
Abstract
Cholangiocarcinoma (CCA) is the as the most frequently observed biliary tract malignancy, which has low survival rate in addition to constrained treatment options; nevertheless, the fundamental molecular phenomenon underlying malignant progression of CCA is quite ambiguous. Recently long non-coding RNAs (lncRNAs) have been found to have significant regulatory functions in several human cancers. Herein, we have figured out that lncRNA SNHG1, with substantially enhanced expression in CCA, is capable of acting as the oncogenic molecule of CCA. As revealed by our data, SNHG1 knockdown extensively inhibited CCA cell migration as well as proliferation in vitro and in vivo. In addition, in accordance with the findings of the RNA-Seq analysis, SNHG1 knockdown exhibited a significant impact on the target genes that were linked to cell migration and regulation of cell proliferation, in addition to the apoptotic phenomenon. In a mechanistic manner, we also showed that SNHG1 bound to the histone methyltransferase enhancer of the zeste homolog 2 (EZH2, which is regarded as the catalytic subunit of the polycomb repressive complex 2 (PRC2), which is an extremely conserved protein complex regulating gene expression with the help of methylating lysine 27 on histone H3), specifying the histone alteration pattern on the target genes, including CDKN1A, and, as a result, altered the CCA cell biology. These data verified a major function of the epigenetic regulation of SNHG1 in CCA oncogenesis, in addition to its likely function as a target for CCA interruption.
Similar content being viewed by others


Introduction
Cholangiocarcinoma (CCA), the most frequently observed biliary tract cancer, encompasses 3% of all the gastrointestinal malignancies, in addition to being suggested as the malignancy that stems from ductal epithelial cells1,2. Improved knowledge of the carcinogenesis is critically important for the advancement of diagnostic markers, together with developing the innovative and productive therapies for CCA patients. The current modalities meant to establish a CCA diagnosis are insufficient, since it is still considered to be an uphill task to detect the ailment at an an early primary phase for enabling possibly therapeutic surgical treatments. Developing innovative biomarkers requires additional research on the DNA-methylation markers, in addition to peptide panels and non-coding RNAs.
Owing to the recent developments in deep-sequencing technologies, there are a number of previously unknown transcripts that have resulted in the identification. The majority (>99%) of these transcripts are regarded as long non-coding RNAs (lncRNAs), which are defined by their length of >200 nucleotides, which is found in a large number of RNA families; moreover, lncRNAs possess constrained protein-coding potential, while lacking the identifiable open reading frames, which are considered quite essential for assessing the protein-coding potential3,4,5,6,7. Recently, lncRNAs have been demonstrated to be capable of serving as pivotal regulators in numerous biological developments, for instance, cellular proliferation, development, differentiation, etc. Nonetheless, several lncRNAs have been uncharacterized; moreover, lncRNAs in CCA continue to a growing sphere of study, since few numbers of lncRNAs have been characterized in CCA tumorigenesis.
Abnormal expression of lncRNAs is likely to be involved in multiple aspects associated with human health and diseases, which include cancer, in particular, CCA8,9,10,11,12,13,14. Among them, small nucleolar RNA host gene 1 (SNHG1, also called UHG; U22HG; lncRNA16; LINC00057; NCRNA00057) has garnered our attention. Studying the invasive pathophysiology of SNHG1 together with its overexpression is considered to be an effective predictor of oncogenesis in multifarious kinds of cancer, including esophageal squamous cell carcinoma15, lung squamous cell carcinoma16, hepatocellular carcinoma17,18, colorectal cancer19,20, and gastric cancer21. Nonetheless, finding out whether SNHG1 is capable of serving as an “oncogene” in CCA is considered worthy enough.
In this research work, we have detected the lncRNA SNHG1, which, owing to its substantially enhanced expression in CCA, is capable of acting as an oncogenic molecule of CCA. In addition, SNHG1 bound to the histone methyltransferase enhancer of the zeste homolog 2 (EZH2, which is a catalytic subunit of polycomb repressive complex 2 (PRC2), an extremely conserved protein complex regulating gene expression with the help of methylating lysine 27 on histone H3), followed by epigenetically suppressing CDKN1A (an inhibitor of the cyclin-dependent kinase, in addition to being a tumor-suppressive gene, which is used in treatment of several cancers22,23,24) expression in CCA cells, and, as a result, altered the CCA cell biology. Altogether, we shed light on the fact that SNHG1 is capable of acting as an oncogenic molecule of CCA.
Results
SNHG1 is upregulated in human CCA tissues
When a detailed characterization of expressed lncRNAs in CCA, analysis of The Cancer Genome Atlas CCA, as well as typical tissue RNA-Sequencing data (including 9 normal as well as 36 cancer specimens) in addition to one independent microarray dataset from the Gene Expression Omnibus (GSE76297; 92 cancer tissue specimens as well as 91 normal tissue specimens), revealed the fact that SNHG1 expression is augmented in tumor tissues in comparison with the nearby tissues (Fig. 1a, b). For the verification of the informatic data, SNHG1 expression in a group consisting of 17 pairs of CCA tumor and adjacent tissues was figured out using quantitative real time -PCR (qRT-PCR), whereby it was validated that SNHG1 exhibited remarkable expression levels in carcinoma tissues when compared with adjacent tissues (Fig. 1c). When compared with normal human intrahepatic biliary epithelial cells (HIBEpiC), SNHG1 expression was higher in CCA cell lines (Fig. 1d). As suggested by these findings, SNHG1 might have the potential to act as an “oncogene” for the promotion of CCA growth.

a Hierarchical clustering analysis of lncRNAs that were differentially expressed (fold change > 2; P < 0.05) in CCA tissues and normal tissues from TGCA database. b SNHG1 is overexpressed in GEO datasets (GSE76297). c SNHG1 was detected in 17 pairs of CCA tissues by qRT-PCR. The levels of SNHG1 in CCA tissues are significantly higher than those in non-tumorous tissues. d SNHG1 expression was analyzed by qRT-PCR in two CCA cell lines (HuCCT1 and RBE), compared with the normal human intrahepatic biliary epithelial cells (HIBEpiC). Error bars indicate means ± SD.**P < 0.01;****P < 0.0001.
Knockdown of SNHG1 in CCA cell lines inhibits cell proliferation and migration
While investigating the impact of increased SNHG1 in CCA, at first, as evident from Fig. 2a, qRT-PCR findings suggested that the expression of SNHG1 in the small interfering RNA (siRNA)-mediated knockdown group was substantially lowered, in comparison with the si-SC (scramble negative control) group, in the HuCCT1 and RBE cell lines (Fig. 2a). The clonogenic formation number exhibited a significant decline with the knockdown of SNHG1 in the two cell lines (Fig. 2b). In addition to that, as suggested by the CCK8 assays, the knockdown of SNHG1expression disallowed the cell feasibility in HuCCT1 and RBE cell lines, when compared with the control cells (Fig. 2c). Thereafter, as revealed by the Transwell assays, the knockdown of SNHG1 exhibited dramatic repression of the migration of cells (Fig. 2d). As suggested by these data, the SNHG1 performs a quintessential function in CCA cancer cell proliferation as well as migration.

a qRT-PCR detected the expression of SNHG1 after siRNA transfection in HuCCT1 and RBE cell lines. b Colony formation assays were used to determine the cell colony formation ability of si-SNHG1-transfected cells. c CCK8 assays were used to determine the cell viability of si-SNHG1-transfected cholangiocarcinoma cells. d Transwell assays showed that SNHG1 knockdown inhibited cholangiocarcinoma cell migration.Error bars indicate means ± SD. *P < 0.05; **P < 0.01;***P < 0.001;****P < 0.0001.
Depletion of SNHG1 leads to increased cell apoptosis and delayed cell cycle in CCA cell lines
For the purpose of additionally investigating whether the SNHG1 is capable of affecting the apoptosis of CCA cell lines, flow cytometry was carried out as well. As suggested by key observations, HuCCT1 and RBE cell lines transfected with SNHG1 siRNA manifested higher apoptotic rate in comparison with the control group (Fig. 3a). Subsequent to that, for the purpose of deciding on whether the impacts of SNHG1 on CCA cell proliferation and apoptosis are the key observations of the SNHG1-mediated alternations in the progression of cell cycle, we carried out the flow cytometry assay in HuCCT1 and RBE cell lines. As highlighted by the flow cytometry assay, the SNHG1 knockdown gave rise to increased G0/G1 phase together with decreased S and G2/M phases, as compared with the control cells (Fig. 3b). Considered collectively, SNHG1 is capable of accelerating cell proliferation, in addition to inhibiting apoptosis and regulating cell cycle progression of CCA cancer cell lines.

a FACS analysis of the effect of SNHG1 on cell apoptosis. b FACS analysis of the effect of SNHG1on cell cycle analysis. Error bars indicate means ± SD.*P < 0.05; **P < 0.01;***P < 0.001;n.s. not significant.
Knockdown of SNHG1 inhibits CCA cell tumorigenesis in vivo
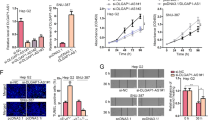
For the purpose of additionally confirming whether the SNHG1 exerts an impact on the tumorigenesis of CCA in vivo, HuCCT1 cells transfected with sh-SNHG1 or control vector were injected into nude mice. On the 16th day following the injection, tumors that developed in the sh-SNHG1 cohort were remarkably smaller in comparison with those in the control cohort (Fig. 4a, b). Likewise, the average tumor volumes together with the respective weights in the ultimate experiment were apparently lower in the sh-SNHG1cohort as compared with the control vector cohort (Fig. 4c, d). As revealed by our findings, the silencing of SNHG1 is capable of repressing the CCA tumor progression in vivo, suggesting the fact that SNHG1 performs a substantial function in CCA tumor proliferation.

a, b Scramble or sh-SNHG1was stably transfected into HuCCT1 cells, which were injected into the nude mice. c Tumor volumes were calculated after injection every 4 days. Bars indicate standard deviation (S.D.). d Tumor weights are represented as means of tumor weights ± SD.*P < 0.05.
Related target genes of SNHG1 in CCA
For the purpose of specifying the target genes that are capable of being regulated by SNHG1 in CCA, RNA transcriptome sequencing was carried out in controls or siRNAs. A frequent set of 971 mRNAs revealed ≥1.5-fold increased abundance; conversely, 546 genes exhibited a decline in abundance (≤1.5-fold) owing to the silencing SNHG1 (Fig. 5a, Supplementary Table S2). A thorough research of the ontology analysis highlighted the most distinct over-represented biological phenomena with the involvement of pathways in cell migration together with regulation of cell growth and apoptotic phenomenon (Fig. 5b). For the purpose of prioritizing most SNHG1-associated genes, attention was given to the genes that were most extensively expressed with knockdown of SNHG1. Prospectively, among the most highly expressed genes, a number of renowned genes associated with proliferation and migration (e.g., IL32, IL11, LRIG1, CMTM3, CDKN1A, PIK3IP1, CD82, G0S2, GDF15, and ADAM19, et al.) are included. Verification of some of these genes was carried out using qRT-PCR, subsequent to the knockdown of SNHG1 in HuCCT1 and RBE cells (Fig. 5c, d).

a Mean-centered, hierarchical clustering of 1295 transcripts altered (≥1.5-fold change) in siNC-treated cells and siRNA SNHG1-treated cells with three repeats. b Gene ontology analysis for all genes with altered expressions. c, d The altered mRNA levels of genes were selectively confirmed by qRT-PCR in knockdown SNHG1. Error bars indicate means ± SD.*P < 0.05; **P < 0.01;***P< 0.001;****P < 0.0001.
SNHG1 epigenetically silenced CDKN1A transcription through EZH2-mediated H3K27me3 demethylation
There are reports that a number of lncRNAs have been validated for working through the co-operation with chromatin-modifying enzymes for the purpose of accelerating epigenetic activation and, accordingly, silencing of the target gene expression25. In particular, PRC2, which is a classical methyltransferase, constituted with EZH2, in addition to EED and SUZ12, is capable of serving as a catalyst not only in the di- but also in the trimethylation of lysine residue 27 of histone 3 (H3K27me3), and, accordingly, performing epigenetic repression of the expression of the target genes26,27. In our research work, for the purpose of investigating the system for SNHG1-mediated regulation, first, subcellular fractionation location assays were performed to verify the localization of SNHG1 mainly in the nucleus mainly (Fig. 6a). Additionally, the interation probabilies of EZH2 and SNHG1 were identified on the website (http://pridb.gdcb.iastate.edu/RPISeq/index.html); moreover, as revealed by the findings, EZH2 is capable of binding with the SNHG1 well (Fig. 6b). As evident from Fig. 6c, amplification of endogenous SNHG1was observed in the anti-EZH2 RNA immunoprecipitation (RIP) fraction associated with the input, when compared with the IgG fraction in the HuCCT1 and RBE cell lines. Then, RNA pull-down assays demonstrated that labeled SNHG1 RNA, but not non-labeled RNA, specially retrieved EZH2 from HuCCT1 nuclear extract (Fig. 6d, e). Considered collectively, these results validated the fact that SNHG1 was capable of interacting with EZH2.

a After nuclear and cytosolic separation, RNA expression levels were measured by qRT-PCR. GAPDH was used as a cytosol marker and U1 was used as a nucleus marker. b The interation probabilies of EZH2 and SNHG1 were detected on the website, and the results showed that EZH2 could well bind with SNHG1 well (http://pridb.gdcb.iastate.edu/RPISeq/index.html). c RIP experiment for EZH2 was performed and the co-precipitated RNA was subjected to qRT-PCR for SNHG1. d Schematic of the RNA pull-down experiment for the identification of proteins associated with SNHG1. e In vitro transcribed, pull-down assays showed that desthiobiotinylated SNHG1 could retrieve EZH2 in HuCCT1 cells, but not GAPDH. GAPDH was a negative control. f The methylation-related genes were detected by qRT-PCR in HuCCT1 and RBE cell lines after knockdown of EZH2. g The correlation between EZH2 and CDKN1A expression was detected by analyzing GSE76297 data. h The correlation between SNHG1 and CDKN1A expression was detected by analyzing GSE76297 data. i The altered protein levels of CDKN1A were selectively confirmed by western blot in knockdown of SNHG1. j The altered protein level of CDKN1A was selectively confirmed by western blot in knockdown EZH2. k Immunofluorescence was done to explore if the expression of CDKN1A was changed by knockdown of SNHG1 in vivo. l, m ChIP of EZH2 and H3K27me3 of the promoter region of CDKN1A locus after siRNA treatment targeting si-SC and si-SNHG1 1# in HuCCT1 cells; qPCR was performed to detect the quantitation of ChIP assays. Enrichment was quantified relative to input controls. Antibody directed against IgG was used as a negative control. Error bars indicate means ± SD. *P < 0.05; **P < 0.01;***P < 0.001;****P < 0.0001; n.s. not significant.
Subsequent to that, we tested whether EZH2 was capable of coregulating the suppression of these SNHG1-suppressed genes through a bond with SNHG1. Primarily, we investigated the expression of SNHG1-suppressed genes with by knockdown or not of EZH2, using RT-qPCR. In addition, as revealed by the findings, the SNHG1-suppressed genes exhibited an increase through the knockdown of EZH2, as well (Fig. 6f), in the HuCCT1 and RBE cell lines.
Correlation analysis of the dataset GSE76297 (92 pairs of cancer and 91 normal tissue samples) from the Molecular Signature Database suggested that CDKN1A was substantially negatively correlated with EZH2 (Fig. 6g) and SNHG1 (Fig. 6h). In addition, the protein level of CDKN1A exhibited an increase through the knockdown of SNHG1 (Fig. 6i) and EZH2 (Fig. 6j). Simultaneously, immunofluorescence was done to explore whether the expressions of EZH2 and CDKN1A were changed with the knockdown of SNHG1 in vivo. The results showed that when SNHG1 was knocked down the expression of CDKN1A was improved (Fig. 6k), while the expression of EZH2 showed no obvious change (Figure S1 A).
To summarize, we shed light on one of the coregulating genes by SNHG1 as well as EZH2 in CCA, CDKN1A, based on the report that the hypermethylation of the CDKN1A promoter region contributed to CDKN1A transcription inactivation in breast cancer28. In addition, as revealed by more and more evidences, EZH2 directly targets the CDKN1A promoter in the neural progenitors; moreover, its activity exhibits correlation with the modifications in the H3K27me3 levels29. The EZH2 blockade by RNA interruption hampers the proliferation of the ovarian cancer through the facilitation of re-expression of p2130,31.
Furthermore, to additionally determine whether SNHG1 suppressed the expression of CDKN1A through interaction with the EZH2, chromatin immunoprecipitation (ChIP) analysis was conducted. As suggested by the ChIP assays, knockdown of SNHG1 lowered the binding of EZH2, together with the H3K27me3 levels all through the promoters of CDKN1A (Fig. 6l, m). These findings validated the fact that EZH2 is capable of directly binding to the promoter of CDKN1A, followed by repressing the CDKN1A expression directly through mediation of the H3K27me3 demethylation modification.
As discovered by our findings, knockdown of the SNHG1 lowered the binding of EZH2 as well as H3K27 trimethylation levels all through the promoter of the CDKN1A, resulting in the augmented level of the CDKN1A that was capable of decelerating the growth of CCA. As suggested by our findings, SNHG1 promotes CCA malignancy through a bond with EZH2, followed by repressing the expression of the CDKN1A epigenetically in the nucleus.
CDKN1A is a bona target of SNHG1, and CDKN1A overexpression suppresses CCA cell proliferation and metastasis
CDKN1A, considered to be among the most CKIs, is an integral checkpoint of the P53 signaling pathway with respect to G1/S transition through the inhibition of the function of kinases, for instance, CyclinD/CDK4, CyclinD/CDK6, as well as CyclinE/CDK232,33, playing different kinds of function to inhibit the cell progression in typical as well as cancer cells, in addition to being downregulated in various cancers34,35,36,37. However, no report has proved that CDKN1A is a tumor suppression gene of CCA.
First, for the assessment of CDKN1A expression in CCA tissues, we carried out the analysis of the dataset GSE76297 (92 pairs of cancer and 91 normal tissue samples) and figured out that CDKN1A had lower expression in the cancer tissues in comparison with the typical tissues in CCA (Fig. 7a). Subsequent to that, by qRT-PCR, we found that the CDKN1A expression is lower in CCA tumor tissues in comparison with the neighboring ones through the detection of the expression in a cohort of 17 pairs of CCA tumor tissues in comparison with the nearby tissues using qRT-PCR (Fig. 7b). In addition, we also found that overexpression of CDKN1A is capable of substantially suppressing proliferation and invasion of HuCCT1 and RBE cell lines; moreover, overexpression of CDKN1A is capable of partially reversing the SNHG1-mediated progression and migration promotion (Fig. 7c–f).

a Expression level of CDKN1A in cholangiocarcinoma by analysis of GES76297 data. b CDKN1A was detected in 17 pairs of CCA tissues by qRT-PCR. c–f HuCCT1 and RBE cells transfected with vector/pcDNA-CDKN1A/pcDNA-SNHG1 and with SNHG1 followed by transfection with CDKN1A. After transfection, cells were analyzed by CCK8 assays (c, d) and Transwell assays (e, f). g Proposed model of SNHG1 regulating CDKN1A expression to promote CCA tumor growth. Error bars indicate means ± SD. *P < 0.05; **P < 0.01; n.s. not significant.
In summary, our study demonstrated the regulatory mechanism in tumorigenesis of SNHG1, by which SNHG1 could promote malignancy of CCA by binding to histone methyltransferase EZH2 (the catalytic subunit of the PRC2, a highly conserved protein complex that regulates gene expression by methylating lysine 27 on histone H3), and then specified the histone modification pattern on the target genes, including CDKN1A, thus explaining cell survival and metastasis of CCA (Fig. 7g).
Discussion
During the previous decades, more and more evidence has laid emphasis on the rising importance of lncRNAs in diversified human cancers, which include CCA9,10,11,12,14. LncRNAs exhibit an evident benefit associated with their relative tissue-specific expression, as well as the functional structure at the transcriptional levels. Nevertheless, lncRNAs in CCA are still considered to be part of a growing sphere, since there are just few lncRNAs that have been linked to CCA tumorigenesis, requiring further investigations to be considered. By this study we have discovered that the expression of SNHG1, an lncRNA, leads to substantial upregulation of the CCA tissues.
The dysregulation of lncRNAs is linked with an extensive array of pathological mechanisms; conversely, the mechanisms of lncRNA expression are quite unclear and require additional investigation. As indicated by our data, knockdown of SNHG1 expression substantially inhibits cell growth in vitro and vivo. Moreover, SNHG1 has manifested oncogenic attribute in different kinds of cancers; nevertheless, the genes influenced by the SNHG1 continue to remain uncertain. With the help of RNA-Seq, we not only observed the fact that the findings of gene ontology analysis were, in particular, associated with cell migration and regulation of cell progression, together with apoptotic mechanism, but also figured out several target genes that were regulated by the SNHG1. The latest research works have highlighted that some lncRNAs perform the epigenetic regulation of gene expression with the help of DNA methylation as well as histone modifications, containing the methylation, in addition to functions such as acetylation and phosphorylation38,39,40. As discovered by our findings, SNHG1 is capable of interacting with EZH2, which is considered to be a type of histone methylation modification complex in the nucleus, thus regulating an array of the target gene’s expression, as shown by the mounting evidences.
CDKN1A, one of the genes regulated by SNHG1 in accordance with the RNA-Seq, is capable of exhibiting the role of tumor suppressor genes in different kinds of cancer35,36,37. Furthermore, there are reports that hypermethylation of the CDKN1A promoter region contributes to the CDKN1A transcription inactivation in breast cancer28. Nevertheless, the function of CDKN1Ain the tumorigenesis of CCA continues to be uncertain. As discovered by our findings, SNHG1is capable of binding to EZH2, which is a kind of histone methylation modification complex in the nucleus, thus suppressing the target gene’s expression, which includes CDKN1A, an innovative tumor suppressor in CCA.
To summarize, as suggested by our research works, the regulatory mechanism in the tumorigenesis of SNHG1, by which SNHG1 could promote malignancy of CCA through regulating the transcription of CDKN1A epigenetically in the nucleus, facilitates cell survival and metastasis of CCA. Ultimately, fast-paced developments in the oligonucleotide/nanoparticle technology bring forth optimism for the delivery of siRNA-oriented therapeutics for the regulation of the lncRNA levels in vivo. As highlighted by our key observations, there is a need of additional research dealing with the capability of SNHG1 as an informative biomarker as well as a therapeutic target in patients with CCA.
Materials and methods
Tissue gathering and ethics statement
Altogether 17 samples were analyzed in this research, and all were subjected to resection of the CCA at the Second Affiliated Hospital of Nanjing Medical University. All the specimens were instantly frozen in tubes with RNAlater preservation liquid after being removed and were kept in liquid nitrogen till the extraction of RNA. Our research was permitted by the Ethics Committee of Nanjing Medical University (Nanjing, Jiangsu, PR China) with written consent from every patient.
RNA extraction as well as qRT-PCR analyses
All RNAs were obtained from the cultured cells or specimens with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). For RT-qPCR, 1 μg of RNA was reverse transcribed with cDNA using a Reverse Transcription Kit (Takara, Tokyo, Japan). Real-time PCR analyses were carried out with SYBR Green (Takara, Tokyo, Japan). The findings were regulated to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences are shown in Supplementary Table S1.
Cell culture
CCA cell lines (HuCCT1 and RBE) and normal HIBEpiC were obtained from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China). All the cell lines were maintained in Dulbecco’s modified Eagle medium (Life Technologies Corporation Attn, Grand Island, USA) coupled with 10% fetal bovine serum (FBS; Sciencell, Carlsbad, CA), 100 mg/ml streptomycin, and 100 U/ml penicillin (Invitrogen, Shanghai, China) in humidified air at 37 °C with 5% CO2.
Transfection of cell lines
CCA cells were planted at six-well plates as well as transfected with particular siRNA (100 nM) or scramble negative control siRNA (100 nM) the next day using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) on the basis of the producer’s protocol. Scrambled negative control siRNA (si-SC) was bought from Invitrogen (Invitrogen, Carlsbad, CA, USA). sh-SNHG1 was cloned into pENTRTM/U6 vector; and the orders of siRNAs and shRNA are summed up in Supplementary Table S1.
Subcellular fractionation address
The division of nuclear as well as cytosolic fractions was constructed with the PARIS Kit (Life Technologies, Carlsbad, CA, USA) following the producer’s guides.
Cell proliferation analysis
Cell viability was monitored with CCK8 kit (Houston TX, USA) following the producer’s suggestions. The HuCCT1 and RBE cells transfected with si-SNHG1 or si-SC (scramble negative control) (3000 cells/well) were cultivated in five 96-well plates with six replicate wells. In terms of the colony formation assay, altogether 500 transfected cells were addressed in a 6-well plate and kept in a medium with 10% FBS for 2 weeks with the replacement of the medium every 4 days. Then, colonies were confirmed with methanol and dyed with 0.1% crystal violet (Sigma-Aldrich, St Louis, MO) for 15 min. The quantity of visibly stained colonies was counted in colony formation. In different treatment groups, wells were independently measured in triplicate.
Assays of cell migration
In terms of the migration assays, after 24 h of transfection, 3.5 × 104 cells in medium with 1% FBS were put into the upper chamber of an insert (Millipore, Billerica, MA, USA), while medium with 10% FBS was put into the lower chamber. After 24 h of incubation, the remaining cells on the upper level of the membrane were cleaned, while those cells that had migrated through the membrane were dyed with methanol as well as 0.1% crystal violet and then imaged with an IX71 inverted microscope (Olympus, Tokyo, Japan). The experiments were done in triplicate.
Flow cytometry analysis
Flow cytometry assays were performed as previously reported by Xu et al.41. After the cells were transfected with siRNAs for 48 h, we harvested the cells and then performed fluorescein isothiocyanate (FITC)-Annexin V and propidium iodide staining by using the FITC-Annexin V Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s instruction. Cell cycle analysis was stained with propidium oxide by the Cycle TEST PLUS DNA Reagent Kit (BD Biosciences) by following the manual and then evaluated by FACScan. The count of the cells in each phase was assessed.
Western blot assay as well as antibodies
Cell protein lysates were divided by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to 0.22-m nitrocellulose membranes (Sigma-Aldrich, St Louis, MO), and cultivated with particular antibodies. Densitometry (Quantity One software; Bio-Rad) was used to quantify the autoradiograms. GAPDH antibody was employed as control. Anti-EZH2 was from proteintech (Wuhan, China) and anti-CDKN1A was from Abcam (Cambridge, UK).
In vivo tumor formation assay
Athymic male mice, 4 weeks old and bought from the Animal Center of the Nanjing University (Nanjing, China), were kept in particular in pathogen-free situations. HuCCT1 cells were stably transfected with shRNA or empty vector and obtained from cell culture plates, cleaned with phosphate-buffered saline, and re-suspended at 2 × 107 cells/ml. The cells were then xenografted into BALB/c male nude mice. Besides, the size of the tumor, length × width2 × 0.5, was calculated every 3 days. At 16 days post injection, the mice were sacrificed by CO2 asphyxiation and the tumor weights were weighed and examined. The study was performed in coincidence with the Instruction for the Care and Use of Laboratory Animals of the National Institutes of Health strictly. The agreement was licensed by the Committee on the Ethics of Animal Experiments of Nanjing Medical University.
Deep sequencing of whole transcriptome
Total RNA from the HuCCT1 cells with SNHG1 knockdown as well as control HuCCT1 cells were separated and quantified. The concentration of each specimen was measured with NanoDrop 2000 (Thermo Scientific, USA). The amount was evaluated with Agilent2200 (Agilent, USA). The ordering library of each RNA specimen was carried out with Ion Proton Total RNA-Seq Kit v2 following the conditions recommended by the manufacturer (Life technologies, USA). Data can be accessed in Supplementary Table S2.
RIP assays
RIP experiment was carried out to study whether SNHG1 could interact with EZH2, using a Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA) following the manufacturer’s guidelines. The antibody used for RIP assays of EZH2 was obtained from Millipore (Billerica, MA, USA).
ChIP assays
ChIP assays were carried out with EZ-CHIP Kit following the manufacturer’s recommendations (Millipore, Billerica, MA, USA). EZH2 antibody was purchased from Millipore (Billerica, MA, USA) and H3 trimethyl Lys 27(H3K27me3) antibody was from Abcam (Cambridge, UK). The sequences of ChIP primer are shown in Supplementary Table S1. We calculated the ChIP data as a proportion of the added DNA using the equation 2[input Ct − target Ct] × 0.1 × 100.
Immunofluorescence
CCA cells were seeded on chamber slides and were fixed with 4% paraformaldehyde for 10 min at room temperature. Then, the cells were incubated with antibodies against EZH2 (proteintech, Wuhan, China) or CDKN1A (Abcam, Cambridge, UK) at 4 °C overnight. Then, the slides were incubated with matched secondary antibodies at room temperature for 50 min in dark condition. The nuclei of the cells were stained with 4′,6-diamidino-2-phenylindole (Sigma) at room temperature for 10 min. Fluorescence images were captured using a Pannoramic MIDI/250 (3D HISTECH, Hungary).
In vitro transcription assays and RNA pull-down assays
In vitro translation assays were performed using mMessage mMachine KIT following the manufacturer’s recommendations (Ambion, USA). SNHG1 RNAs were labeled by desthiobiotinylation, by using the Pierce RNA 3′ End Desthio-biotinylation Kit (Pierce, Thermo). The Pierce Magnetic RNA-Protein Pull-Down Kit was used to perform the RNA pull-down assays according to the manufacturer’s instructions (Pierce, Thermo).
Statistical analysis
Statistical analyses were carried out with GraphPad Prism5 (GraphPad Software, La Jolla, USA). The statistical significance of distinctions between various groups was calculated by Student’s t-test or a Chi-square test, as suitable. All data were represented as means ± S.D., were counted, and two-sided P-values of 0.05 were considered for statistical significance.
References
Wang, J. G. et al. Coding-noncoding gene expression in intrahepatic cholangiocarcinoma. Transl. Res. 168, 107–121 (2016).
Rizvi, S., Khan, S. A., Hallemeier, C. L., Kelley, R. K. & Gores, G. J. Cholangiocarcinoma—evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 15, 95–111 (2018).
Lee, S. et al. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell 164, 69–80 (2016).
Niazi, F. & Valadkhan, S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3′ UTRs. RNA 18, 825–843 (2012).
Zhao, J. et al. Long non-coding RNA Linc00152 is involved in cell cycle arrest, apoptosis, epithelial to mesenchymal transition, cell migration and invasion in gastric cancer. Cell Cycle 14, 3112–3123 (2015).
Jia, H. et al. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 16, 1478–1487 (2010).
Saha, A., Bhattacharya, S. & Bhattacharya, A. Serum stress responsive gene EhslncRNA of Entamoeba histolytica is a novel long noncoding RNA. Sci. Rep. 6, 27476 (2016).
Ma, S. L., Li, A. J., Hu, Z. Y., Shang, F. S. & Wu, M. C. Coexpression of the carbamoylphosphate synthase 1 gene and its long noncoding RNA correlates with poor prognosis of patients with intrahepatic cholangiocarcinoma. Mol. Med. Rep. 12, 7915–7926 (2015).
Lu, X. et al. Long noncoding RNA AFAP1-AS1 promoted tumor growth and invasion in cholangiocarcinoma. Cell. Physiol. Biochem. 42, 222–230 (2017).
Jiang, X. M. et al. LncRNA CCAT1 as the unfavorable prognostic biomarker for cholangiocarcinoma. Eur. Rev. Med. Pharmacol. Sci. 21, 1242–1247 (2017).
Zhang, F. et al. Long noncoding RNA PCAT1 regulates extrahepatic cholangiocarcinoma progression via the Wnt/beta-catenin-signaling pathway. Biomed. Pharmacother. 94, 55–62 (2017).
Tan, X., Huang, Z. & Li, X. Long non-coding RNA MALAT1 interacts with miR-204 to modulate human hilar cholangiocarcinoma proliferation, migration, and invasion by targeting CXCR4. J. Cell. Biochem. 118, 3643–3653 (2017).
Wang, C. et al. Long non-coding RNA MALAT1 promotes cholangiocarcinoma cell proliferation and invasion by activating PI3K/Akt pathway. Neoplasma 64, 725–731 (2017).
Zhang, S. et al. LncRNA-CCAT1 promotes migration, invasion, and EMT in intrahepatic cholangiocarcinoma through suppressing miR-152. Dig. Dis. Sci. 62, 3050–3058 (2017).
Zhang, Y. et al. Downregulation of SNHG1 suppresses cell proliferation and invasion by regulating Notch signaling pathway in esophageal squamous cell cancer. Cancer Biomark. 21, 89–96 (2017).
Zhang, H. Y., Yang, W., Zheng, F. S., Wang, Y. B. & Lu, J. B. Long non-coding RNA SNHG1 regulates zinc finger E-box binding homeobox 1 expression by interacting with TAp63 and promotes cell metastasis and invasion in Lung squamous cell carcinoma. Biomed. Pharmacother. 90, 650–658 (2017).
Zhang, H. et al. Expression of long non-coding RNA (lncRNA) small nucleolar RNA host gene 1 (SNHG1) exacerbates hepatocellular carcinoma through suppressing miR-195. Med. Sci. Monit. 22, 4820–4829 (2016).
Zhang, M. et al. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomed. Pharmacother. 80, 73–79 (2016).
Tian, T., Qiu, R. & Qiu, X. SNHG1 promotes cell proliferation by acting as a sponge of miR-145 in colorectal cancer. Oncotarget 9, 2128–2139 (2018).
Zhu, Y. et al. Up-regulation of lncRNA SNHG1 indicates poor prognosis and promotes cell proliferation and metastasis of colorectal cancer by activation of the Wnt/beta-catenin signaling pathway. Oncotarget 8, 111715–111727 (2017).
Hu, Y. et al. LncRNA-SNHG1 contributes to gastric cancer cell proliferation by regulating DNMT1. Biochem. Biophys. Res. Commun. 491, 926–931 (2017).
Gupta, R. et al. Synergistic tumor suppression by combined inhibition of telomerase and CDKN1A. Proc. Natl Acad. Sci. USA 111, E3062–E3071 (2014).
Gulappa, T., Reddy, R. S., Suman, S., Nyakeriga, A. M. & Damodaran, C. Molecular interplay between cdk4 and p21 dictates G0/G1 cell cycle arrest in prostate cancer cells. Cancer Lett. 337, 177–183 (2013).
Liu, Y. W. et al. LincRNAFEZF1-AS1 represses p21 expression to promote gastric cancer proliferation through LSD1-Mediated H3K4me2 demethylation. Mol. Cancer 16, 39 (2017).
Marchese, F. P. & Huarte, M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics 9, 21–26 (2014).
Shen, X. et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 32, 491–502 (2008).
Yamaguchi, H. & Hung, M. C. Regulation and role of EZH2 in cancer. Cancer Res. Treat. 46, 209–222 (2014).
Askari, M., Sobti, R. C., Nikbakht, M. & Sharma, S. C. Aberrant promoter hypermethylation ofp21 (WAF1/CIP1) gene and its impact on expression and role of polymorphism in the risk of breast cancer. Mol. Cell. Biochem. 382, 19–26 (2013).
Akizu, N. et al. EZH2 regulates neuroepithelium structure and neuroblast proliferation by repressingp21. Open Biol. 6, 150227 (2016).
Seward, S. et al. EZH2 blockade by RNA interference inhibits growth of ovarian cancer by facilitating re-expression ofp21(waf1/cip1) and by inhibiting mutant p53. Cancer Lett. 336, 53–60 (2013).
Fan, T. et al. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol. Cancer Res. 9, 418–429 (2011).
Sherr, C. J. & Roberts, J. M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512 (1999).
Gartel, A. L. & Radhakrishnan, S. K. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 65, 3980–3985 (2005).
Xia, X. et al. Cytoplasmic p21 is responsible for paclitaxel resistance in ovarian cancer A2780 cells. Eur. J. Gynaecol. Oncol. 36, 662–666 (2015).
Jalili, A. et al. Dual suppression of the cyclin-dependent kinase inhibitors CDKN2C and CDKN1A in human melanoma. J. Natl. Cancer Inst. 104, 1673–1679 (2012).
Wei, C. Y. et al. Expression of CDKN1A/p21 and TGFBR2 in breast cancer and their prognostic significance. Int. J. Clin. Exp. Pathol. 8, 14619–14629 (2015).
Guo, H., Xu, Y. & Fu, Q. Curcumin inhibits growth of prostate carcinoma via miR-208-mediated CDKN1A activation. Tumor Biol. 36, 8511–8517 (2015).
Kotake, Y. et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing ofp15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962 (2011).
Wu, Y. et al. Long non-coding RNA HOTAIR promotes tumor cell invasion and metastasis by recruiting EZH2 and repressing E-cadherin in oral squamous cell carcinoma. Int. J. Oncol. 46, 2586–2594 (2015).
Merry, C. R. et al. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 24, 6240–6253 (2015).
Xu, Y. et al. The lncRNA TUG1 modulates proliferation in trophoblast cells via epigenetic suppression of RND3. Cell Death Dis. 8, e3104 (2017).
Acknowledgements
This study was supported by the Project of Standard Diagnosis and Treatment of Key Disease of Jiangsu Province (BE2015722); Project of the peak of the six talents of Jiangsu Province (WSN-018); and Scientific Research Foundation for Health of Jiangsu Province (H201408). We also thank NovelBio Bio-Pharm Technology Co., Ltd for the support of next-generation sequencing and bioinformatics analysis with NovelBrain Cloud Analysis Platform (www.novelbrain.com)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by J. Chipuk
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, Y., Zhang, M., Wang, N. et al. Epigenetic silencing of tumor suppressor gene CDKN1A by oncogenic long non-coding RNA SNHG1 in cholangiocarcinoma. Cell Death Dis 9, 746 (2018). https://doi.org/10.1038/s41419-018-0768-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0768-6
This article is cited by
-
LncRNA SNHG1: role in tumorigenesis of multiple human cancers
Cancer Cell International (2023)
-
Upregulation of TTYH3 promotes epithelial-to-mesenchymal transition through Wnt/β-catenin signaling and inhibits apoptosis in cholangiocarcinoma
Cellular Oncology (2021)
-
Epigenetic silencing of KLF2 by long non-coding RNA SNHG1 inhibits periodontal ligament stem cell osteogenesis differentiation
Stem Cell Research & Therapy (2020)
-
The long non-coding RNA SNHG1 promotes glioma progression by competitively binding to miR-194 to regulate PHLDA1 expression
Cell Death & Disease (2019)
-
The transcriptional landscape of lncRNAs reveals the oncogenic function of LINC00511 in ER-negative breast cancer
Cell Death & Disease (2019)

{kind=link}