
Abstract
Ceramide synthases (CERS) produce ceramides which are key intermediators in the biosynthesis of complex sphingolipids and play an important role in cell proliferation, differentiation, apoptosis and senescence. CERS6 is an isoform of ceramide synthases known to generate ceramides with C16 acyl chain (C16-Cer). CERS6 and C16-Cer levels were significantly higher in acute lymphoblastic leukemia (ALL) cells in comparison to peripheral blood mononuclear cells and T lymphocytes derived from healthy human volunteers. We investigated the role of CERS6 in chemo-resistance in T-ALL cell lines. Stable knockdown of CERS6 in CCRF-CEM and MOLT-4 cells resulted in increased sensitivity to ABT-737, a pan-BCL-2 inhibitor, while CCRF-CEM cells with exogenous CERS6 expression showed resistance to ABT-737 relative to the vector control. The cytotoxic activity of ABT-737 in CERS6 knockdown cells was significantly reduced by the addition of a caspase-8 inhibitor Z-IETD, suggesting that CERS6 alters the cytotoxicity via extrinsic pathway of apoptosis. By co-immunoprecipitation of CERS6 in CCRF-CEM cells, we identified CD95/Fas, a mediator of extrinsic apoptotic pathway, as a novel CERS6 binding partner. In Fas pull-down samples, FADD (Fas-associated protein with death domain) was detected at higher levels in cells with CERS6 knockdown compared with control cells when treated with ABT-737, and this was reversed by the overexpression of CERS6, demonstrating that CERS6 interferes with Fas–FADD DISC assembly. CERS6 may serve as a biomarker in determining the effectiveness of anticancer agents acting via the extrinsic pathway in T-ALL.
Similar content being viewed by others


Introduction
Acute lymphoblastic leukemia (ALL) is the most common childhood and adolescent cancer, and approximately 2900 new cases of pediatric ALL are diagnosed annually in the United States1. Sixty percent of the cases occur at less than 20 years2 and the survival rate of childhood ALL is close to 90%3,4. Although much progress has been made in understanding cellular responses to standard chemotherapeutic agents, approximately 10% of pediatric patients with ALL do not respond to treatment, and ultimately die of the disease3,4. As cancer cells continue to evolve mechanisms to circumvent stressors leading to development of intrinsic or acquired drug resistance, a significant percentage of patients with standard-risk and high-risk ALL relapse, and post-relapse treatment rarely results in long-term survival5. Mortality in high-risk disease is about 35%6 and the treatment of infants and adults still needs improvement4.
Sphingolipids are bioactive lipids and primarily include sphingosines, ceramides and sphingomyelins that control a variety of cellular functions7. Many chemotherapeutic drugs are reported to modulate sphingolipid pathways, though their contribution to cytotoxicity is controversial. Ceramides, key intermediates in the biosynthesis of all the complex sphingolipids, have a significant role in the regulation of cell growth, differentiation, apoptosis and senescence8. Six mammalian ceramide synthase (CERS1–6) homologs use a relatively restricted subset of acyl-Coenzyme A for ceramide synthesis by N-acylation of the sphingoid base9. CERSs produce ceramides using either the de novo synthesized sphinganine10 or by acylation of sphingosines from the salvage pathway (Fig. 1a). Ceramides are also generated by breakdown of sphingomyelins using sphingomyelinases.

a Biosynthesis and metabolism of ceramides. Ceramides are generated either de novo or via the salvage pathway. They are metabolized to sphingosine or may serve as substrates for the synthesis of glucosyl ceramides, lactosyl ceramides or sphingomyelins. Sphingomyelins degrade to ceramides via the sphingomyelinase pathway and are synthesized from ceramides via sphingomyelin synthase. b mRNA expression of the six ceramide synthase isoforms in the NCI PPTP panel of 23 cell lines comprising six different pediatric cancers. RBD rhabdomyosarcoma, BT brain tumor, EFT Ewing’s family of tumors, NB neuroblastoma, ALL acute lymphoblastic leukemia, LYM lymphoma. The mRNA expression was determined within the NCI Pediatric Preclinical Testing Program. c CERS6 protein expression in acute lymphoblastic leukemia (ALL) cell lines in comparison to peripheral blood mononuclear cells (PBMCs) and T lymphocytes obtained from blood of healthy human volunteers. GAPDH was used as a loading control. d C16-Ceramides in T-cell ALL cells in comparison to PBMCs and T lymphocytes. Ceramide levels were quantitated by HPLC/MS/MS (PBMC: 0.28 ± 0.04, n = 18; T lymphocytes: 0.36 ± 0.01, n = 3; T-ALL: 0.54 ± 0.09, n = 9). e CERS6 protein expression in T lymphocytes isolated from primary lymphoid malignancy samples in comparison to normal T lymphocytes *** p < 0.001
The expression levels of CERSs vary between different tissues. CERS1 is predominantly expressed in the brain and skeletal muscles in mouse11. While CERS2 is ubiquitously expressed, kidney, liver and intestine are the major tissues with high CERS2 expression11. CERS3 shows highest expression in testis and CERS4 is not specific to any particular tissue. CERS5 has been extensively studied and is the main CERS found in the lung epithelia7. CERS6 is expressed in almost all tissues, but shows a very low expression profile in each of them, except for small intestine11. CERS6 predominantly synthesizes C16-ceramide (C16-Cer) and is subcellularly localized mainly in the endoplasmic reticulum12, and also in the plasma membrane13. CERS6 displays homology with CERS514 and CERS6 knockout mouse shows a high reduction in C16-Cer levels in most tissues15. Knockdown of CERS6 in colon adenocarcinoma cells caused a decrease in C16-Cer and protected the cells against tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)16, whereas C16-Cer generated by CERS6 in human head and neck squamous cell carcinoma (HNSCC) cells increased tumor development and growth17, suggesting an anti-apoptotic role of CERS6 in tumor cells.
Treatment of pediatric ALL includes remission induction therapy for 4–5 weeks, consolidation therapy for 4–8 weeks, delayed intensification therapy for 8–9 weeks sandwiched between two interim maintenance therapies for 8 weeks each, followed by maintenance therapy for 2–3 years, and is the longest phase of treatment18,19. Glucocorticoid is one of the backbone regimen for the treatment of pediatric ALL. ABT-737 is a small molecule inhibitor of B-cell lymphoma 2 (BCL-2) family of proteins20, which binds directly to the hydrophobic groove of anti-apoptotic BCL-2, B-cell lymphoma-extra large (BCL-XL) or BCL-w promoting the oligomerization of BAX and BAK to induce apoptosis21. Although ABT-737 as a single agent has shown substantial activity in multiple myeloma22, it can significantly enhance the activity of ALL standard-of-care treatment drugs like vincristine, L-asparaginase and dexamethasone in vitro and in vivo23. A recently developed modified BCl-2 inhibitor, ABT-199, shows high selectivity towards BCL-2 without inhibiting BCL-XL or BCL-w and is currently approved for chronic lymphocytic leukemia.
The purpose of this study is to understand the roles of ceramides in cancer and to define sphingolipids as potential targets in cancer chemotherapy. We hypothesized that high levels of CERS6 interfere with apoptosis and render ALL cells resistant to drug treatment. CERS6 knockdown increased T-ALL cell sensitivity to ABT-737, while CERS6 overexpression rendered the cells resistant to ABT-737. Finally, we studied whether CERS6 binds to CD95/Fas and interferes with association of FADD to Fas, and thus inhibiting the extrinsic apoptotic pathway upon drug treatment.
Materials and methods
Chemicals and reagents
Sphingolipid standards includingC14:0-, C16:0-, C17:0-, C18:0-, C18:1-, C20:0-, C24:0-, C24:1-ceramide and C18:0-, C18:1-, C24:0-, C24:1-dihydroceramide were purchased from Avanti Polar Lipids (Alabaster, AL); ammonium formate and formic acid were obtained from Fisher Scientific (Pittsburg, PA); chloroform, ethyl acetate, methanol, 2-propanol, NaF, NaHCO3, Na3VO4, Tris-HCl, Triton X-100, pepstatin A, aprotinin, leupeptin, 200 proof ethanol, isopropanol, puromycin, dexamethasone, and anti-FLAG-M2 (1 μg/ml) antibody from Sigma-Aldrich (St. Louis, MO); ABT-737 from Cayman Chemical (Ann Arbor, MI); DTT, EDTA, NaCl, PMSF, SDS, TBE, trypsin/EDTA, Lipofectamine®, PLUSTM reagent, Superscript® III first-strand synthesis system for RT-PCR from Thermo Fisher Scientific (Waltham, MA); Triton X-114 from Acros Organics (Morris, NJ); Z-IETD from R&D Systems (Minneapolis, MN); Fas ligand from GeneTex (Irvine, CA); anti-CERS6, anti-FLIP and anti-GAPDH antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); anti-Fas and anti-FADD from BD Transduction Laboratories (San Jose, CA); anti-HA antibody from Roche (Indianapolis, IN); anti-caspase-3, anti-cleaved-caspase-3, anti-caspase-8 and anti-PARP from Cell Signaling Technology (Danvers, MA); Age1-HF, BamH1-HF, EcoR1-HF, Mlu1-HF, Pme1 and Sgf1 restriction enzymes from New England Biolabs (Ipswich, MA); bovine serum albumin from Jackson ImmunoResearch Laboratories (West Grove, PA); all oligos were synthesized from Integrated DNA Technologies (Coralville, IA).
mRNA expression of CERS isoforms in various cancers
The expression of messenger RNA (mRNA) in the NCI PPTP cell lines were determined in the previous study using Affymetrix U133 Microarray system24. The data were used to determine mRNA expression of CERS1-CERS6. CERS6 expression levels in various cancers and normal tissue samples were obtained from UCSC Xena (https://xenabrowser.net)
Extraction of PBMCs and T lymphocytes
Human blood from normal healthy volunteers was obtained from United Blood Services (Lubbock TX). Peripheral blood mononuclear cells (PBMCs) were extracted using LSM-Lymphocyte Separation Medium (MP Biomedicals, Santa Ana, CA) as per the manufacturer’s protocol. T lymphocytes were separated from PBMCs using Dynabeads Untouched Human T Cells (Thermo Fisher Scientific, Baltics UAB) extraction kit following the manufacturer’s protocol.
Cell culture
ALL cell lines (Supplemental Table S1) used in the study were cultured in RPMI (GE Lifesciences) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Life Technologies) or Iscove’s modified Dulbecco’s medium (Life Technologies) with insulin–transferrin–selenium (Sigma-Aldrich) (10 µg/ml insulin, 5.5 µg/ml transferrin (substantially iron-free), 5 ng/ml sodium selenite), and 20% heat-inactivated FBS. All cell lines were cultured in physical hypoxia (5% CO2, and 5% O2). Cell lines were tested for and free of mycoplasma, and cell line identities were verified using short tandem repeat genotyping as compared with the original primary sample material within COGcell database: www.COGcell.org.
T lymphocytes from primary clinical samples
Ficoll-separated mononuclear cells from peripheral blood of patients diagnosed with lymphoid malignancies were obtained from the Texas Cancer Cell Repository (TXCCR) with informed consent from all patients. T lymphocytes were separated using Dynabeads Untouched Human T Cells (Thermo fisher Scientific, Baltics UAB) extraction kit following the manufacturer’s protocol.
Immunoblotting
Cells grown in T75 or T150 flask were washed once with ice-cold 1× phosphate-buffered saline (PBS). Cells lysis was performed on ice with modified RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 (Triton X-114 for co-immunoprecipitation), 1 µg/ml Leupeptin, 1 µg/ml aprotinin, 1 µg/ml pepstatin A, 1 mM PMSF, 1 mM Na3VO4 and 1 mM NaF), followed by centrifugation at 14,000 × g for 15 min at 4 °C. Protein concentration was determined by BCA assay (Pierce, Rockford, IL). Equal amount of protein samples were loaded and electrophoretically separated on a 4–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to Hybond membrane (GE Healthcare, Piscataway, NJ), blocked with 1% bovine serum albumin or 5% skim milk, immunoblotted with the indicated primary antibodies and incubated with 1:3000 horseradish peroxidase-conjugated mouse or rabbit immunoglobulin G (IgG) secondary antibodies followed by detection with enhanced chemiluminescence (GE Healthcare). The membrane was stripped and re-probed with anti-GAPDH antibody to confirm equal loading.
Determination of ceramides by LC-MS/MS
The sphingolipids were analyzed using previously reported method25 with modification. Briefly, cell pellet (7.5 × 106 cells) added with 50 µl of internal standard (C17:0-ceramide, 1 pM) was extracted twice with ethyl acetate/2-propanol/water (60/28/12; v/v/v). Sphingolipids were separated using gradient elution with mobile phase A (2 mM ammonium formate and 0.2% formic acid (v/v) in water) and B (1 mM ammonium formate and 0.2% formic acid (v/v) in methanol) on an Agilent 1200 series high-performance liquid chromatography (HPLC) system with Spectra C8SR column (3 µm, 150 × 3.0 mm, Peeke Scientific, Redwood, CA). Mass spectrometric detection was performed by multiple reaction monitoring (MRM) mode on a Sciex 4000 QTRAP mass spectrometer (AB Sciex, Framingham, MA) operating in positive ion mode. Analyst software 1.6.3 (AB Sciex) was used for the data acquisition as well as processing. Sphingolipid data generated from liquid chromatography-tandem mass spectrometry (LC-MS/MS) were normalized to lipid phosphate as previously described26.
CERS6 knockdown by lentiviral transduction
CERS6 short hairpin RNA (shRNA; TRCN0000128836) in pLKO.1-puro lentiviral vector was from Dharmacon (GE Healthcare Bio-Sciences, Pittsburgh, PA). pLKO.1-puro eGFP shRNA sequence (Sigma-Aldrich), which targets enhanced green fluorescent protein (eGFP), was used as a non-targeting control (NT-shRNA). Knockdown of CERS6 in CCRF-CEM or MOLT-4 cells was carried out as described under “Cell transduction” section below.
Cloning and mutagenesis
CERS6 complementary DNA (cDNA) was amplified by the Expand High Fidelity PCR System (Roche) using pLX304-CERS6 (DNASU Plasmid Repository, Arizona State University) as a template and primers as specified (Supplemental Table S3). The PCR-amplified CERS6 gene was subcloned into Sgf1 and Mlu1 sites of pCMV6-Entry-mycDDK (OriGene, Rockville, MD) to create pCMV6-CERS6-mycDDK using LigaFastTM Rapid DNA Ligation System (Promega, Madison WI). CERS6 was ligated into EcoR1 and Mlu1 sites of pLenti-HTBH-DDK27,28 (a kind gift from Dr. Lan Huang, UC Davis, and modified by S-JW) to be used for CERS6 overexpression by lentiviral transduction. FAS cDNA was synthesized using RNA extracted from CCRF-CEM cells and amplified as mentioned above. FAS gene was subcloned into Sgf1 and Mlu1 sites of pCMV6-AC-HA (OriGene, Rockville, MD) to create pCMV6-AC-FAS-HA. The FAS mutant clones of deletion and point substitutions were generated with specific primers (Supplemental Table S3) using QuikChange Site-Directed Mutagenesis Kit (Agilent, Clara, CA). The DNA sequences of all constructs were verified by MacroGen USA (Rockville, MD) using an automated ABI-3730xl DNA Analyzer and ABI PRISM® BigDye™ Terminator v3.0 Ready Reaction Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). All plasmid DNAs were prepared using purification kits from Qiagen (Valencia, CA) and were endotoxin-free when used for transfection into mammalian cells.
Cell transduction
HEK293FT (Life Technologies) cells were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific) supplemented with 10% FBS, 2 mM glutamine, 100 units/ml penicillin, 100 µg/ml streptomycin sulfate and 1 mM sodium pyruvate (Life Technologies). Ten million cells were plated at cell on a 10 cm tissue culture dish and incubated at 37 °C 5% CO2 incubator until the cells reach 80% confluence. The HEK293FT cells were co-transfected with either lentiviral open reading frames or shRNAs along with Lenti-vpak Packaging Kit (OriGene) using the transfection reagent MegaTran 1.0 (OriGene). After 48–72 h of transfection, the virus-containing medium was collected, spun down, filtered (0.45 µm) and used for targeting into CCRF-CEM or MOLT-4 cells by infection. The virus-infected stable clones were obtained after at least 2–3 weeks of selection in 10% FBS/RPMI-1640 with 0.5 µg/ml of puromycin (Sigma-Aldrich).
Cell transfection
HEK293FT cells at 80% confluency were transfected with recombinant DNA constructs (Fig. 6a) along with Plus reagent and Lipofectamine (both from Invitrogen, Carlsbad, CA). Cells were harvested after 48 h of transfection and samples prepared as described under “Immunoblotting” section above.
DIMSCAN cytotoxicity assay
Cells were plated (3000 cells/well for CCRF-CEM and 4500 cells/well for MOLT-4) in a 96-well plate using 150 µl of culture medium and incubated for at least 12 h followed by 50 µl of drug treatment at the following concentrations: 0.001 to 1 µM for ABT-737 and 0.0001 to 1 µM for dexamethasone. Stock solutions for ABT-737 was prepared using dimethyl sulfoxide and dexamethasone was prepared using sterile water. DIMSCAN assay was performed for 72 h post treatment as previously described29.
Apoptosis assay by flow cytometry
Apoptosis was determined using Annexin-V surface positivity by flow cytometry. CCRF-CEM or MOLT-4 cells were incubated with ABT-737, Z-IETD or combination, washed twice with PBS and subjected to Annexin-V assay using Apo AlertTM Annexin-V-FITC Apoptosis kit (Takara Clontech, Mountain View, CA) as previously described23.
Immunoprecipitation (IP)
The cell lines samples were prepared for IP as indicated in the “Immunoblotting” section. For purification of CERS6 (Fig. 5a), 2000 µg of protein lysates as prepared above were pulled down at 4 °C overnight with Ni-NTA His-Bind Resin (EMD-Millipore, Billerica, MA), washed 4 times with modified RIPA and then eluted with 350 mM imidazole. Further, the eluate was incubated with EZview Red Streptavidin Affinity Gel (Sigma) for 2 h, washed 4 times with modified RIPA, and digested with AcTEV Protease (Invitrogen, Carlsbad, CA) for 3 h. Immunoprecipitation of Fas was performed using 1000 µg of protein lysates incubated with 2 µg of anti-Fas antibody Rb (Proteintech, Rosemont, IL) overnight at 4 °C. Fas and associated proteins were pulled down using 50 µl of Protein G Plus Agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) and sample prepared using 4× NuPAGE lithium dodecyl sulfate loading buffer and 100 mM DTT, followed by heating at 70 °C for 10 min. For immunoprecipitation of CERS6 (Fig. 6b), 1000 µg of protein lysates were pulled down at 4 °C overnight with 50 µl EZview Red anti-FLAG-M2 affinity gels (Sigma-Aldrich), washed 4 times with modified RIPA and then eluted with an excess of 3× FLAG peptide (100 μg/ml). Immuno-complexes were resolved by 4–12% SDS-PAGE and immunoblotted with the indicated antibodies.
Cellular fractionation
Cytoplasmic and plasma membrane fractions were isolated from 293FT whole cells using Minute™ Plasma Membrane Protein Isolation and Cell Fractionation Kit (Invent Biotechologies, Plymouth, MN) as specified by the manufacturer's protocol.
FAS surface expression
FAS expression levels were determined in CERS6 knockdown and overexpressing CCRF-CEM cells by flow cytometry using CD95-FITC and FITC Mouse Anti-human IgG (both from BD Biosciences, San Jose, CA) as described previously30. Briefly, cells at logarithmic growth phase were washed twice with PBS, and resuspended in 100 μl of binding buffer (BD Biosciences) at a final concentration of 0.5 × 106 cells per 50 μl. Either anti-human IgG-FITC or CD95-FITC (20 μl per sample) was added to the cell suspension, and the mixure was indubated for 10 min at room temperature. The cells were washed and resuspended in 390 μl of binding buffer. The cells were analyzed by flow cytometry (BD LSR II operated by FACS DIVA) with band-pass filters of 525 ± 25 mm.
Statistical analysis
Assessment of significance were performed using Student’s t-test and test results were considered significant at p < 0.05. Data were plotted and analyzed using GraphPad Prism 6 and SigmaPlot v11.
Results
CERS6 is highly expressed in T-ALL cells in comparison to normal cells
The mRNA expression of six CERS isoforms was analyzed in the NCI PPTP (National Cancer Institute Pediatric Preclinical Testing Program) panel of 23 cell lines29. CERS2, CERS5 and CERS6 expression levels were significantly higher than CERS1, CERS3 and CERS4 (8.7 ± 0.4, 7.9 ± 0.3 and 8.6 ±1.0 vs 5.0 ± 0.3, 3.2 ± 0.1 and 4.7 ± 0.7, p < 0.0001, Fig. 1b). Moreover, the relative expression was consistent (CV for CERS1: 5.6%, CERS2: 4.2%, CERS3: 2.2%, CERS4: 14.1%, CERS5: 3.9% and CERS6: 12.1%) in all the 23 cell lines analyzed, but different from the tissue distribution in normal cells11. CERS2 has been reported to be the most abundant enzyme with highest expression in most tissues, while mRNA expression of CERS6 is low in major tissues11. Here, we observed that the levels of CERS6, responsible for the generation of C16-Cer, were as high in cancer cells and similar to the levels of CERS2 (8.7 ± 0.4) among all cell lines tested. CERS6 was also found to be overexpressed in several other adult cancers when compared to normal tissue samples (Supplemental Fig. S1).
Next, we compared CERS6 protein levels in 10 leukemia cell lines (Supplemental Table S1)31 to PBMCs and T lymphocytes isolated from blood of healthy human volunteers, representing normal cells. CERS6 levels were significantly higher in ALL cells compared to PBMCs or T lymphocytes (Fig. 1c). To verify the functionality of CERS6, the levels of C16-Cer, the product of CERS6, were quantitated in ALL and normal cells by LC/MS/MS. As anticipated, C16-Cer levels were significantly higher in ALL cell lines in comparison to PBMCs or T lymphocytes (0.54 ± 0.09 vs 0.28 ± 0.04, p < 0.001 or 0.36 ± 0.01, p < 0.001, Fig. 1d). To investigate whether CERS6 levels are elevated in primary T-lymphoid malignancies, we employed clinical samples from four lymphoid malignancy patients (Supplemental Table S2) and found that the levels of CERS6 were higher in T lymphocytes isolated from the four patient samples in comparison to normal T lymphocytes (Fig. 1e).
CERS6 alters sensitivity of ALL cells to ABT-737, a pan-BCL-2 family of protein inhibitor
To determine whether higher levels of CERS6 in ALL contributes to resistance in cytotoxicity of chemotherapeutic drugs, we carried out loss-of-function (knockdown of CERS6) and gain-of-function (CERS6 overexpression) experiments in ALL cells. CERS6 was stably knocked down using shRNA by lentiviral transduction in two T-ALL cell lines, CCRF-CEM and MOLT-4. The decrease in CERS6 levels after CERS6 knockdown was confirmed by immunoblotting (Fig. 2a). C16-Cer levels were significantly reduced in CERS6 knockdown cells compared with cells transduced with non-targeted shRNA (1.5 ± 0.1 vs 7.3 ± 0.9 pmole/nmole Pi, p < 0.01 for CCRF-CEM and 7.4 ± 0.4 vs 10.9 ± 0.3 pmole/nmole Pi, p < 0.01 for MOLT-4, Fig. 2b), demonstrating that the CERS6 activity was also reduced with the decrease in CERS6 levels in both cell lines. Then, the effect of CERS6 knockdown on the cytotoxicity of ABT-737, a pan-BCL-2 inhibitor, was evaluated. Both CCRF-CEM and MOLT-4 cells with CERS6 knockdown displayed a significant increase in cytotoxicity upon ABT-737 treatment (5.2 ± 2.7% vs 86.6 ± 4.6% survival at 100 nM ABT-737, p < 0.001 for CCRF-CEM and 26.1 ± 3.8% vs 73.6 ± 4.6% survival at 100 nM ABT-737, p < 0.001 for MOLT-4, Fig. 2c). In both cell lines with CERS6 knockdown, the percentage of apoptotic cells in response to ABT-737 treatment was significantly higher compared with the cells transduced with non-targeted shRNA (98.0 ± 0.4% vs 38.0 ± 0.4%, p < 0.0001 for CCRF-CEM and 90.8 ± 0.3% vs 72.1 ± 0.3%, p < 0.0001 for MOLT-4, Fig. 2d and Supplemental Fig. S2). Immunoblotting showed that cleaved poly (ADP-ribose) polymerase (PARP) and cleaved caspase-3, markers for apoptotic cell death, were increased in both cell lines with CERS6 knockdown upon ABT-737 treatment in comparison to cells transduced with non-targeted shRNA (Fig. 2e).

a CERS6 protein levels in CCRF-CEM and MOLT-4 cells after stably knocking down CERS6 using shRNA (NT-shRNA non-targeted shRNA, shCERS6-Mixed shRNA against CERS6 and selected with puromycin, shCERS6-Single shRNA against CERS6 and selected with puromycin followed by repopulation from a single cell). GAPDH was used as a loading control. b C16-Ceramide levels (pmole/nmole of inorganic phosphate) in ALL cells after CERS6 knockdown. Ceramide levels were quantitated by HPLC/MS/MS (1.5 ± 0.1 vs 7.3 ± 0.9 pmole/nmole Pi for CCRF-CEM and 7.4 ± 0.4 vs 10.9 ± 0.3 pmole/nmole Pi for MOLT-4; n = 3). c Knockdown of CERS6 in CCRF-CEM and MOLT-4 sensitized the cells to ABT-737. Dose response curves showing concentration of ABT-737 on x-axis and survival fraction on y-axis on a log10 scale. Bar graphs depict survival fractions at 100 nM of ABT-737 in both cell lines (5.2 ± 2.7% vs 86.6 ± 4.6% survival at 100 nM ABT-737 for CCRF-CEM and 26.1 ± 3.8% vs 73.6 ± 4.6% survival at 100 nM ABT-737 for MOLT-4; n = 6). d Annexin-V apoptosis assay by flow cytometry showing the percentage of live and apoptotic cells upon ABT-737 treatment (1 µM for 16 h) in CCRF-CEM or MOLT-4 cells with CERS6 knockdown in comparison to cells transduced with NT-shRNA (98.0 ± 0.4% vs 38.0 ± 0.4% for CCRF-CEM and 90.8 ± 0.3% vs 72.1 ± 0.3% for MOLT-4; n = 3). e CERS6 knockdown in CCRF-CEM or MOLT-4 cells show higher levels of cleaved PARP and cleaved caspase-3 on ABT-737 treatment (1 µM for 16 h). GAPDH was used as a loading control * p < 0.05, ** p < 0.01, *** p < 0.001
Based on the expression of CERS6 in ALL cells, CCRF-CEM cells with relatively low CERS6 expression was selected for exogenous expression of CERS6. After stably expressing CERS6 in CCRF-CEM cells, the increase in CERS6 protein was confirmed in comparison to cells transduced with control vector (Fig. 3a). C16-Cer levels in CCRF-CEM cells with exogenous CERS6 expression were significantly higher relative to its control cells (10.1 ± 0.5 vs 5.7 ± 0.1 pmole/nmole Pi, p < 0.001, Fig. 3b). Increase in CERS6 levels rendered CCRF-CEM cells resistant to ABT-737 in comparison to control cells (39.5 ± 2.2% vs 2.3 ± 1.8% survival at 300 nM ABT-737, p < 0.001, Fig. 3c). The percentage of apoptotic cells were significantly lower (36.8 ± 3.8% vs 85.2 ± 0.6%, p < 0.01, Fig. 3d), and cleaved PARP and caspase-3 were less (Fig. 3e) in CCRF-CEM cells overexpressing CERS6 in comparison to cells transduced with control vector when treated with ABT-737.

a CERS6 levels in CCRF-CEM cells upon CERS6 overexpression (Control cells transduced with empty vector, CERS6-Mixed CERS6 overexpressing cells and selected with puromycin, CERS6-Single CERS6 overexpressing cells and selected with puromycin followed by repopulation from a single cell; 10.1 ± 0.5 vs 5.7 ± 0.1 pmole/nmole Pi; n = 3). b C16-Cer levels (pmole/nmole of inorganic phosphate) in CCRF-CEM cells after CERS6 overexpression. Ceramide levels were quantitated by HPLC/MS/MS. c CERS6 overexpression in CCRF-CEM cells rendered the cells resistant to ABT-737. Dose response curves showing concentration of ABT-737 on x-axis and survival fraction on y-axis on a log10 scale. Bar graphs depict survival fractions at 300 nM of ABT-737.(39.5 ± 2.2% vs 2.3 ± 1.8% survival at 300 nM ABT-737; n = 6) (d) Annexin-V apoptosis assay by flow cytometry showing the percentage of live and apoptotic cells upon ABT-737 treatment (2 µM for 16 h) in CERS6 overexpressing CCRF-CEM cells in comparison to cells transduced with empty vector (36.8 ± 3.8% vs 85.2 ± 0.6%; n = 3). e CERS6 overexpressing CCRF-CEM cells show lower levels of cleaved PARP and cleaved Caspase-3 on ABT-737 treatment (2 µM for 16 h) in comparison to control cells. The far right lane (shCERS6-Single) is a sample from ABT-737-treated CERS6 knockdown CCRF-CEM cells (from Fig. 2e) and represents positive control for cleaved PARP and cleaved caspase-3. GAPDH was used as a loading control * p < 0.05, ** p < 0.01, *** p < 0.001
CERS6-mediated resistance to ABT-737 occurs via the extrinsic pathway of apoptosis
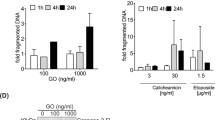
ABT-737-inducing apoptosis via mitochondrial (intrinsic) pathway of apoptosis is well documented. However, the extent of changes in anti- and pro-apoptotic BCL-2 family of proteins in ALL cells with CERS6 knockdown or exogenous expression was not consistent with the differences in cytotoxicity of ABT-737 (Supplemental Fig. S3a). Ceramides in plasma membranes are reported to form “ceramide-enriched platforms” which aid in clustering of death receptors in the extrinsic pathway of apoptosis32. Thus, the effect of CERS6 modulation in extrinsic apoptotic pathway in response to ABT-737 treatment was investigated. The involvement of extrinsic apoptotic pathway was assessed by measuring the levels of cleaved caspase-8 in CCRF-CEM and MOLT-4 cells with CERS6 knockdown upon treatment of ABT-737. Higher levels of cleaved caspase-8 were observed in both cell lines with CERS6 knockdown compared with their respective controls (Fig. 4a), demonstrating that CERS6 is associated with ABT-737 resistance via the extrinsic pathway of apoptosis. Then, CCRF-CEM and MOLT-4 with CERS6 knockdown were treated with Z-IETD, a caspase-8 inhibitor, 1 h prior to ABT-737 treatment. The percentage of apoptotic cells was significantly decreased in cells with CERS6 knockdown pretreated with Z-IETD in response to ABT-737 in comparison to cells without Z-IETD pretreatment (23.4 ± 1.7% vs 37.5 ± 1.5%, p < 0.01 for CCRF-CEM and 8.2 ± 0.1% vs 20.8 ± 0.3%, p < 0.01 for MOLT-4, Fig. 4b). Cleaved caspase-8, PARP and caspase-3 were reduced by the addition of Z-IETD in CERS6 knockdown cells treated with ABT-737 compared with cells without the caspase-8 inhibitor (Fig. 4c). We measured changes in soluble Fas ligand (FasL) in culture medium upon treatment of ABT-737 and found that ABT-737 induced the release of soluble FasL into the medium and that FasL release by ABT-737 was earlier and greater in CERS6 knockdown cells relative to its empty vector control (Fig. 4d). Further, when CCRF-CEM cells were treated with soluble FasL, cells with CERS6 knockdown showed significantly higher percentage of apoptotic cells compared with cells transduced with non-targeted shRNA (70.9 ± 0.6% vs 14.1 ± 0.1%, p < 0.0001, Fig. 4e). CCRF-CEM cells with CERS6 knockdown were sensitized to dexamethasone, part of ALL standard treatment, in comparison to control cells (32.5 ± 11.9% vs 2.2 ± 1.1% survival at 100 nM, p < 0.001, Supplemental Figs. S3b, c).

a Higher caspase-8 activity seen with CCRF-CEM and MOLT-4 CERS6 knockdown cells in comparison to cells transduced with non-targeted shRNA upon ABT-737 treatment. GAPDH was used as a loading control. b Annexin-V apoptosis assay by flow cytometry showing the percentage of apoptotic cells in ABT-737-treated CERS6 knockdown cells (CCRF-CEM and MOLT-4), with or without caspase-8 inhibitor, Z-IETD (23.4 ± 1.7% vs 37.5 ± 1.5% for CCRF-CEM and 8.2 ± 0.1% vs 20.8 ± 0.3% for MOLT-4; n = 3). c ABT-737-treated CERS6 knockdown cells show decreased levels of cleaved Caspase-8, cleaved PARP and cleaved caspase-3 on addition of Z-IETD in comparison to ABT-737-treated CERS6 knockdown cells without Z-IETD. d Levels of soluble FasL released in cultured medium upon treatment of ABT-737 in CERS6 knockdown cells compared to cells transduced with non-targeted shRNA. e Left: Caspase-8 activity was increased in a dose-dependent manner upon treatment of varying concentrations of Fas ligand (FasL) in CCRF-CEM cells with CERS6 knockdown. GAPDH was used as a loading control. e Right: Annexin-V apoptosis assay by flow cytometry showing increase in apoptotic cells in CCRF-CEM cells with CERS6 knockdown in comparison to cells transduced with NT-shRNA upon treatment of 1000 ng/ml of FasL (70.9 ± 0.6% vs 14.1 ± 0.1%; n = 3) ** p < 0.01, *** p < 0.001
CERS6 binds to CD95/Fas and interferes with FADD assembly to Fas in the extrinsic pathway of apoptosis
After showing that CERS6 interferes with the extrinsic pathway of apoptosis, we sought to determine the mechanism by which caspase-8 activation is attenuated by CERS6. We wanted to see if CERS6 interacted with any of the key players involved in the extrinsic pathway of apoptosis, namely Fas death receptor, FADD (Fas-associated protein with death domain), or caspase-8. Co-immunoprecipitation of CERS6 in CERS6 overexpressing CCRF-CEM cells using a two-step purification protocol (Fig. 5a) was conducted. The efficiency of the pull-down was confirmed by detecting CERS6 at different stages of purification (Fig. 5b). The results showed that CERS6 binds to Fas death receptor, also known as CD95, when the final pull-down sample was probed with Fas antibody (Fig. 5c). Fas ligand binds to Fas death receptor to form the death-inducing signaling complex (DISC) composed of Fas, FADD and pro-caspase-833 which initiates caspase-8 cleavage, further transducing a downstream signaling cascade of caspase activation resulting in apoptosis. Then, Fas was pulled down in ABT-737-treated CCRF-CEM cells to assess the effect of CERS6 on FADD association with Fas. We found that higher levels of FADD were associated with Fas in cells with CERS6 knockdown as compared to control cells on treatment with ABT-737 (Fig. 5d). We also found a decrease in FADD–Fas association in CCRF-CEM cells overexpressing CERS6 on ABT-737 treatment in comparison to ABT-737-treated CCRF-CEM CERS6 knocked down cells (Fig. 5e). Together, these results suggest that CERS6 binds to Fas and inhibits its assembly with FADD leading to decreased apoptosis via the extrinsic pathway.

a Schematic diagram listing the steps involved in the purification of CERS6 along with associated proteins from CERS6 overexpressing CCRF-CEM cells. HTBK-DDK (His6-TEV-Biotinylated region-His6-DDK) is a tag placed at the C-terminal site on CERS6 overexpressing vector. X, Y and Z represent unknown proteins associated with CERS6. b Confirmation of CERS6 protein expression at various purification steps. (I) total lysate sample; (II) pull-down with Ni-NTA beads; (III) pull-down with streptavidin beads; and (IV) sample after AcTEV digestion (the TBH-DDK part of the tag is cleaved off). c Detection of Fas as a binding partner of CERS6. Other proteins involved in the DISC assembly of the extrinsic pathway did not bind to CERS6. d Higher FADD–Fas association in CERS6 knockdown cells treated with ABT-737. CERS6 and FADD were detected in Fas pull-down samples. Normal IgG was used as a control. e FADD–Fas association is decreased in ABT-737-treated CERS6 overexpressing CCRF-CEM cells in comparison to cells with CERS6 knockdown. Cleaved caspase-8, cleaved PARP and cleaved caspase-3 levels were compared between CERS6 knockdown and overexpressing cells treated with ABT-737
CERS6 binds to intracellular domains of CD95/Fas
In order to determine the binding site of CERS6 on Fas, CERS6 and Fas were exogenously expressed in HEK293FT cells using two distinct tags, CERS6 with mycDDK and Fas with HA tag. To determine if any particular domain was critical for CERS6-Fas binding, deletion mutants of Fas intracellular region were constructed as shown in Fig. 6a. The binding was anticipated to occur in the death domain, the region where FADD binds to Fas to form DISC34, but, the deletion of the death domain only, did not affect CERS6 binding to Fas (Fig. 6b, left). Cysteine 199 residue adjacent to the transmembrane domain of Fas has been reported to be critical for the ability of Fas to trigger apoptosis35. The binding persisted even with the C199V mutation on Fas, suggesting that this residue is not critical for CERS6 binding to Fas. The binding of CERS6 to each of the deletion mutants in the intracellular region of Fas indicates that CERS6 binding to Fas involves multiple sites of Fas. To confirm this, deletion mutants (∆174–314 and ∆174–236 and 315–335, Fig. 6a) of the Fas intracellular region were constructed. The binding was lost with the deletion of amino acids 174 to 314, the region comprising the transmembrane domain to death domain, while the deletion of the entire cytoplasmic region except for the death domain did not affect the binding, suggesting that the binding occurs at more than one region from amino acids 174 to 314 (Fig. 6b, right). Further, we also confirmed that the binding of CERS6 and FAS mainly occurs in in plasma membrane relative to cytoplasmic fraction (which includes endoplasmic reticulum (ER) and other organelles) even with exogenous expression of CERS6 in cytoplasm (Fig. 6c). To conclude, our data show that CERS6 binds to Fas and inhibits the formation of DISC thereby blocking apoptosis upon treatment of drugs acting via extrinsic pathway of apoptosis (Fig. 7).

a Constructs (pCMV6-mycDDK or pCMV6-AC-HA) encoding a human wild-type CERS6 tagged with mycDDK at COOH terminus, FAS full-length and FAS mutants tagged with HA epitopes. HD homeobox (DNA binding) domain, TLC TRAM/LAG/CLN8 homology domain, CRD cysteine-rich domain, TD transmembrane domain, DD death domain. b Co-immunoprecipitation of full-length CERS6 and Fas and Fas mutants to detect direct binding. c Co-immunoprecipitation of full-length CERS6 and Fas/Fas mutants in cellular fractions (Cyt cytosolic fraction that includes ER and other organelles, PM plasma membrane). GAPDH (right) is used as a marker for cytosolic fraction while E-cadherin is the marker for plasma membrane

Proposed mechanism of CERS6-mediated resistance to chemotherapy via extrinsic pathway of apoptosis. Left: FasL binds to Fas and recruits FADD to form DISC complex which activates caspase-8 to induce apoptosis upon drug treatment. Right: CERS6 bound to Fas inhibits the formation of DISC complex and inhibits extrinsic apoptosis, and thus it inhibits downstream activation of apoptosis upon drug treatment, resulting in resistance
Discussion
Sphingolipid profiles are frequently altered between cancer cells and normal cells, which may be an indication of prevailing mechanisms used by tumor cells to overcome stressors. Here we report (1) that ceramide synthase-6 (CERS6), an enzyme responsible for the generation of C16 ceramides, is overexpressed in T-cell ALL cells compared with T lymphocytes and PBMCs; (2) that genetic modification of CERS6 in T-cell ALL cell lines resulted in significant changes in sensitivity to chemotherapeutic drugs; and (3) that CERS6 binds to Fas and renders resistance to chemotherapy via the extrinsic apoptotic pathway by interfering with the Fas–FADD association in ALL cells.
Several studies have examined how the metabolism of bioactive sphingolipids is reconfigured in cancer and whether modulating the balance between pro-apoptotic and pro-survival sphingolipids can overcome resistance to anticancer drugs12. For instance, treatment of chemo-resistant chronic myeloid leukemia cells with a BCR-ABL tyrosine kinase inhibitor demonstrated that increase in ceramide synthases preceded cell death36. Treatment of HL-60/VCR, the multidrug-resistant variant of HL-60 leukemia cells, with curcumin induced an increase in ceramide generation and accumulation, leading to apoptosis37. However, a majority of these studies consider ceramide species and corresponding ceramide synthases to be pro-apoptotic, without species-specific differences in activity or the mechanisms of inducing or interfering apoptosis in cancer cells in response to chemotherapy. Moreover, no studies have addressed the biological function of ceramide synthases apart from acylation of sphinganine to form ceramides. Our study, for the first time, elucidates the non-enzymatic role of CERS6, wherein its binding to Fas blocks the activation of the extrinsic apoptotic pathway in ALL cells upon treatment of anticancer drugs.
Our observation on higher levels of CERS6 and C16-Cer in T-ALL cell lines is consistent with a few other cancers as reported in the literature. C16-Cer levels were significantly higher in breast cancer relative to normal tissues38, while CERS6 mRNA levels were found to be expressively increased in ER-positive tumors in comparison to ER-negative tumors39. In non-small cell lung cancer, CERS6 levels were markedly higher in comparison to controls and was found to be associated with poor prognosis and increased metastasis40. C16-Cer levels were significantly increased in HNSCC which correlated with higher mRNA expression of CERS6, when 12 pairs of HNSCC tumors and normal tissues were compared41. In our study, we have shown higher expression of CERS6 in T-ALL in comparison to normal cells is associated with T-ALL drug resistance.
Previously, it has been shown that knockdown of CERS6 in HNSCC cells induced stress-mediated apoptosis which could be reversed by overexpressing CERS6 in these cells17. We knocked down CERS6 as well as overexpressed it in ALL cell lines and showed that these genetic alterations changed the sensitivity of ALL cells to chemotherapy. Our studies further demonstrated that CERS6-altered ALL sensitivity to ABT-737 occurs via the extrinsic pathway, by using a caspase-8 inhibitor. Although ABT-737 is anticipated to primarily act via the mitochondrial pathway of apoptosis, it has also been reported to act through the extrinsic pathway, where it enhances TRAIL-mediated cytotoxicity in renal, prostate and lung cancer cells by upregulating TRAIL receptor, death receptor 542. Our study suggests the expression of CERS6 as the mediator in inducing resistance to chemotherapy acting via extrinsic pathway of apoptosis.
We have demonstrated that there is clear processing of caspase-8 upon ABT-737 treatment, in order to show the involvement of extrinsic apoptotic pathway. However, caspase cleavage during late-stage apoptosis does not faithfully indicate one pathway or another43. To further confirm the involvement of extrinsic pathway, we conducted experiments by treating the cells with FasL, which is exclusive to the extrinsic pathway of apoptosis and showed that caspase-8 is activated in CERS6 knockdown cells (Fig. 4e). In addition, we have studied the effect of knockdown or overexpression of CERS6 on the cell surface expression of FAS. We conducted flow cytometric analysis of FAS cell surface expression in CERS6 knockdown and exogenous expressed cells. Interestingly, CERS6 knockdown resulted in increased FAS cell surface expression (Supplemental Fig. S4). However, CERS6 overexpression did not affect the expression of FAS. The mechanism of FAS increase with the knockdown of CERS6 warrants further investigation in future studies.
Finally, we show that CERS6 binds to Fas and interferes with Fas–FADD association to form DISC complex which activates caspase-8 in the extrinsic pathway of apoptosis. Fas has been reported to interact with a palmitoyl transferase, DHHC7 (aspartate-histidine-histidine-cysteine family of acyl transferases) which stabilizes Fas by palmitoylation at C19935. Being an acyl transferase, CERS6 was thought to interfere with binding of DHHC7 thereby destabilizing Fas and its ability to cause apoptosis. Binding of CERS6 to Fas persisted with C199V mutation, suggesting that C199 is not critical for CERS6 binding. BCL2L13 and SIRT3 are two proteins reported to be interacting with CERS6, where the former is shown to inhibit the activity of CERS644, while the latter deacetylates CERS6 to cause an increase in its activity45. These interactions affect the activity of CERS6, but there are no reports showing that CERS6 affects the function of other proteins. Here, we show that CERS6 affects the function of Fas by directly interfering with its assembly with FADD.
In summary, our study shows that CERS6 plays an important role in ALL resistance to chemotherapy by interfering with the Fas–FADD assembly in the extrinsic pathway of apoptosis. CERS6 may serve as a biomarker to stratify ALL patients to determine whether they were likely to respond to drugs acting via the extrinsic pathway of apoptosis. As cancer cells continue to develop resistance to chemotherapeutic drugs and evolve new mechanisms to evade apoptosis, future studies on determining CERS6 as a biomarker for drug resistance in cancer are warranted.
References
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 62, 10–29 (2012).
Pui, C. H., Robison, L. L. & Look, A. T. Acute lymphoblastic leukaemia. Lancet 371, 1030–1043 (2008).
Hunger, S. P. et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the Children’s Oncology Group. J. Clin. Oncol. 30, 1663–1669 (2012).
Pieters, R. et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370, 240–250 (2007).
Oriol, A. et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica 95, 589–596 (2010).
Pui, C. H. et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med. 360, 2730–2741 (2009).
Hannun, Y. A. & Obeid, L. M. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 (2008).
Ogretmen, B. & Hannun, Y. A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 4, 604–616 (2004).
Mullen, T. D., Hannun, Y. A. & Obeid, L. M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 441, 789–802 (2012).
Reynolds, C. P., Maurer, B. J. & Kolesnick, R. N. Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett. 206, 169–180 (2004).
Levy, M. & Futerman, A. H. Mammalian ceramide synthases. IUBMB Life 62, 347–356 (2010).
Kitatani, K., Taniguchi, M. & Okazaki, T. Role of sphingolipids and metabolizing enzymes in hematological malignancies. Mol. Cells 38, 482–495 (2015).
Ghosh, D., Lippert, D., Krokhin, O., Cortens, J. P. & Wilkins, J. A. Defining the membrane proteome of NK cells. J. Mass Spectrom. 45, 1–25 (2010).
Weinmann, A., Galle, P. R. & Teufel, A. LASS6, an additional member of the longevity assurance gene family. Int. J. Mol. Med. 16, 905–910 (2005).
Ebel, P. et al. Inactivation of ceramide synthase 6 in mice results in an altered sphingolipid metabolism and behavioral abnormalities. J. Biol. Chem. 288, 21433–21447 (2013).
White-Gilbertson, S. et al. Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 28, 1132–1141 (2009).
Senkal, C. E., Ponnusamy, S., Bielawski, J., Hannun, Y. A. & Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 24, 296–308 (2010).
Hunger, S. P. & Mullighan, C. G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 373, 1541–1552 (2015).
Pui, C. H. & Evans, W. E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354, 166–178 (2006).
Oltersdorf, T. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005).
Kang, M. H. & Reynolds, C. P. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 15, 1126–1132 (2009).
Chauhan, D. et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene 26, 2374–2380 (2007).
Kang, M. H. et al. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood 110, 2057–2066 (2007).
Neale, G. et al. Molecular characterization of the pediatric preclinical testing panel. Clin. Cancer Res. 14, 4572–4583 (2008).
Holliday, M. W. Jr, Cox, S. B., Kang, M. H. & Maurer, B. J. C22:0- and C24:0-dihydroceramides confer mixed cytotoxicity in T-cell acute lymphoblastic leukemia cell lines. PLoS One 8, e74768 (2013).
Van Veldhoven, P. P. & Bell, R. M. Effect of harvesting methods, growth conditions and growth phase on diacylglycerol levels in cultured human adherent cells. Biochim. Biophys. Acta 959, 185–196 (1988).
Wang, X. et al. Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565 (2007).
Zhang, Y. et al. DSSylation, a novel protein modification targets proteins induced by oxidative stress, and facilitates their degradation in cells. Protein Cell 5, 124–140 (2014).
Kang, M. H. et al. National Cancer Institute pediatric preclinical testing program: model description for in vitro cytotoxicity testing. Pediatr. Blood Cancer 56, 239–249 (2011).
Kang, M. H., Wan, Z., Kang, Y. H., Sposto, R. & Reynolds, C. P. Mechanism of synergy of N-(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J. Natl. Cancer Inst. 100, 580–595 (2008).
Makena, M. R., Koneru, B., Nguyen, T. H., Kang, M. H. & Reynolds, C. P. Reactive oxygen species-mediated synergism of fenretinide and romidepsin in preclinical models of T-cell lymphoid malignancies. Mol. Cancer Ther. 16, 649–661 (2017).
Morad, S. A. & Cabot, M. C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 13, 51–65 (2013).
Kischkel, F. C. et al. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 12, 611–620 (2000).
Kischkel, F. C. et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14, 5579–5588 (1995).
Rossin, A. et al. Fas palmitoylation by the palmitoyl acyltransferase DHHC7 regulates Fas stability. Cell Death Differ. 22, 643–653 (2015).
Camgoz, A., Gencer, E. B., Ural, A. U., Avcu, F. & Baran, Y. Roles of ceramide synthase and ceramide clearence genes in nilotinib-induced cell death in chronic myeloid leukemia cells. Leuk. Lymphoma 52, 1574–1584 (2011).
Shakor, A. B. et al. Curcumin induces apoptosis of multidrug-resistant human leukemia HL60 cells by complex pathways leading to ceramide accumulation. Biochim. Biophys. Acta 1841, 1672–1682 (2014).
Schiffmann, S. et al. Ceramide synthases and ceramide levels are increased in breast cancer tissue. Carcinogenesis 30, 745–752 (2009).
Ruckhaberle, E. et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 112, 41–52 (2008).
Suzuki, M. et al. Targeting ceramide synthase 6-dependent metastasis-prone phenotype in lung cancer cells. J. Clin. Invest. 126, 254–265 (2016).
Karahatay, S. et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): attenuation of C(18)-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 256, 101–111 (2007).
Song, J. H., Kandasamy, K. & Kraft, A. S. ABT-737 induces expression of the death receptor 5 and sensitizes human cancer cells to TRAIL-induced apoptosis. J. Biol. Chem. 283, 25003–25013 (2008).
Woo, M. et al. In vivo evidence that caspase-3 is required for Fas-mediated apoptosis of hepatocytes. J. Immunol. 163, 4909–4916 (1999).
Jensen, S. A. et al. Bcl2L13 is a ceramide synthase inhibitor in glioblastoma. Proc. Natl Acad. Sci. USA 111, 5682–5687 (2014).
Novgorodov, S. A. et al. SIRT3 deacetylates ceramide synthases: implications for mitochondrial dysfunction and brain injury. J. Biol. Chem. 291, 1957–1973 (2016).
Acknowledgements
This work was funded by the National Cancer Institute, NIH (R15CA159308 to MHK), and by Cancer Prevention and Research Institute of Texas (individual investigator awards RP101042 to MHK). We would like to thank Dr. C. Patrick Reynolds, Director of Cancer Center, TTUHSC and the Cancer Center staff for providing us with clinical samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Herold
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verlekar, D., Wei, SJ., Cho, H. et al. Ceramide synthase-6 confers resistance to chemotherapy by binding to CD95/Fas in T-cell acute lymphoblastic leukemia. Cell Death Dis 9, 925 (2018). https://doi.org/10.1038/s41419-018-0964-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0964-4
This article is cited by
-
The ceramide synthase (CERS/LASS) family: Functions involved in cancer progression
Cellular Oncology (2023)
-
Comprehensive analysis of LASS6 expression and prognostic value in ovarian cancer
Journal of Ovarian Research (2021)
-
Alteration of payload in extracellular vesicles by crosstalk with mesenchymal stem cells from different origin
Journal of Nanobiotechnology (2021)
-
Ribosome profiling at isoform level reveals evolutionary conserved impacts of differential splicing on the proteome
Nature Communications (2020)
