
Abstract
Currently, immunotherapy is attracting a lot of attention and may potentially become a leading approach in the treatment of cancer. One emerging therapeutic, the chimeric-antigen receptor T-cell adoptive immunotherapy (CAR-T) is showing remarkable efficacy in the treatment of several B-cell malignancies. The popularity of CAR-T has been founded on two CAR T-cell products recently approved by FDA (during 2017) in the treatment of relapsed/refractory B-cell acute lymphoblastic leukemia and B-cell lymphoma. However, their toxicities observed in clinical trials were extremely significant and in some cases even fatal with no approved algorithms for toxicity prediction being available to date. A deeper understanding of the biological basis of such complications is the key to prompt and comprehensive clinical management. Here we review the wide spectrum of effects associated with CAR T cell therapy with a major focus on the pathogenesis of cytokine release syndrome and neurotoxicity as the most common, potentially life-threatening effects of this treatment. We discuss the basis of clinical management and the existing models that predict the severity of toxicity, as well as the key factors that modulate this event. Finally, we will summarize the literature detailing universal allogenic CAR T-cells and their toxicity profile.
Similar content being viewed by others

Facts
-
1.
The chimeric-antigen receptor T-cell adoptive immunotherapy (CAR-T) is a potent instrument for treating several hematological malignancies, not only those expressing the CD19 receptor.
-
2.
There is a pressing need to make this therapy available to a wider spectrum of patients.
-
3.
However, although the safety levels of CAR-T therapy are generally acceptable, several fatal outcomes due to severe cytotoxicity have been reported in clinical trials of CAR-T therapies.
-
4.
Therefore, better understanding of the spectrum of toxicities, their etiology and pathogenesis as well as the knowledge of toxicity-promoting factors may help develop and validate the predictive scales and define better prophylactic strategies for high-risk patients.
Open questions
It is known that some of the factors that worsen the toxicity of CAR-T therapy (higher CAR T-cell dose, intensive lymphodepletion) also positively affect its efficacy. How can one achieve the proper balance between these?
What kind of predictive model one should use for the toxicity risk assessment and which group of patients should be given the treatment for prophylaxis of such toxicity?
Would universal allogenic CAR T-cells be as safe and effective as the autologous CAR T-cells?
Introduction
Adoptive immunotherapy is a rapidly evolving field in modern cancer biology and its treatment. This approach is based on the ex vivo modification and expansion of patient-derived antigen-presenting cells (APCs) or T-cells followed by their subsequent re-introduction back into the patient. One of the most promising modalities within this field is genetically modified T-cell expressing chimeric-antigen receptor (CAR-T), which is able to specifically recognize the target antigen (e.g., the CD19 receptor of B-cells) and eliminate the target cancer cells.
Although the safety profile of CAR-T therapy is generally acceptable and the treatment-related mortality is low, several fatal outcomes have been reported in CAR-T clinical trials. Better understanding of the spectrum of toxicities, their etiology and pathogenesis as well as the knowledge of toxicity-promoting factors may aid in the development and clinical validation of the predictive scales and define better prophylactic strategies for high-risk patients.
Car design and therapeutic efficacy
CAR structure
In general, CARs are transmembrane molecules composed of several functional elements. An extracellular single-chain variable fragment (scFv), derived from the antigen-recognizing sequence of an antibody, is fused to a hinge/spacer module and a transmembrane domain, which is further linked to the intracellular domain. The latter is critical for the transmission of the activation signal. CAR-T cells can been divided into four generations (as seen in Fig. 1). Whereas the term ‘generation’ was initially used to describe the CAR structure, it now more broadly (and collectively) refers to the CAR and CAR-T cells bearing it. The clinical trials that have yielded the most success include an FDA-approved second-generation CAR-T products from Novartis and Kite Pharma1,2. Third-generation CAR-Ts have also been explored and so far have failed to show additional benefits during clinical trials and consequently, further clinical studies are eagerly awaited3. More recently, a 4th generation of CAR-Ts has been developed and contains a genetic construct, encoding a separate co-stimulation molecule4 or suicide genes5,6, as well as cytokine genes7. The initial pre-clinical success8 of TRUCKs7, designed to additionally secrete IL12, could not be reproduced in clinical trials (NCT01236573, NCT01457131) and resulted in their termination due to unexpected toxicity and lack of effect.

а The basic design of CAR includes two functional parts: the extracellular domain (derived from the variable region of the monoclonal antibody (mAb) and reformatted into the single-chain variable Fragment (scFv) consisting of the linked variable regions of both heavy (VH) and light (VL) chains that recognize the target antigen, and the intracellular domain (derived from the ITAMs of the CD3 complex ζ-chain), providing the first activation signal. b The first-generation CAR consisted of scFv, transmembrane domain and CD3 ζ ITAMs as an intercellular activation signal. The second-generation CAR bears an additional co-stimulatory intracellular domain (*) such as CD28, CD137 signal domains. The third-generation CAR includes two or more different co-stimulatory domains (* and **) and the fourth generation CAR-T may be generally defined as the T-cell, bearing CAR of any design (2nd or 3rd generation) with expression of additional molecules, such as cytokines, antibodies (#) or separate co-stimulation molecules (***)
Therapeutic efficacy
CAR-T therapy has demonstrated the highest efficacy in the context of B-cell neoplasms. Despite the high rates of complete remission in acute lymphoblastic leukemia (ALL, up to 94% in large trials1,6,9,10,11), in one of the trials9 (phase I, CTL019, reviewed in Table 1), the probability of event-free survival was 67% at 6 months, and sustained remission was reported in 19 patients within a follow-up period of 1–24 months (with a median of 7 months). Most of the patients (15 of 19) received no further therapy. Of note, the results reported by Lee et al.12 and Davila et al.13, in the context of ALL, do not assess the curative potential of the therapy, as most of the patients proceeded to allogenic hematopoietic stem-cell transplantation (allo-HSCT) after achieving complete remission (CR). The latest articles improve our understanding of long-term outcomes reporting an overall survival rate of 52% at 18 months (ZUMA-1, median follow-up of 15.4 months; 42% of PTS showed a stable response)2 and 76 % at 12 months (ELIANA, median follow-up of 13.1 months)1. At the longest median follow-up of 29 months (range: 1–65), Park et al. (n = 53, ALL) reported the median event-free survival of 6.1 months (95% CI: 5.0–11.5) in which the median overall survival was 12.9 months (95% CI: 8.7–23.4). Importantly, a high disease burden, defined as ≥5% of bone marrow blasts or extramedullary disease, resulted in a significantly worse long-term prognosis14.
In relapsed or refractory acute myeloid leukemia (AML), the results were also quite promising15,16, although the absence of the ‘ideal’ AML-specific targets that are not expressed by normal hematopoietic progenitors may limit the scope of the CAR-T approach. Recent results of the BCMA-specific CAR-T therapy in heavily pretreated multiple myeloma (MM) patients showed particularly encouraging results. In this trial 95% CR/near CR with a median follow-up of 6 months17 were observed, although no data addressing the long-term survival is available.
In contrast to hematological malignancies, the efficacy of CAR-T cells is much lower for solid cancers. To date, the best results for CAR-T therapy in solid tumors have been reported for the treatment of HER2-positive sarcomas, with 4 of 17 PTS achieving disease stabilization18. Moreover, 3 of 11 neuroblastoma patients showed CR and two of them displayed prolonged CR19.
As there are several excellent reviews summarizing the recent information about clinical trials in the field of CAR-T20,21,22,23, we will omit the detailed description of clinical trial data in this review, to avoid duplication.
Despite the spectacular success of CAR-T therapy in treating B-cell malignancies, serious side effects have been associated with this therapy. As seen below, we will detail these adverse reactions and outline the clinical approaches currently being used to alleviate these complications.
Toxicities of Car-T therapy
CAR-T therapy has a unique toxicity profile that is not easy to predict and evaluate as it may differ when distinct CAR-T designs are used. Consequently, unexpected toxicities were the reasons for why the early termination of several trials (e.g., NCT01236573, NCT01457131, and NCT02535364) had occurred. While this is a hindrance, many excellent studies are ongoing with a view to overcome this hurdle.
Cytokine release syndrome (CRS)
CRS is the most common adverse event seen across the trials incorporating CAR-T cell therapy (74–100% in the anti-CD19 setting)1,6,10,24,25,26,27,28. This complication is thought to have arisen due to the expansion and activation of CAR-T cells which leads to a massive over-production of cytokines by a number of immune cell types that result in an elevated systemic inflammatory response.
Clinical manifestations of CRS
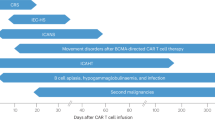
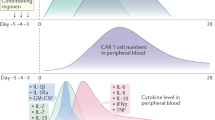
As shown in Fig. 2, clinical symptoms of CRS can range from mild (fever, myalgias, fatigue, and mild hypotension) to serious symptoms, such as hypotension requiring vasopressors, respiratory failure, coagulopathy, and multi-organ system failure. CRS can manifest within the first week after CAR-T infusion and progress further within 1–2 weeks. Fever is usually the first and most obligatory sign of CRS28. Patients with more severe CRS experience the fever earlier which is prolonged and where the peak of temperature is higher28,29. Thus, the ‘time to fever’ and its peak have been adequately exploited in the CRS predictive scale28.

Activated CAR T-cells (CAR, green) release effector cytokines that in turn activate (1) Endothelial cells (E, pink) and (2) Antigen-presenting cells and macrophages (APC, gray). These cells produce IL6 driving CRS onset. (3) CAR-T cells are able to penetrate the blood–brain barrier (BBB). All the above-mentioned as well as other factors (see CRES pathogenesis) probably contribute to the BBB disruption and the passive passage of cytokines into the CNS (4) resulting in CRES. Pericytes (P, yellow) as well as endothelial cells exposed to effector cytokines produce IL6 (5) driving CRES further
CRS grading
For prompt and efficient CRS management, accurate and robust clinical grading scales have recently been introduced, each with minor differences, where all of them categorize CRS from grade 1 toxicity that requires only symptomatic management to grade 4, which is potentially life-threatening, and grade 5, where lethal complications are evident. In clinical experiences, grade 4–5 CRS was noted as ‘severe CRS’ (sCRS).
Whereas, the grading of CRS originated from The National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE v4.03), then Lee et al.30 proposed the updated grading, and finally MD Anderson Cancer Centre CAR-T Therapy-Associated Toxicity Working Group (CARTOX) grading was developed31. The CTCAE grading was designed for CRS associated with antibody therapeutics, whereas Lee et al. developed the grading specifically for CAR-T therapy and defined low-dose requirement for vasopressors as grade 2 CRS. By contrast, the need for vasopressors was defined as grade 4 CRS in CTCAE. Furthermore, Lee et al.30 provided treatment guidelines based on their grading system and this grading system is most commonly used across the trials. Although the CARTOX scale seems to be nearly identical to that of Lee et al., it does have an advantage of it being easier to use (Table 2).
Pathogenesis of CRS
The first event driving CRS is the target-dependent activation of CAR-T cells, which release effector cytokines, such as IFNγ, TNFα, and IL2. These molecules in turn are able to activate macrophages that produce a broad spectrum of pro-inflammatory cytokines, resulting in hypercytokinemia and progression of CRS32,33. During CAR-associated CRS, the three most significantly elevated cytokines are IL6, IFN-γ, and IL1034. Their levels in CRS grade ≥4 increase ~2–3 logs in comparison to ‘no CRS’ cases28. Increased IL6 levels are associated with key clinical features of sCRS (which include hypoxia, hypotension, impaired coagulation and organ system failure)35. In clinical trials, anti-IL6-directed therapy was highly effective in CRS management9,31,36,37. Taken together, these data suggest that IL6 has a significant pathological role in CRS.
The IL 6 signaling pathway is exerted via the interaction of IL6 with its specific receptor (IL6R). The latter is present either in the membrane-bound form or in the soluble state (sIL6R). IL6R complexes the gp130 receptor, which serves a signal transducing component of the complex by activating JAK/STAT transcription pathway. Notably, IL6 signaling via the membrane-bound IL6R is restricted to hepatocytes, certain types of epithelial cells, and some leukocytes. This process is known as cis-signaling. On the contrary, during trans-signaling, sIL6R is excreted to serum where it binds circulating IL6 and, being recruited by the ubiquitously expressed gp130 component, may affect many tissues (reviewed in refs. 38,39).
Significant efforts have also been made to uncover the exact source(s) of IL6 during CAR-T cell activation32. Using co-culture assays (CAR T-cells and APCs) and data from the patients participating in clinical trials of CAR-T therapy, monocyte-lineage APCs were demonstrated to be exclusive IL6-producing cell type (among CAR T-cells, bystander T-cells). Based on the results of trans-well co-cultivation assays, it was importantly concluded that although occurring in response to CAR-T mediated recognition of leukemic cells IL6 production by APCs was independent of direct contact between CART19 and APCs32. Recent studies have explored endothelial activation in CRS and found that it may significantly modulate CRS severity. The markers for endothelial activation (VWF and Ang2) were elevated in patients developing sCRS, either before starting the CAR-T therapy or while sCRS developed and persisted28. A case study of a patient who succumbed to sCRS revealed endothelial cells as being one of the principle sources of IL640, thus implicating a direct link between the activated endothelium to overall IL6 production. Notably, there is no evidence to date that T-cells or CAR-T cells may be a significant source of IL632.
Importantly, IL6 was shown to influence only naive CD4+41,42 and CD8+ T-cells43. Upon activation, T-cells undergo a significant loss of IL6R density41, partly due to its increased shedding and hence formation of sIL6R44. Thus, although a problem of potential influence of IL6 on CAR T-cells cytotoxic function does exist, the likelihood of it is minimal32. This is because CD4+ and CD8+ CAR-T-cells function as activated T-lymphocytes that are no longer IL6- dependent. Indeed, IL6 was shown to have no impact on CAR-T-cell transcriptional profiling or cytotoxicity32. Moreover, it was demonstrated that CART19 treatment in an immune-deficient ALL mice model failed to mimic clinically observed CRS32, but was able to induce durable remissions in mice lacking APCs45, thus underlining the importance of immune cells (including APCs) interaction in pathogenesis of CRS. That blocking IL6 with the anti-IL6R antibody Tocilizumab did not significantly compromise therapeutic efficacy in clinics31, together with the above-mentioned data, suggest that IL6 is dispensable for CAR-T cell function. However, it should be mentioned that in the research by Singh et al.32 the level of sIL6R in the medium was not assessed thereby the absence of sIL6R-dependent trans-signaling in CAR-T should not be completely ruled out.
At present, it is not clear whether CD4+ CAR-T cells, similar to their normal CD4+ counterparts, are able to produce sIL6R and thus worsen the course of CRS. It should be noted however, that according to the work of Yang et al.46 the behavior of CD4+ CAR cells activated by CAR signaling was shown to be substantially different from CD4+ T-cells activated by TCR signaling. Based on this notion, it may be assumed that sIL6R secretion in CD4+ CAR-T cells also differs from the one in regular CD4+ T-cells. Clearly, this important question requires further experimental investigation.
As seen in Fig. 3, such events are depicted in the context of CRS.

Clinical features of CRS according to the published data
Predicting CRS severity
Recently, Teachey et al.47 measured the levels of cytokines and biomarkers in 51 patients (of which 39 were pediatric), who were treated with CART19 (CTL019) for ALL. Therein, none of the standard clinical laboratory tests could predict CRS severity. Furthermore, many of the tests such as ferritin, CRP, LDH, AST, ALT, and BUN, showed abnormal levels only after CRS onset. Cytokine analysis has allowed the development of robust predictive models based on measuring 2–3 cytokines in children (such as IFNγ, IL13, and MIP1α) and adults (IFNγ, sgp130, eotaxin). Similarly, another prediction system has also been developed (from a study of 133 patients, JCAR014)28 based on the persistence of fever >38.9 °C during the first 36 h after therapeutic infusion. Here, sensitivity in sCRS prediction was 100% (with 84% specificity). Analysis of the information on a single cytokine (MCP-1) for patients with fever >38.9 °C allowed specificities of around 95% to be observed. In comparison to the model proposed by Teachey et al., this approach is more reliable, feasible and easier to follow as the measurement of MCP-1 values was needed from just 30 of the 133 patients, to safely classify the rest with a fever measurement. Although very encouraging, these approaches need further exploration through clinical trials. To ensure greater robustness one may suggest different CAR-T designs may ultimately also need design-specific CRS prediction scales.
HLH/MAS as a complication of CRS
Patients with sCRS may develop a macrophage activation syndrome (MAS), which also referred to as hemophagocytic lymphohistiocytosis (HLH). Identification of CRS-related MAS is complicated due to the commonality in pathogenesis of both conditions based on high serum cytokines such as IFNγ, which results in macrophage activation48. Outside the setting of CAR-T therapy, fever, cytopenia, hyperferritinemia as well as bone marrow hemophagocytosis are the key diagnostic features. To support the diagnosis, the exceptional diagnostic value of glycosylated ferritin for HLH/MAS has also been shown in several studies49,50,51. Following CART19 therapy, ferritin >10000 mg/dL (a typical diagnostic parameter for HLH in pediatric, but not in adult patients52) was invariably observed in all patients with grade 4–5 CRS and in 51 and 83% CRS grade 0–3 patients in pediatric and adult cohorts, respectively47. Thus, for patients treated with CAR-T, the only reliable indicators for HLH/MAS are hemophagocytosis, hypofibrinogenemia and probably hypertriglyceridemia, as the whole range of other above-mentioned diagnostic features is observed during CAR-T mediated CRS. Interestingly, hemophagocytosis in the absence of other HLH features has been noted to be non-specific for HLH diagnosis in adults53,54.
Recently, the frequency of HLH/MAS in CAR-T- treated patients has been reported to be as low as ~1%. The diagnostic criteria for this condition have been based upon unexplainable severity of liver, kidney and pulmonary organ toxicity and hemophagocytosis31. The treatment of HLH/MAS requires more active immune suppression and includes etoposide/cyclosporine-based regimens31,55. In this context, the diagnostic value of glycosylated ferritin has not been studied and may hold some potential.
CAR-T-related neurotoxicity
Neurotoxicity, also known as CAR-T Related Encephalopathy Syndrome (CRES) is reportedly very common in CART19 trials6,9,10,24,25. The underlying cause is unknown and is likely unrelated to the recognition of cryptic CNS antigens by CART19 cells and off-target cytotoxicity. There is conflicting data about CD19 expression levels in brain tissue with more evidence for the absence of this antigen in the CNS56,57,58. Notably, neurotoxicity was reported in clinical trials of another CD19-targeting agent, Blinatumomab. CRES has not yet been clearly observed upon the targeting of other tumor antigens, except for CNS tumors59 and CD22 in ALL patients60. In the latter case, only mild subjective impairments were observed that might have been related to fever or comorbidity. Importantly, it must be noted that there were no cases of sCRS observed in this trial60 since only CRS grades 1 and 2 were reported, yet sCRS is known as high-risk factor for neurotoxicity (grade ≥3)61.
Clinical diagnosis and grading of neurotoxicity
Clinical symptoms of CRES (summarized in Fig. 4) commonly include headache, seizures, delirium, anxiety, tremor and impaired writing ability, aphasia, decreased consciousness and even coma with cerebral edema. The median time for onset of CRES is 4 days after infusion and the median duration is 5 days61. Grading of CRES is often performed according to CTCAE v. 4.03, but this system is imperfect, as it was not customized specifically for CAR-T neurotoxicity. Recently, an MD Anderson Cancer Centre CARTOX group has developed a new grading system (Table 3) based on a 10-point neurological assessment tool (CARTOX-10)31. It is very convenient and patients can be promptly assessed at high frequency. In brief, it evaluates patients’ ability to orient themselves in space, time and in their personality, as well it assesses their speech and writing abilities.

Clinical features of CRES according to the published data
Pathogenesis and prediction of neurotoxicity
Several mechanisms for trafficking cytokines into the CNS have been previously described62. These include their passive leakage through the blood–brain–barrier (BBB), through active uptake mechanisms, via activation of endothelial cells and perivascular macrophages and on-site cytokine production downstream of the accumulation of immune cells in the CNS.
Following CART19 therapy, several of these mechanisms are engaged. First of all, CART19 cells penetrate the CNS as they have been detected in CSF9,11,12,37,63,64 either by PCR63 or by flow cytometry11. This is in line with the observations that CART19 therapy successfully eliminates CNS disease in patients with no subsequent CNS relapse9,64,65. Gust et al.61 found the concentrations of several cytokines (including IL6 and IFNγ) in CSF to be comparable with serum levels. This was interpreted as either a failure in the functionality of the BBB and/or due to the formation of local sources of cytokines in the CNS. Consistent with this idea, primary human BBB pericytes were demonstrated to secrete IL6 and VEGF upon exposure to IFNγ and TNFα. Furthermore, CNS toxicity was associated with increased BBB permeability mediated by the higher Ang2:Ang1 ratio in which Ang1 stabilizes endothelial cells of the BBB, while Ang2 has an opposing effect61. Interestingly, treatment of mice harboring experimental cerebral malaria (which is associated with a deregulated inflammatory response and high levels of TNFα, IL6, and IFNγ66,67,68,69,70,71), with fingolimod (a sphingosine-1-phosphate receptor modulator) was accompanied by higher Ang1 serum levels and decreased BBB permeability, which resulted in improved outcomes72. Similar observations were made for mice administered with recombinant Ang173. Thus, pharmacologic modulation of the Ang2:Ang1 ratio could be explored further for acceptable CAR-T-associated CRES management61. In support of these findings, human autopsy results revealed the breakdown of the BBB in two patients who succumbed to CRES through cerebral edema after JCAR015 treatment74, highlighting the importance of this mechanism impact in severe CRES.
Neelapu et al.31 reported that the manifestation of CRES is biphasic with the onset of the first phase occurring simultaneously with CRS symptoms and usually within the first 5 days after CAR-T treatment. This ‘acute’ type of CRES had a tendency to be shorter and milder (grade 1–2) and may have benefitted from anti-IL6 therapy. The authors proposed that this may have been attributable to greater BBB permeability, which allowed the therapeutic antibodies to reach the CNS. An alternative explanation for the effectivity of anti-IL6 therapy was the lack of BBB damage during the ‘first phase’ of CRES, as the endothelium and pericytes were inactive and did not produce cytokines locally. In this instance, the elimination of IL6 after prompt therapy and resolution of CRS would prevent the progression of neurotoxicity by breaking the cytokine ‘loop’. Subsequently, the ‘second phase’ of CRES, which may happen even 3–4 weeks after cellular therapy (as seen in 10% of patients) may arise due to the formation of a local cytokine source within the CNS.
While CRS and CRES probably have certain commonalities in their pathogenesis, such as (1) IL6 production by APCs (and pericytes or endothelial cells) and (2) a deregulated endothelium (due to increased Ang2:Ang1 ratio and VWF), CRS and CRES are different pathological processes. In general, CRES has been rarely observed in the absence of any CRS, as evidenced in the study of CRES in the ZUMA-1 trial, in which 5 of 90 patients had CRES without CRS27 and in the Gust et al.61 patient cohort (where 5 of 53 patients had CRES with no CRS). These cases are usually mild and subjective (grade 1). However, there was also a case report of a patient with severe CRES with highly elevated CSF-cytokines in the absence of sCRS63. Taking into account the simultaneous prevalence of CRS and CRES grade ≥261, CRS may appear to trigger the development of CRES, which may continue to develop independently (as highlighted in Fig. 3).
Gust et al.61 suggested that it would be advisable to prevent CRES development, as once established, it can be less likely to resolve after IL6-directed therapy. They proposed a predictive model for grade ≥4 CRES, based on a fever ≥38.9 °C and elevated serum IL6 and MCP1 levels in the first 36 h after CAR-T therapy. Here, the model had a sensitivity and specificity of 100 and 94%61. Alternatively, another group analyzing grade 3–4 CRES in 53 adults proposed a model based on the baseline platelet count of <60 (or a mean corpuscular hemoglobin concentration of >33.2%) and a morphological presence of ALL in bone marrow, which predicted grade 3–4 CRES with 95% sensitivity and 70% specificity75.
Management of CRS and CRES
Generally, the successful management of these significantly adverse events include addressing three key points. Firstly, careful patient evaluation before CAR-T infusion is necessary as there are some patient-related risk factors, such as thrombocytopenia for CRS28,75 and pre-existing neurological conditions61 for CRES (as well as a high tumor burden for both of them28,61,75. Secondly, close attention to fever as the first sign of CRS along with frequent monitoring for CRES clinical signs using the CARTOX-10 tool for both outpatients30 and inpatients are strongly recommended. Thirdly, a supporting treatment with anti-IL6 or steroid therapy according to refs.30,31 has to be prompt. In agreement with Neelapu et al.31, it is reasonable to start anti-IL6 therapy early in CRES when accompanied by CRS of any grade in order to prevent toxicity progression. In the prescription information for FDA-approved CAR-Ts that are listed in Table 1, the only IL6-directed therapy mentioned for the treatment of CAR-T-induced CRS is Tocilizumab (anti-IL6R). It should be noted that other agents, such as anti-IL6 (siltuximab)76, anti-TNFα mAbs (infliximab), soluble TNFα receptor (etanercept), and IL-1R-based inhibitors (anakinra)30 were also used. However, the current algorithm that employs Tocilizumab (with or without steroids treatment), allows the reversal of most cases of CRS within FDA-approved products, as no CRS-related deaths were reported in ELIANA trial and a low figure of 4% for the ZUMA-1 trial. Administration of Tocilizumab demonstrated prompt attenuation of CRS clinical symptoms (including fever and hypotension)77. However, this agent does not penetrate the CNS and may conversely increase CSF levels of IL6 and thus contributing to CRES development. Consequently, the anti-IL6 agent Siltuximab may be of preference (for neurotoxicity management), as it does not increase serum IL6 levels61. Thus current strategies for the treatment of CRS and CRES include anti-IL6 therapy which is generally more effective in CRS management, and steroids, which are often used to treat CRES.
The modern option for toxicity control may also include the use of suicide genes that are introduced into CAR-T cells for their quick elimination (if needed) and CAR T-cells of such design have already been tested in a clinical setting6,11,78,79,80. Additionally, a number of novel target-therapy approaches may also soon hold great potential in improving efficacy and toxicity management. For example, there is a pre-clinical study of CAR-T with specificity to diverse antigens secreting anti-PD1 antibody for efficacy improvement purposes81. As IL-6 is a cytokine associated with high toxicity (which apparently does not interfere with CAR-T cell function and therapy efficacy), developing CAR T-cells secreting the IL6 receptor with impaired function or an anti-IL6 antibody may therefore be key to safe and effective therapy, while simultaneously by-passing the need for anti-IL6 prophylaxis.
In summary, management of either CRS or CRES is challenging because both these complications are associated with high peak levels of CAR T-cells in the blood28,61. On the other hand, high concentration of CAR-T is required for efficient therapy and hence, decreasing CAR T-cell levels would compromise the treatment efficacy28.
Other side effects of CAR T-cell therapy are summarized in the Table 4.
Main factors affecting toxicity
Analyzing the main factors that contribute to toxicity in CAR-T is complex, as previous trials have differed with respect to several features, such as CAR construct, disease type and lymphodepletion type. Consequently, we must be mindful of these differences particularly when comparing the outcomes from different trials. The factors affecting toxicity are summarized in Table 5.
Future perspectives of Car-T approach
One important area of CAR-T research, which requires further development, stems from the limited clinical availability of CAR-T therapy. Considerations here include high costs of therapy and administration delays related to the time-periods required for manufacturing CAR-T. Due to the production failure that occurs in 5–10% of patients and other reasons mentioned above, the idea of designing a universal CAR-T (known as UCART) is an area of rapid development. For example, in 2012, Torikai et al. reported a plausible approach to designing UCART by modifying T-cells in such a way that the latter not only expressed the second-generation CAR, but also lacked the expression of the endogenous T-cell receptor. Here, the UCART was able to kill CD19+ cells and proliferate in vitro82. Another successful approach for generating UCARTs was reported in 2017 by Qasim et al.83 These UCARTs were provided by Celectis (France) and were designed to silence the expression of the TCRA and CD52 genes that allowed using Alemtuzumab (an anti-CD52 antibody) during the lymphodepletion regimen to prevent host immunity-dependent killing of UCARTs. The efficacy of this approach was observed in two infants with relapsed ALL. They were treated with these UCARTs without any signs of serious CRS and neurotoxicity. Encouragingly, both children reached CR and were negative for minimal residual disease and remained in CR after 18 and 12 months post-UCART administration.
Expanding the repertoire of antigen targets for CAR-T is another viable approach. In this respect, universal anti-CD123 CAR-T (UCART123) therapy initiated by Cellectis (2017) was not as successful as the CD19 one. The first two patients treated in two clinical trials for AML and blastic plasmacytoid dendritic cell neoplasm (BPDCN) experienced sCRS and capillary leak syndrome (CLS), which contributed to the death of one of the patients84. A clinical trial by Stemline Therapeutics, targeting the same CD123 with a monoclonal antibody demonstrated a similar toxicity profile as 3 of 32 patients experienced grade 5 CLS (resulting in their death). However, the therapy was hailed as generally effective85. At the same time, the phase I trial targeting CD123 with auto CAR-T cells in AML and BPDCN patients did not reveal any CLS cases16 and is therefore possible that the UCART123 toxicity profile (in CLS) could be attributable either to the allogenic origin of cells or to other yet unknown factors.
Alternatively, Celyad is developing a CAR-T bearing Natural Killer Receptor-2 that can potentially target several antigens both in solid and hematological malignancies. In a phase I clinical trial designed to treat AML and MM (NCT02203825), an acceptable safety profile was observed with no evidence of CRS, TLS or off-tumor/on-target toxicity during the first two (out of four) dose levels. At the same time, no objective responses were reported, although there was an improvement in overall survival after the therapy86,87. Currently, a phase I clinical trial which enrolled patients with different cancers is ongoing (NCT03018405) and indicates encouraging initial efficacy giving disease stabilisation for 2 out of 3 patients with colon cancer, whereas 1 AML patient experienced morphological CR88,89.
Furthermore, other ‘universal’ targets for CAR-T cells include VEGF-1 (and VEGFR2), which are not only expressed on vascular endothelial cells, but also on the tumor cells of different lineages90. While positive pre-clinical91,92, including suppression of metastasis92 were reported for CAR-T VEGF-1, the trials using CAR-T anti-VEGFR2 (NCT01218867) failed to show significant clinical activity.
Conclusions
Undoubtedly, CAR-T therapy is one of the biggest recent breakthroughs in cancer therapy as it holds great potential and promise in the treatment of hematological malignancies and which may be an alternative to allo-HSCT for some patients93. In support of this, the long-term outcomes of patients who haven’t undergone allo-HSCT after CAR-Ts are eagerly awaited. At this moment, CAR-T therapy is still not fully functional against solid tumors and unfortunately, studies show little clinical benefit when extended into the clinical setting. As most of the pre-clinical models utilize immune-deficient animals, which fail to recapitulate the entire spectrum of interactions between immune cells, toxicities are therefore often observed in clinical trials for the therapies that were found to be safe in pre-clinical studies. To this end, great progress in understanding the molecular basis of toxicity has been instigated and which await further clinical validation. In support of this, experts in the field also agree on the necessity of developing customized prognostic scales for CAR-T-specific toxicity94, with several excellent scales having recently been reported28,47,61,75.
Looking forwards, indeed more work needs to be done to unravel the full potential of CAR-T-based anti-cancer therapy, but the clinical results of this therapy, achieved to date, offer great optimism and therefore further investigations are certainly justified and the outcomes of which are eagerly awaited.
References
Maude, S. L. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Till, B. G. et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 119, 3940–3950 (2012).
Brentjens, R. J. & Curran, K. J. Novel cellular therapies for leukemia: CAR-modified T cells targeted to the CD19 antigen. Hematol. Am. Soc. Hematol. Educ. Progr. 2012, 143–151 (2012).
Chang, L.-J. et al. 4SCAR19 chimeric antigen receptor-modified T cells as a breakthrough therapy for highly chemotherapy-resistant late-stage B cell lymphoma patients with bulky tumor mass. Blood 126, 264 (2015).
Chang, L.-J. et al. Safety and efficacy evaluation of 4SCAR19 chimeric antigen receptor-modified T cells targeting B Cell acute lymphoblastic leukemia—three-year follow-up of a multicenter phase I/II study. Blood 128, 587 (2016).
Chmielewski, M. & Abken, H. TRUCKs: the fourth generation of CARs. Expert Opin. Biol. Ther. 15, 1145–1154 (2015).
Chmielewski, M., Hombach, A. A. & Abken, H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 257, 83–90 (2014).
Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014).
Pan, J. et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia 31, 2587–2593 (2017).
Turtle, C. J. et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 126, 2123–2138 (2016).
Lee, D. W. et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 385, 517–528 (2015).
Davila, M. L. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B-cell acute lymphoblastic leukemia. Sci. Transl. Med. 6, 224ra25 (2014).
Park, J. H. et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 378, 449–459 (2018).
Ritchie, D. S. et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 21, 2122–2129 (2013).
Budde, L. et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial [oral abstract]. In ASH 2017 Proceedings. https://ash.confex.com/ash/2017/webprogram/Paper108199.html. Accessed 24 February 2018 (2017).
Fan, F. et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J. Clin. Oncol. 35, LBA3001 (2017).
Ahmed, N. et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T sells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 33, 1688–1696 (2015).
Louis, C. U. et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056 (2011).
Yong, C. S. M. et al. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 95, 356–363 (2017).
Holzinger, A., Barden, M. & Abken, H. The growing world of CAR T cell trials: a systematic review. Cancer Immunol. Immunother. 65, 1433–1450 (2016).
Hartmann, J., Schüßler‐Lenz, M., Bondanza, A. & Buchholz, C. J. Clinical development of CAR T cells—challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 9, 1183–1197 (2017).
Gauthier, J. & Yakoub-Agha, I. Chimeric antigen-receptor T-cell therapy for hematological malignancies and solid tumors: clinical data to date, current limitations and perspectives. Curr. Res. Transl. Med. 65, 93–102 (2017).
Schuster, S. J. et al. Primary analysis of Juliet: a global, pivotal, phase 2 trial of CTL019 in adult patients with pelapsed or pefractory diffuse large B-cell lymphoma. Blood 130(Suppl 1):577 (2017).
Abramson, J. S. et al. High durable CR rates in relapsed/refractory (R/R) aggressive B-NHL treated with the CD19-directed CAR T cell product JCAR017 (TRANSCEND NHL 001): defined composition allows for dose-finding and definition of pivotal cohort. In 2017 Annual American Society for Hematology Meeting, 9–12 Dec, Atlanta, GA (2017).
FDA. FDA briefing document oncologic drugs advisory committee meeting. BLA 125646. Tisagenlecleucel. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM566166.pdf. Accessed 22 Nov 2017.
BLA Clinical review memorandum. Yescarta. https://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM585388.pdf.
Hay, K. A. et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 130, 2295–2306 (2017).
Fitzgerald, J. C. et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit. Care Med. 45, e124–e131 (2017).
Lee, D. W. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195 (2014).
Neelapu, S. S. et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 151, 47–62 (2018).
Singh, N. et al. Monocyte lineage–derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy 19, 867–880 (2017).
Mosser, D. M. & Edwards, J. P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
Maude, S. L., Barrett, D., Teachey, D. T. & Grupp, S. A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 20, 119–122 (2014).
Tanaka, T., Narazaki, M. & Kishimoto, T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 8, 959–970 (2016).
Bonifant, C. L., Jackson, H. J., Brentjens, R. J. & Curran, K. J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 3, 16011 (2016).
Grupp, Sa et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518 (2013).
Tanaka, T., Narazaki, M., Masuda, K. & Kishimoto, T. Interleukin-6; pathogenesis and treatment of autoimmune inflammatory diseases. Inflamm. Regen. 33, 54–65 (2013).
Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247 (2012).
Obstfeld, A. E. et al. Cytokine release syndrome associated with chimeric-antigen receptor T-cell therapy: clinicopathological insights. Blood 130, 2569–2572 (2017).
Jones, G. W. et al. Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J. Immunol. 184, 2130–2139 (2010).
Dienz, O. & Rincon, M. The effects of IL-6 on CD4 T cell responses. Clin. Immunol. 130, 27–33 (2009).
Böttcher, J. P. et al. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep. 8, 1318–1327 (2014).
Briso, E. M., Dienz, O. & Rincon, M. Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J. Immunol. 180, 7102–7106 (2008).
Barrett, D. M. et al. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum. Gene Ther. 24, 717–727 (2013).
Yang, Y. et al. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 9, eaag1209 (2017).
Teachey, D. T. et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 6, 664–679 (2016).
Arico, M., Danesino, C., Pende, D. & Moretta, L. Pathogenesis of haemophagocytic lymphohistiocytosis. Br. J. Haematol. 114, 761–769 (2001).
Wang, Z. et al. Early diagnostic value of low percentage of glycosylated ferritin in secondary hemophagocytic lymphohistiocytosis. Int. J. Hematol. 90, 501–505 (2009).
Nabergoj, M. et al. Diagnostic and prognostic value of low percentage of glycosylated ferritin in acquired hemophagocytic lymphohistiocytosis: a single-center study. Int. J. Lab. Hematol. 39, 620–624 (2017).
Lambotte, O. et al. High ferritin and low glycosylated ferritin may also be a marker of excessive macrophage activation. J. Rheumatol. 30, 1027–1028 (2003).
Ma, A. D., Fedoriw, Y. D. & Roehrs, P. Hyperferritinemia and hemophagocytic lymphohistiocytosis. Single institution experience in adult and pediatric patients. Blood 120, 2135 (2012).
Ho, C. et al. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am. J. Clin. Pathol. 141, 62–71 (2014).
George, M. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J. Blood Med. 5, 69–86 (2014).
La Rosee, P. Treatment of hemophagocytic lymphohistiocytosis in adults. Hematology 2015, 190–196 (2015).
Kranick, S., Phan, G., Kochenderfer, J., Rosenberg, S. & Nath, A. Aphasia as a complication of CD19-targeted chimeric antigen receptor immunotherapy (S52.006). Neurology 82, S52.006 (2014).
Uckun, F. F. M. et al. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood 71, 13–29 (1988).
Tissue expression of CD19—Summary: the human protein atlas. https://www.proteinatlas.org/ENSG00000177455-CD19/tissue. Accessed 27 Nov 2017.
Brown, C. E. et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 21, 4062–4072 (2015).
Fry, T. J. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 24, 20–28 (2017).
Gust, J. et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 7, 1404–1419 (2017).
Felger, J. C. & Lotrich, F. E. Inflammatory cytokines in depression: neruobiological mechanisms and therapeutic implications. Neuroscience 246, 199–229 (2013).
Hu, Y. et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J. Hematol. Oncol. 9, 70 (2016).
Abramson, J. S. et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. N. Engl. J. Med. 377, 783–784 (2017).
Johnson, K. Another win for CAR-T therapy | Medpage today. https://www.medpagetoday.com/meetingcoverage/additionalmeetings/64895. Accessed 25 Oct 2017 (2017).
van der Heyde, H. C., Nolan, J., Combes, V., Gramaglia, I. & Grau, G. E. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22, 503–508 (2006).
Grau, G. E. et al. Tumor necrosis factor and disease severity in children with falciparum malaria. N. Engl. J. Med. 320, 1586–1591 (1989).
Grau, G. E. et al. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc. Natl Acad. Sci. USA 86, 5572–5574 (1989).
Grau, G. E. et al. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 237, 1210–1212 (1987).
Lyke, K. E. et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1b), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in malian children with severe plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect. Immun. 72, 5630–5637 (2004).
Bauer, P. R., Heyde, H. C., Sun, G., Specian, R. D. & Granger, D. N. Regulation of endothelial cell adhesion molecule expression in an experimental model of cerebral malaria. Microcirculation 9, 463–470 (2002).
Finney, C. A. et al. S1P is associated with protection in human and experimental cerebral malaria. Mol. Med. 17, 717–725 (2011).
Higgins, S. J. et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci. Transl. Med. 8, 358ra128 (2016).
Gilbert, M. J. Severe neurotoxicity in the phase 2 trial of JCAR015 in adult B-ALL (ROCKET Study): analysis of patient, protocol and product attributes [oral report]. In 32nd Annual Meeting and Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2017): Part One National Harbor, MD, USA. 8–12 November 2017. http://www.junotherapeutics.com/wp-content/uploads/2017/12/Gilbert-2017-SITC-Annual-Meeting-Oral-Plenary-Presentation_FINAL.pdf.
Park, J. H. et al. Baseline and early post-treatment clinical and laboratory factors associated with severe neurotoxicity following 19-28z CAR T cells in adult patients with relapsed B-ALL. J. Clin. Oncol. 35, 7024–7024 (2017).
Chen, F. et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J. Immunol. Methods 434, 1–8 (2016).
Kroschinsky, F. et al. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care. 21, 89 (2017).
Turtle, C. J. et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J. Clin. Oncol. 35, 3010–3020 (2017).
Turtle, C. J. et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 8, 355ra116 (2016).
Abramson, J. S. et al. CR rates in relapsed/refractory (R/R) aggressive B-NHL treated with the CD19-directed CAR T-cell product JCAR017 (TRANSCEND NHL 001). J. Clin. Oncol. 35, 7513 (2017).
Li, S. et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin. Cancer Res. 23, 6982–6992 (2017).
Torikai, H. et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119, 5697–5705 (2012).
Qasim, W. et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 9, eaaj2013 (2017).
Taylor, N. P. FDA hits Cellectis’ off-the-shelf CAR-T program with clinical hold after first patient treated in phase 1 trial dies | FierceBiotech. http://www.fiercebiotech.com/biotech/fda-hits-cellectis-off-shelf-car-t-program-clinical-hold-after-first-patient-treated-phase. Accessed 20 Oct 2017.
Pemmaraju, N. et al. Ongoing phase 2 clinical trial of SL-401 in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN): stage 1 and stage 2 results safety and tolerability clinical activity. In EHA Meet (2017).
Demoulin, B. et al. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Futur. Oncol. 13, 1593–1605 (2017).
Nikiforow, S. et al. A first-in-human phase I trial of NKG2D chimeric antigen receptor-T cells in AML/MDS and multiple myeloma. J. Clin. Oncol. 34 (15_suppl), TPS3102 (2017).
Fernández, C. R. Celyad reports impressive preliminary CAR-T results in solid tumors. https://labiotech.eu/celyad-car-t-solid-tumor/. Accessed 25 Oct 2017 (2017).
Celyad reports a first complete response in a relapsed refractory AML patient in the THINK trial. https://globenewswire.com/news-release/2017/10/03/1139875/0/en/Celyad-reports-a-first-complete-response-in-a-relapsed-refractory-AML-patient-in-the-THINK-trial.html. Accessed 30 Dec 2017.
Schwartz, J. D., Rowinsky, E. K., Youssoufian, H., Pytowski, B. & Wu, Y. Vascular endothelial growth factor receptor-1 in human cancer. Cancer 116, 1027–1032 (2010).
Chinnasamy, D. et al. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 73, 3371–3380 (2013).
Wang, W. et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. 20, 970–978 (2013).
Kate, J. CAR T-cell therapy: getting a handle on toxicity. https://www.medscape.com/viewarticle/864780. Accessed 31 Dec 2017.
Predicting the risk and severity of cytokine release syndrome in patients receiving CAR T-cell therapy—ASH clinical news. https://www.ashclinicalnews.org/news/predicting-risk-severity-cytokine-release-syndrome-patients-receiving-car-t-cell-therapy/. Accessed 31 Dec 2017.
Hill, J. A. et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T cell immunotherapy. Blood 131, 121–130 (2017).
Inman, S. JCAR015 experience informs future CAR-T studies. http://www.onclive.com/web-exclusives/jcar015-experience-informs-future-cart-studies. Accessed 8 Feb 2018.
DeAngelo, D. J. et al. Clinical outcomes for the phase 2, single‐arm, multicenter trial of JCAR015 in adult B‐ALL (ROCKET Study). In 32nd Annual Meeting and Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2017): Part One National Harbor, MD, USA. 8–12 November 2017. Annual meeting abstract book 305 (2017).
Frisén, L. Swelling of the optic nerve head: a staging scheme. J. Neurol. Neurosurg. Psychiatry 45, 13–18 (1982).
Brudno, J. N. et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J. Clin. Oncol. 34, 1112–1121 (2016).
Donahue, R. E. et al. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J. Exp. Med. 176, 1125–1135 (1992).
Cooper, A. R. et al. Cytoreductive conditioning intensity predicts clonal diversity in ADA-SCID retroviral gene therapy patients. Blood 129, 2624–2635 (2017).
Braun, C. J. et al. Gene therapy for Wiskott-Aldrich syndrome—long-term efficacy and genotoxicity. Sci. Transl. Med. 6, 227ra33–227ra33 (2014).
Ott, M. G. et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 12, 401–409 (2006).
Mukherjee, S. & Thrasher, A. J. Gene therapy for PIDs: progress, pitfalls and prospects. Gene 525, 174–181 (2013).
Six, E. et al. LMO2 associated clonal T cell proliferation 15 years after gamma-retrovirus mediated gene therapy for SCIDX1. In 20th ASGCT Annual Meeting (Washington, DC) (2017).
Zhang, Y. et al. An analytical biomarker for treatment of patients with recurrent B-ALL after remission induced by infusion of anti-CD19 chimeric antigen receptor T (CAR-T) cells. Sci. China Life Sci. 59, 379–385 (2016).
Porter, D. L. et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7, 303ra139 (2015).
Dai, H. et al. Tolerance and efficacy of autologous or donor-derived T cells expressing CD19 chimeric antigen receptors in adult B-ALL with extramedullary leukemia. Oncoimmunology 4, e1027469 (2015).
Namuduri, M. & Brentjens, R. J. Medical management of side effects related to CAR T cell therapy in hematologic malignancies. Expert Rev. Hematol. 9, 511–513 (2016).
Rasool, M. et al. Development of tumor lysis syndrome (TLS): A potential risk factor in cancer patients receiving anticancer therapy. Bioinformation 10, 703–707 (2014).
Mirrakhimov, A. E., Voore, P., Khan, M. & Ali, A. M. Tumor lysis syndrome: a clinical review. World J. Crit. Care Med. 4, 130–138 (2015).
Coiffier, B., Altman, A., Pui, C.-H., Younes, A. & Cairo, M. S. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J. Clin. Oncol. 26, 2767–2778 (2008).
Jones, G. L., Will, A., Jackson, G. H., Webb, N. J. A. & Rule, S. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br. J. Haematol. 169, 661–671 (2015).
Rouce, R. H. et al. Safety of multiple doses of CAR T cells. Blood 126, 4425 (2015).
Maus, M. V. et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 1, 26–31 (2013).
Kershaw, M. H. et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 12, 6106–6115 (2006).
Jensen, M. C. et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 16, 1245–1256 (2010).
Kochenderfer, J. N. et al. Donor-derived anti-CD19 chimeric-antigen-receptor-expressing T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 122, 151 (2013).
Lamers, C. H. J. et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol. 24, e20–e22 (2006).
Turtle, C. J. et al. Anti-CD19 chimeric antigen receptor-modified T cell therapy for B cell non-Hodgkin lymphoma and chronic lymphocytic leukemia: fludarabine and cyclophosphamide lymphodepletion improves in vivo expansion and persistence of CAR-T cells and clinical outcomes. Blood 126, 184 (2015).
Morgan, R. A. et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010).
Cameron, B. J. et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3 directed Tcells. Sci. Transl. Med. 5, 197ra103 (2013).
Long, A. H. et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 21, 581–590 (2015).
Abramson, J. et al. High CR rates in relapsed/refractory (R/R) agressive B-NHL treated with the CD19-directed CAR T cell product JCAR017 (TRANSCEND NHL 001). Hematol. Oncol. 35, 138–138 (2017).
Frey, N. V. et al. Optimizing chimeric antigen receptor T cell therapy for adults patients with relapsed or refractory acute lymphoblastic leukemia. In ASCO Meeting Abstracts Abstract 7002. doi:10.1200/JCO.2016.34.15_suppl.7002 (2016).
Stenger, M. Interleukin-15 levels associated with lymphoma response to CAR T-cell treatment. http://www.ascopost.com/News/55461.
Acknowledgements
We thank Andrey Gorchakov, Elena Vasileva, and Marina Konopleva for their help and productive discussion. The study was funded by the research grant #17-34-50167 mol_nr from the Russian Foundation for Basic Research (RFBR), RSCF #14-15-00816 to A.P and N.B. O.S., S.S., G.M., and M.P. also acknowledge the grant support from the Russian Government Program for the Recruitment of the leading scientists into the Russian Institutions of Higher Education 14.W03.31.0029.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by I. Amelio
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Titov, A., Petukhov, A., Staliarova, A. et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis 9, 897 (2018). https://doi.org/10.1038/s41419-018-0918-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0918-x
This article is cited by
-
Molecular understanding and clinical outcomes of CAR T cell therapy in the treatment of urological tumors
Cell Death & Disease (2024)
-
Current Insights into CAR T-Cell-Based Therapies for Myelodysplastic Syndrome
Pharmaceutical Research (2024)
-
Predictive value of pre-infusion tumor growth rate for the occurrence and severity of CRS and ICANS in lymphoma under CAR T-cell therapy
Annals of Hematology (2024)
-
Molecular and therapeutic effect of CRISPR in treating cancer
Medical Oncology (2023)
-
CAR-T Cell Therapy: the Efficacy and Toxicity Balance
Current Hematologic Malignancy Reports (2023)
