
Abstract
NRLP3 inflammasome is a protein complex involved in the maturation of IL1β. In the onset of type 1 diabetes as well as in islet transplantation, IL-1β is one of the cytokines involved in the recruitment of immune cells in islets and eventually in islet destruction. Whether IL-1β is produced by islet cells is still under debate and NLRP3 inflammasome-dependent IL-1β production has not been yet determined in human islets. The aim of this study was to determine the expression and the regulation of the NRLP3 inflammasome in human islets. Human islets were stimulated with LPS and successively with ATP (LPS + ATP) in the presence or absence of the inflammasome inhibitor glyburide. Islets were also incubated in hypoxic or normoxic conditions for 24 h in the presence or absence of glyburide. Then, IL1B and NLRP3 expression was studied by real time PCR, protein expression by western blot, protein localization by immunofluorescence and protein secretion by ELISA. LPS + ATP increased gene expression of NRLP3 and IL1B. Glyburide partially prevented this effect. IL-1β protein was localized in β and non-β cells. Moreover, LPS + ATP increased IL-1β protein expression and production, which were prevented by glyburide. Hypoxia increased gene expression of NRLP3 and IL1B and induced IL-1β and caspase-1 production. Finally, hypoxia-induced cell death which was not prevented by inhibition of NLRP3 inflammasome. NRLP3 inflammasome is expressed and plays a role in IL-1β production by human islets. By contrast, NRLP3 inflammasome activation is not involved in islet cell death induced by hypoxia.
Similar content being viewed by others

Introduction
Inflammasomes are multi-protein complexes activated in response to infections, inflammations and in autoimmune processes. The best characterized inflammasome is composed of a NOD-like receptor family, pyrin domain containing 3 (NLRP3), an apoptosis-associated speck (ASC)-like protein and pro-caspase 1, and is called NLRP3 inflammasome. The main function of NLRP3 inflammasome is the maturation and the production of interleukin-1 beta (IL-1β). To this end, two distinct signals are involved. The first (priming) signal induces transcription and traduction of pro-IL-1β and NLRP3 via a Toll-like receptor- nuclear factor-kappa B (TLR-NF-κB) pathway. The second signal (activation) results in the NLRP3 inflammasome formation and activation of caspase-1. Activated caspase-1 will then cleave the inactive pro-IL-1β into the active IL-1β form, promoting its secretion1. IL-1β is a mediator of inflammation and immune responses playing a central role in the host defence against many pathogens. However, IL-1β overproduction leads to a chronic inflammation and contributes to the pathogenesis of many inflammatory and autoimmune diseases2. In type 1 diabetes (T1D), it participates to the recruitment of immune cells in islets, alters insulin secretion and induces β-cell apoptosis3,4. IL-1β also plays a pivotal role in cell destruction that occurs after islet transplantation. Studies have shown that an increased production of IL-1β, TNFα, and IFNγ after islet transplantation induces activation of macrophages, then a local production of IL-1β leading to dysfunction and destruction of the graft5,6,7,8. In solid organ graft, inflammasomes have been shown to play an important role in acute and chronic rejection9. However, their expression and role in grafted islets have not been determined. Furthermore, inflammasome-dependent IL-1β production in transplanted islets may be induced by hypoxia. Indeed, islet isolation procedure disrupts vascularization, and even if after transplantation, the process of neovascularization is quickly activated10, the restoration of a functional and partial blood flow lasts for weeks11 or months for a complete functional vascularization to occur12. Hypoxia has been shown to be linked to oxydative stress, process responsible for reactive oxygen species (ROS) generation13. Interestingly, studies have shown that ROS are activators of inflammasome and subsequently IL-1β production14,15. However, hypoxia as an enhancer of inflammasome has not been yet demonstrated in islets.
A better understanding of mechanisms involved in islet destruction by IL-1β is primordial to try to improve islet viability in T1D and after islet transplantation. Therefore, in this study, we assessed whether NLRP3 inflammasome is expressed and regulated in human islets in physiological and pathophysiological (hypoxia) conditions.
Materials and methods
Human islet isolation and culture
Human islets were isolated from brain-dead multiorgan donors using a method previously described16,17. Islets were used for research only if not suitable for transplantation. The use of human islets for research was approved by our local institutional ethical committee. After purification, islets were transferred in CMRL 1066 medium containing 5.6 mmol/l glucose and supplemented with penicillin, streptomycin, glutamin, HEPES, and 10% FCS (hereafter referred to as complete CMRL). In all preparations, islets were incubated first at 37 °C for 18–24 h and subsequently at 24 °C for maximum 48 h when required, before to be used in experiments.
Human islet and islet cells treatments
For immunostaining, human islets were dissociated into single cells as described18. After incubation overnight at 37 °C in complete CMRL, 50,000 cells were attached on 35-mm-diameter Petri dishes coated with 0.1 mg/ml of poly-L-lysine and incubated overnight at 37 °C.
Aliquots of 1000 human islets and 50’000 islet cells were incubated 1 h in complete CMRL, in the presence or absence of 200 μM glyburide, used here as an inflammasome inhibitor (Santa Cruz biotechnology, Dallas, Texas, USA). Then, human islets and islet cells were exposed or not to 1 μg/ml LPS (Sigma, Saint-Louis, Missouri, USA) for 4 h or to 5 mM ATP (Sigma, Saint-Louis, Missouri, USA) for 30 min or to 1 μg/ml LPS for 4 h and successively to 5 mM ATP for 30 min (LPS + ATP).
In hypoxia experiments, 1000 human islets were incubated in complete CMRL in hypoxia chamber (1% O2) (Stemcell Technologies, Grenoble, France) or in normoxia conditions (21% O2) at 37 °C for 24 h in presence or absence of 200 μM glyburide.
Real-time quantitative PCR analysis
RNA was extracted from human islets using the RNeasy minikit (Qiagen, Courtaboeuf, France). RNA was reverse transcribed using the High Capacity cDNA Reverse transcription kit (ThermoFischer Scientific, Waltham, MA, USA). Gene amplification was achieved with the RT-PCR method using the TaqMan Fast Advance Master Mix (ThermoFischer Scientific). Primers used for amplification were purchased from ThermoFischer Scientific: human Rplp0 (Hs9999902_m1), human IL-1β (Hs01555410-m1), human NLRP3 (Hs00918082-m1). Gene expressions values were normalized to the value of the housekeeping gene RPLP0 and calculated based on the comparative cycle threshold Ct method (2−ΔCt method).
Immunoblot analysis
Islets were solubilized in lysis buffer (20 mM Tris, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 0.5% Triton X-100 and proteases inhibitors) at 4 °C for 10 min. Proteins were separated by SDS-PAGE using a 10% or 15% resolving gel and transferred to a polyvinylidene difluoride membrane (Merck Millipore). The membrane was blocked with saline buffer (10 mm Tris, pH 7.4, 320 mm NaCl) containing 5% bovine serum albumin (BSA) for 1 h at room temperature and blotted overnight at 4 °C with the following antibodies at the dilution indicated by the manufacturer’s instructions: anti-IL-1β (sc:7884, Santa Cruz), anti-HIF (04-1006, Merck Millipore), anti-actin (MAB1501R, Merck Millipore). An ECL protein detection kit (Amersham Biosciences, Piscataway, USA) and a Molecular Omager ChemiDoc XRS+System (Bio-Rad, Hercules, CA, USA) were used for visualization of the bands.
Immunofluorescence staining
Islet cells, attached on Petri dishes as described above, were fixed in 10% methanol-free formalin, permeabilized with 0.1% Triton X-100 in phosphate buffered saline (PBS), rinsed, incubated in 0.5% BSA in PBS, and then exposed for 2 h to a combination of primary antibodies: a rabbit anti-IL-1β (1:50, Santa Cruz, sc:7884), a guinea pig anti-insulin (1:100, DakoCytomation, Carpinteria, CA, USA), a mouse anti-glucagon (1:4000, Sigma-Aldrich, St Louis, MO, USA) or a rat anti-somatostatin (1:100, Abcam, Cambridge, UK). After rinsing in PBS, islet cells were exposed for 1 h to an adequate combination of fluorescence-labeled secondary antibodies (Jackson ImmunoResearch Laboratories, Rheinfelden, Switzerland). Islet cells were rinsed in PBS and coverslipped before being observed and photographed using a confocal laser scanning microscope, LSM510 META (Zeiss, Feldbach, Germany). Images acquired from the confocal microscope were analyzed for pixel intensity using Image J software. Pixel intensity was quantified separately in manually selected regions of the cells. A region was drawn around each cluster of cells to be measured and the same size region was drawn in an area without fluorescent objects to be used for background subtraction. The net integrated intensity for each cluster of cells was measured by Image J software. To calculate the CTCF (Corrected Total Cell Fluorescence) we used the following formula: CTCF = Integrated density−(Area of selected cell × Mean fluorescence of background readings). In each cluster of cells the number of nuclei were counted and CTCF was normalized to the number of nuclei control.
Release of IL-1β, caspase-1, and VEGF
When required, islet culture supernatants were collected and stored at −20 °C until use. The amount of IL-1β, caspase-1, and VEGF proteins in the culture medium was measured using human-specific enzyme-linked immunosorbent assay (ELISA) kits (R&D systems, Minneapolis, USA) following the manufacturer’s instructions.
Analysis of cell death
Cell death was evaluated measuring intact CK18 and cleaved-CK18 fragment using M65® ELISA and M30 Apoptosense® ELISA kits (PEVIVA, TecoMedical, Switzerland). Intact CK18 is released by necrotic cells. CK18 is also cleaved by caspases during apoptosis generating soluble protein fragments. The M65® ELISA kit detects all the forms of CK18 and measures cell death due to both apoptosis and necrosis. Samples react with a solid phase capture antibody M6 directed against CK18 and the HRP (horseradish peroxidase) conjugated M5 antibody directed against a different epitope of CK18. The M30 Apoptosense® ELISA detects caspases-generated CK18 fragment and measures only apoptosis. Samples react with a solid phase capture antibody M5 directed against CK18 and the HRP-(horseradish peroxidase) conjugated M30 antibody directed against the CK18Asp396 neo-epitope. The difference between the values of M65 and the values of M30 quantifies necrosis. The values were normalized to the total protein concentration in the lysate.
Statistical analysis
Differences between means were assessed either by the Student’s t test or by 1-way ANOVA. Where ANOVA was applied, Tukey post hoc analysis was used to identify significant differences between groups.
Results
NLRP3 inflammasome is expressed and regulated in human islets
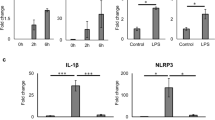
Human islets were incubated with LPS for 4 h and successively with ATP for 30 min (LPS + ATP), and NRLP3 and IL1B expressions analysed by real-time quantitative PCR. In response to this treatment, NRLP3 increased 3.4 ± 1.0 fold (Fig. 1a) and IL1B increased 50.4 ± 1.9 fold (Fig. 1b). The NLRP3 inflammasome inhibitor glyburide partially prevented the increased expression of NLRP3 (Fig. 1a) and IL1B (Fig. 1b) induced by LPS and ATP. Under similar conditions, the IL-1β content was analyzed by western blot, its cell localization by immunofluorescence, and its release in culture supernatant by ELISA. As compared to control, LPS + ATP treatment significantly increased IL-1β content and this effect was partially prevented by glyburide (Fig. 2a, b). Double-immunofluorecence labelling for IL-1β and insulin indicated that IL-1β was present in β-cells. Staining for IL-1β was higher in β-cells treated with LPS + ATP as compared to control. This higher staining was attenuated when cells were treated with glyburide (Fig. 2c, d). IL-1β was also present in non-β-cells (Fig. 2c). By multi-immunofluorescence staining, we showed that α-cells (cells labeled for glucagon) and not δ-cells (cells labeled for somatostatin) expressed IL-1β in addition to β-cells (Figure S1A-B). Interestingly, staining for IL-1β in non-β-cells did not increase after treatment with LPS + ATP. Significant amount of IL-1β (38.1 ± 1.8 pg/ml) was found in the supernatant of islets incubated with LPS + ATP, whereas IL-1β was undetectable in the control or present at low level (0.92 ± 0.28 pg/ml) in the presence of glyburide (Fig. 2e). LPS and ATP alone had a significant effect on IL1B (Figure S2B) similarly to what observed with LPS + ATP. By contrast, neither LPS nor ATP had an effect on IL-1β release (Figure S2C). Both LPS and ATP had a tendency to increase NLRP3 (Figure S3A). Taken together, these results show that NLRP3 inflammasome is expressed and regulated in human islets.

Human islets were stimulated or not (control, CTR) with 1 μg/ml LPS for 4 h and successively with 5 mM ATP for 30 min (LPS + ATP) in the presence or absence of 200 μM glyburide (GLY). NLRP3 (a) and IL1B (b) in human islets were quantified by qRT-PCR and expressed relative to CTR. Data are means ± SEM from four experiments (a) and three experiments (b). *p<0.05, **p<0.01, ****p<0.0001

Human islets were stimulated or not (control, CTR) with 1 μg/ml LPS for 4 h and successively with 5 mM ATP for 30 min (LPS + ATP) in the presence or absence of 200 μM glyburide (GLY). Protein levels of pro- and mature IL-1β forms were analyzed by Western blot (a) and density of IL-1β bands quantified (b). IL-1β cell localization was analyzed by immunofluorescence (c). The red staining (corresponding to IL-1β) was quantified by morphometry and expressed relative to CTR; a total of 79, 88, and 63 cells from three experiments were analyzed for CTR, LPS + ATP and LPS + ATP + GLY, respectively. IL-1β released in the supernatant was quantified by ELISA (e). Data are means ± SEM from three (b and d) and five experiments (e). *p<0.05, **p<0.01, ****p<0.0001
Hypoxia activates NLRP3 inflammasome in human islets
Isolated human islets were incubated under normoxic (control, 21% O2) and hypoxic conditions (1% O2) for 24 h. As expected, Vascular Endothelial Growth Factor (VEGF) gene expression (Fig. 3a), VEGF released into the supernatant (Fig. 3b), and Hypoxia-inducible-factor (HIF) protein expression (Fig. 3c) were increased in response to hypoxia as compared to normoxia. Regarding the inflammasome genes, NRLP3 and IL1B expressions were increased 3.6 ± 0.97 and 5.5 ± 1.1 fold, respectively, in response to hypoxia (Fig. 4a, b). Furthermore, the amounts of caspase-1 and IL-1β found in the supernatant of islets incubated under hypoxia were higher as compared to control (Fig. 4c, d). Glyburide partially prevented the increase in NRLP3 and IL1B expressions and IL-1β and caspase-1 secretion induced by hypoxia (Fig. 5). These results indicate that NLRP3 inflammasome is induced and regulated in human islets in response to hypoxia.

Human islets were incubated either in control condition (Normoxia) or in 1% O2 hypoxia chamber (Hypoxia) for 24 h, and VEGF expression (a), VEGF released (b) and HIF cell content (c) analyzed. a VEGF was evaluated by qRT-PCR. b Secreted VEGF was quantified by ELISA. c Protein level of HIF was performed by Western blot. Data are means ± SEM from three (a) and six experiments (b). **p<0.01

Human islets were incubated in control condition (Normoxia) or in 1% O2 hypoxia chamber (Hypoxia) for 24 h. NLRP3 (a) and IL1B (b) were quantified by qRT-PCR and expressed relative to normoxia. Caspase-1 (c) and IL-1β (d) released were quantified by ELISA. Data are means ± SEM from 6 (a), 9 (b), 4 (c) and four experiments (d). *p<0.05, **p<0.01, ***p<0.001

Human islets were incubated in 1% O2 hypoxia chamber for 24 h in the absence (Hypoxia) or presence or of 200 μM glyburide (Hypoxia + GLY). NLRP3 (a) and IL1B (b) were quantified by qRT-PCR and expressed relative to Hypoxia. Caspase-1 (c) and IL-1β (d) released were quantified by ELISA and results expressed relative to Hypoxia. Data are means ± SEM from three experiments. *p<0.05, **p<0.01
Inhibition of NLRP3 inflammasome does not relieve islet cell death induced by hypoxia
To determine whether NLRP3 inflammasome is involved in islet cell death induced by hypoxia, NLRP3 inflammasome was inhibited by glyburide under hypoxia and cell death analyzed. We analyzed total cell death, necrosis and apoptosis, by quantifying released intact cytokeratin-18 (CK18) and caspases-generated CK18 fragments. As expected, we observed that hypoxiainduced total cell death (Fig. 6a) comprising apoptosis (Fig. 6b) and necrosis (Fig. 6c). However, inhibition of NLRP3 inflammasome by glyburide did not prevent islet cell death, neither necrosis nor apoptosis. These results indicate that NLRP3 inflammasome is not involved in islet cell death induced by hypoxia.

Human islets were incubated in control condition (Normoxia) or in 1% O2 hypoxia chamber (Hypoxia) for 24 h in the presence or absence of 200 μM glyburide (GLY). Cell death was evaluated by Apoptosense® ELISA kits. M65 values express both apoptosis and necrosis (a). M30 values express apoptosis (b). M65-M30 values (difference between the M65 and M30 values) express necrosis (c). NS no significant. Data are means ± SEM from seven experiments. *p<0.05
Discussion
NLRP3 inflammasome is expressed by innate immune cells, mainly macrophages, and its activation requires two distinct signals. The first signal is mediated by the TLR-NF-κB pathway and results in an increase of the cellular contents of the pro-IL-1β and NLRP3 inflammasome. ATP initiates the second signal, promoting the assemblage and the activation of NLRP3 inflammasome, and leading to the maturation and secretion of IL-1β19. In this study, we have investigated the expression and regulation of NLRP3 inflammasome in human islets. NLRP3 inflammasome expression and IL-1β production have been shown to be induced in mouse islet beta cells in response to glucose, even though to a lower level than that observed in macrophages20. With regard to human islets, it is already known that glucose leads to IL-1β secretion21 but NLRP3 inflammasome expression has not yet been demonstrated in human islets. Here, using different approaches, we showed that NLRP3 inflammasome is expressed in human islet cells. In macrophages, NLRP3 inflammasome expression is induced in response to LPS through its receptor TLR4. Studies have shown that TLR4 is also expressed in human islet cells suggesting that NLRP3 inflammasome could be activated in human islets in response to LPS + ATP22,23. In our study, we demonstrated that NLRP3 inflammasome activation in human islets is dependent on the presence of LPS + ATP. Indeed, human islets exposed to LPS + ATP exhibit an increase in IL1B gene expression and secretion. By immunofluorescence, IL-1β is shown in beta and non-beta cells and, interestingly, LPS + ATP treatment increases IL-1β staining in beta cells but not in non-beta cells. Furthermore, inhibition of NLRP3 inflammasome by glyburide prevents the effects of LPS + ATP on gene expression, content and secretion of IL-1β and gene expression of NLRP3, demonstrating that NLRP3 inflammasome is a regulated process. Our results are in agreement with the study of Tschopp et al., who showed that NLRP3 inflammasome is expressed and inhibited by glyburide in murine islet cells20.
NLRP3 inflammasome is involved in metabolic disorders, such as type 2 diabetes and obesity24,25. NLRP3-deficient mice display a greater glucose tolerance and insulin sensitivity20, a reduction of inflammation in adipose tissue and liver, and a protection against obesity-induced insulin resistance26. Moreover, in vitro experiments have shown that saturated fatty acids and IAPP trigger activation of NLRP3 inflammasome leading to production of IL-1β and caspase-127,28.
NLRP3 inflammasome has been also studied in T1D. Genetic studies highlighted an upregulation of TLR4 and TLR2 in patients with T1D29 and two polymorphisms in NLRP3 were identified as a predisposing factor for T1D30. Interestingly, NLRP3-deficient NOD mice were shown to be protected from T1D development31. However, whether local islet production and activation of NLRP3 inflammasome is involved in inflammatory events and/or beta cell destruction in T1D has yet to be investigated. Pancreatic islet transplantation is a cell therapy procedure for T1D. In the pathogenesis of transplant rejection, inflammasome pathways have been shown to be involved in acute and chronic rejection of solid organs9,32,33. However, involvement of inflammasome in grafted islets has not yet been studied. We hypothesized that hypoxic conditions, to which islets are exposed during isolation and after transplantation, could lead to NLRP3 inflammasome activation. Indeed, immediately after transplantation and up to 10 days post-transplant, islets are not vascularized and undergo hypoxia. To assess the effect of hypoxia on inflammasome activation, isolated human islets were incubated 24 h in vitro at 1% O2 to mimic a severe hypoxia. Our results showed that hypoxia increases gene expression of NRLP3 and IL1B and induces IL-1β and caspase-1 production in human islets, demonstrating that hypoxia is an enhancer of NLRP3 inflammasome. These results correlate with studies describing ROS, generated by hypoxia-induced oxidative stress, as activators of NLRP3 inflammasome13,14,15,20. Zhou et al.20 have shown that NLRP3 interacts with thioredoxin (TRX)-interacting protein (TXNIP), leading to inflammasome assembly, in mouse islet cells exposed to oxidative stress. Interestingly, we observed an increase in TXNIP gene expression in response to hypoxia in human islets (data not shown) suggesting that activation of NLRP3 inflammasome could be mediated by TXNIP as observed in mouse islet cells20. We then studied whether NLRP3 inflammasome is involved in hypoxia-induced damage in islets. As expected, hypoxia induces apoptosis and necrosis in human islets; however glyburide does not prevent hypoxia-induced cell death, indicating that NLRP3 inflammasome is not involved in islet cell death induced by hypoxia. To understand the mechanism involved in hypoxia-induced islet cell death is interesting but out of the scope of this study. In other studies, using different cell systems, mechanisms involved in hypoxia-induced cell death have been described. For instance, activation of intrinsic apoptotic pathway characterized by cytochrome c release, activation of caspase-9 and caspase-334,35 and transcription factors, such as HIF-2, p53, and NF-ĸB, were reported to mediate cell death induction in response to hypoxia34,36,37,38,39,40.
Interestingly, a study has shown that activation of NLRP3 inflammasome, induced by high glucose concentrations, endoplasmic reticulum or and oxidative stress, is not involved in mouse islet cell death41. It could be possible that the amount of IL-1β, produced by human islets via NLRP3 inflammasome activation, is insufficient to cause islet cell death. IL-1β may associate with others cytokines to induce cell death42. Apoptosis has been observed only in human islet cells treated with IL-1β plus interferon-γ, not with IL-1β alone43. Furthermore, these cytokines activate p38 involved in β-cell death44. In allograft dysfunction, injury and rejection, NLRP3 inflammasome process occurs very early in the inflammatory cascade9. In islet graft, production of IL-1β alone could play an immunological role in inducing the recruitment of immune cells, such as dendritic cells and T cells. The activation of these immune cells by IL-1β in islet graft could stimulate the production of inflammatory cytokines such as TNFα and IL-1β leading to dysfunction and destruction of islet graft. Further studies are necessary to understand the role of NLRP3 inflammasome-produced IL-1β in human islet grafts.
References
Gross, O., Thomas, C. J., Guarda, G. & Tschopp, J. The inflammasome: an integrated view. Immunol. Rev. 243, 136–151 (2011).
Lukens, J. R., Dixit, V. D. & Kanneganti, T. D. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov. Med. 12, 65–74 (2011).
Donath, M. Y., Storling, J., Maedler, K. & Mandrup-Poulsen, T. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J. Mol. Med. 81, 455–470 (2003).
Mandrup-Poulsen, T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 39, 1005–1029 (1996).
Bottino, R. et al. Transplantation of allogeneic islets of Langerhans in the rat liver: effects of macrophage depletion on graft survival and microenvironment activation. Diabetes 47, 316–323 (1998).
Arnush, M. et al. IL-1 produced and released endogenously within human islets inhibits beta cell function. J. Clin. Invest. 102, 516–526 (1998).
Kaufman, D. B. et al. Differential roles of Mac-1+ cells, and CD4+ and CD8+ T lymphocytes in primary nonfunction and classic rejection of islet allografts. J. Exp. Med. 172, 291–302 (1990).
Kaufman, D. B. et al. Effect of 15-deoxyspergualin on immediate function and long-term survival of transplanted islets in murine recipients of a marginal islet mass. Diabetes 43, 778–783 (1994).
Weigt, S. S., Palchevskiy, V. & Belperio, J. A. Inflammasomes and IL-1 biology in the pathogenesis of allograft dysfunction. J. Clin. Invest. 127, 2022–2029 (2017).
Jansson, L. & Carlsson, P. O. Graft vascular function after transplantation of pancreatic islets. Diabetologia 45, 749–763 (2002).
Menger, M. D., Yamauchi, J. & Vollmar, B. Revascularization and microcirculation of freely grafted islets of Langerhans. World J. Surg. 25, 509–515 (2001).
Morini, S. et al. Revascularization and remodelling of pancreatic islets grafted under the kidney capsule. J. Anat. 210, 565–577 (2007).
Miyata, T. & Takizawa, S. & van Ypersele de Strihou, C. Hypoxia. 1. Intracellular sensors for oxygen and oxidative stress: novel therapeutic targets. Am. J. Physiol. Cell Physiol. 300, C226–C231 (2011).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008).
Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 40, 616–619 (2010).
Ricordi, C., Lacy, P. E., Finke, E. H., Olack, B. J. & Scharp, D. W. Automated method for isolation of human pancreatic islets. Diabetes 37, 413–420 (1988).
Bucher, P. et al. Assessment of a novel two-component enzyme preparation for human islet isolation and transplantation. Transplantation 79, 91–97 (2005).
Wojtusciszyn, A., Armanet, M., Morel, P., Berney, T. & Bosco, D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia 51, 1843–1852 (2008).
Bauernfeind, F. G. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 (2009).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Maedler, K. et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110, 851–860 (2002).
Schulthess, F. T. et al. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 9, 125–139 (2009).
Vives-Pi, M. et al. Evidence of expression of endotoxin receptors CD14, toll-like receptors TLR4 and TLR2 and associated molecule MD-2 and of sensitivity to endotoxin (LPS) in islet beta cells. Clin. Exp. Immunol. 133, 208–218 (2003).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Vandanmagsar, B. et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 (2011).
Wen, H. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 (2011).
Masters, S. L. et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 11, 897–904 (2010).
Devaraj, S. et al. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J. Clin. Endocrinol. Metab. 93, 578–583 (2008).
Pontillo, A. et al. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity 43, 583–589 (2010).
Hu, C. et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc. Natl Acad. Sci. USA 112, 11318–11323 (2015).
Seto, T. et al. Upregulation of the apoptosis-related inflammasome in cardiac allograft rejection. J. Heart Lung Transplant. 29, 352–359 (2010).
Jankovic, D. et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J. Exp. Med. 210, 1899–1910 (2013).
Lenihan, C. R. & Taylor, C. T. The impact of hypoxia on cell death pathways. Biochem. Soc. Trans. 41, 657–663 (2013).
Weinmann, M. et al. Molecular ordering of hypoxia-induced apoptosis: critical involvement of the mitochondrial death pathway in a FADD/caspase-8 independent manner. Oncogene. 23, 3757–3769 (2004).
Menrad, H. et al. Roles of hypoxia-inducible factor-1alpha (HIF-1alpha) versus HIF-2alpha in the survival of hepatocellular tumor spheroids. Hepatology 51, 2183–2192 (2010).
Roberts, A. M. et al. Suppression of hypoxia-inducible factor 2alpha restores p53 activity via Hdm2 and reverses chemoresistance of renal carcinoma cells. Cancer Res. 69, 9056–9064 (2009).
An, W. G. et al. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature 392, 405–408 (1998).
Koumenis, C. et al. Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol. Cell Biol. 21, 1297–1310 (2001).
Cummins, E. P. et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl Acad. Sci. USA 103, 18154–18159 (2006).
Wali, J. A. et al. Activation of the NLRP3 inflammasome complex is not required for stress-induced death of pancreatic islets. PLoS ONE 9, e113128 (2014).
Grishman, E. K., White, P. C. & Savani, R. C. Toll-like receptors, the NLRP3 inflammasome, and interleukin-1beta in the development and progression of type 1 diabetes. Pediatr. Res. 71, 626–632 (2012).
Gurzov, E. N. et al. Signaling by IL-1beta + IFN-gamma and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ. 16, 1539–1550 (2009).
Makeeva, N., Myers, J. W. & Welsh, N. Role of MKK3 and p38 MAPK in cytokine-induced death of insulin-producing cells. Biochem. J. 393, 129–139 2006).
Acknowledgements
We thank Lisa Perez and David Matthey-Doret for their excellent technical work (Geneva University Hospitals and University of Geneva, Department of Surgery, Geneva, Switzerland). This work was supported by grants from the Société Francophone du Diabète (SFD) and Swiss National Science Foundation (31003A_170090/1 and 310030-173138). Human islets for research were provided thanks to the European Consortium for Islet Transplantation funded by Juvenile Diabetes Research Foundation (3-RSC-2016-162-I-X).
Authors contributions
F.L. researched data and wrote the manuscript. E.B., G.P. researched data and contributed to discussion. D.B. and T.B. contributed to discussion and reviewed the manuscript. C.R. researched data. V.L. designed the study, researched data, contributed to discussion and reviewed the manuscript. In Fig. 1, F.L. generated the data and assembled the figure. In Fig. 2, C.R. generated the western blot data, G.P. the immunofluorescence data and E.B. the ELISA data. V.L. assembled the figure. In Fig. 3, F.L. generated the panel A data, E.B. the ELISA, C.R. the western blot data. V.L. assembled the figure. In Fig. 4, F.L. and C.R. generated the panel A and B data, E.B. the ELISA data. F.L. and V.L. assembled the figure. In Fig. 5, V.L. and C.R. generated the panel A and B data, C.R. the ELISA data. V.L. assembled the figure. In Fig. 6, F.L. and C.R. generated the data. V.L. assembled the figure. F.L., C.R., G.P., and E.B. generated data of supplementary figures. V.L. assembled the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by T. Kaufmann
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lebreton, F., Berishvili, E., Parnaud, G. et al. NLRP3 inflammasome is expressed and regulated in human islets. Cell Death Dis 9, 726 (2018). https://doi.org/10.1038/s41419-018-0764-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0764-x
This article is cited by
-
The downregulation of SCGN induced by lipotoxicity promotes NLRP3-mediated β-cell pyroptosis
Cell Death Discovery (2024)
-
Bax Inhibitor-1 preserves pancreatic β-cell proteostasis by limiting proinsulin misfolding and programmed cell death
Cell Death & Disease (2024)
-
ADP as a novel stimulus for NLRP3-inflammasome activation in mice fails to translate to humans
Purinergic Signalling (2024)
-
The trigger for pancreatic disease: NLRP3 inflammasome
Cell Death Discovery (2023)
-
Inflammasomes and adaptive immune responses
Nature Immunology (2021)
