y-box 2 expression predicts poor prognosis in human ovarian carcinoma. Hum. Pathol. 43, 1405–1412 (2012).
Kansara, M. & Thomas, D. M. Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 26, 1–18 (2007).
Bacci, G. et al. High dose ifosfamide in combination with high dose methotrexate, adriamycin and cisplatin in the neoadjuvant treatment of extremity osteosarcoma: preliminary results of an Italian Sarcoma Group/Scandinavian Sarcoma Group Pilot Study. J. Chemother. 14, 198–206 (2002).
Morishita, M. & Di Luccio, E. Cancers and the NSD family of histone lysine methyltransferases. Biochim. Biophys. Acta 1816, 158–163 (2011).
Lu, M., Fan, M. & Yu, X. NSD2 promotes osteosarcoma cell proliferation and metastasis by inhibiting E-cadherin expression. Eur. Rev. Med. Pharmacol. Sci. 21, 928–936 (2017).
Foster, B., Zaidi, D., Young, T., Mobley, M. & Kerr, B. CD117/c-kit in cancer stem cell-mediated progression and therapeutic resistance. Biomedicines 6, 31 (2018).
Basu-Roy, U. et al. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene 31, 2270–2282 (2012).
Tang, L., Wang, D. & Gu, D. Knockdown of Sox2 inhibits OS cells invasion and migration via modulating Wnt/β-Catenin signaling pathway. Pathol. Oncol. Res. 24, 907–913 (2018).
Maurizi, G., Verma, N., Gadi, A., Mansukhani, A. & Basilico, C. Sox2 is required for tumor development and cancer cell proliferation in osteosarcoma. Oncogene 37, 4626–4632 (2018).
Chen, K. et al. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 170, 492–506.e14 (2017).
He, C., Wu, T. & Hao, Y. Anlotinib induces hepatocellular carcinoma apoptosis and inhibits proliferation via Erk and Akt pathway. Biochem. Biophys. Res. Commun. 503, 3093–3099 (2018).
Gibbs, C. P. et al. Stem-like cells in bone sarcomas: implications for tumorigenesis. Neoplasia 7, 967–976 (2005).
Anders, S., Pyl, P. T. & Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L. & Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22 (2011).
Anders, S. & Huber, W. Differential expression of RNA-Seq data at the gene level – the DESeq package. http://bioconductor.org/packages/release/bioc/vignettes/DESeq/inst/doc/DESeq.pdf (accessed 25 May 2018).
Acknowledgements
This study was supported by grants from the National Key R&D Program of China (2016YFC1100600), the Shanghai Ninth People’s Hospital Lin+ Program (JYLJ012), and the National High Technology Research and Development Program (863 Program) (2015AA033603).
Author information
Authors and Affiliations
Contributions
Y.H. and Y.J. conceived the project and designed the experiments; C.H., Y.J. and L.W. performed the experiments; and C.H. and C.L. acquired and analysed the data. Y.H. and Y.S. revised the manuscript and submitted the final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Peschiaroli
Supplementary information
41419_2019_1347_MOESM3_ESM.tif
Fig. S3. NSD2 enrichment at BCL2 and SOX2 gene loci in control and NSD2-KD 143B cells as assessed by ChIP-qPCR. *P<0.05; **P<0.01
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, C., Liu, C., Wang, L. et al. Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death Dis 10, 65 (2019). https://doi.org/10.1038/s41419-019-1347-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1347-1
This article is cited by
-
Structural and functional specificity of H3K36 methylation
Epigenetics & Chromatin (2022)
-
Identification of Novel Serum Proteins Associated with Myelination and Cholesterol Transport in Neuromyelitis Optica Spectrum Disorders by Mass Spectrometry
Indian Journal of Clinical Biochemistry (2022)
-
The role of NSD1, NSD2, and NSD3 histone methyltransferases in solid tumors
Cellular and Molecular Life Sciences (2022)
-
Circular RNA circ_001422 promotes the progression and metastasis of osteosarcoma via the miR-195-5p/FGF2/PI3K/Akt axis
Journal of Experimental & Clinical Cancer Research (2021)
-
NSD2 promotes tumor angiogenesis through methylating and activating STAT3 protein
Oncogene (2021)
