
Abstract
Non-small-cell lung cancer (NSCLC) is the predominant histological type of lung cancer and is characterized by the highest mortality and incidence rates among these types of malignancies. Cardiac glycosides, a class of natural products, have been identified as a potential type of chemotherapeutic agent. This study aims to investigate the anti-cancer effects and the mechanisms of action of Proscillaridin A (P.A) in NSCLC cells. In vitro sodium–potassium pump (Na+/K+ ATPase) enzyme assays indicated that P.A is a direct Na+/K+ ATPase inhibitor. P.A showed potent cytotoxic effects in NSCLC cells at nanomolar levels. Treatment mechanism studies indicated that P.A elevated Ca2+ levels, activated the AMPK pathway and downregulated phosphorylation of ACC and mTOR. Subsequently, P.A increased death receptor 4 (DR4) expression and downregulated NF–κB. Interestingly, P.A selectively suppressed EGFR activation in EGFR mutant cells but not in EGFR wild-type cells. In vivo, P.A significantly suppressed tumor growth in nude mice compared to vehicle-treated mice. Compared with the Afatinib treatment group, P.A displayed less pharmaceutical toxicity, as the body weight of mice treated with P.A did not decrease as much as those treated with Afatinib. Consistent changes in protein levels were obtained from western blotting analysis of tumors and cell lines. Immunohistochemistry analysis of the tumors from P.A-treated mice showed a significant suppression of EGFR phosphorylation (Tyr 1173) and reduction of the cell proliferation marker Ki-67. Taken together, our results suggest that P.A is a promising anti-cancer therapeutic candidate for NSCLC.
Similar content being viewed by others

Introduction
Cancer is one of the leading causes of death both in China and worldwide1. Lung cancer has the highest incidence and mortality rates among all malignancies2. Over 1.6 million cases of lung cancer are diagnosed each year, accounting for 13% of all new cancer diagnoses. Further, 1.4 million deaths per year are attributed to lung cancer, accounting for 18% of all cancer-related deaths3. Among the various types of lung cancers, non-small-cell lung cancer (NSCLC) comprises 80–85% of all cases4; unfortunately, more than 70% of these cases are diagnosed as unresectable, advanced stage tumors5. Although many medical intervention methods have been proposed, the prognosis for NSCLC patients remains poor, with an 18% 5-year overall survival (OS) rate across all stages2.
To date, the main lung cancer treatment strategy involves the direct inhibition of tumor cell growth by cytotoxic agents and targeted therapies6. However, drug resistance is common and treatments are limited, thus new strategies have been developed including those affecting intracellular calcium (Ca2+) homeostasis. Ca2+, a second messenger, is involved in various fundamental functions, such as the regulation of gene transcription and cellular metabolic activity, which affects both cell proliferation and cell death7. It has been demonstrated that Ca2+ levels could be altered in different tumor types—such as ovarian, prostate, brain, and breast cancer—by altering Ca2+ channels and disrupting pump activity through post-translational modification8,9,10. Thus, inducing cell death by increasing the intracellular Ca2+ levels may be a novel method for the treatment of cancer. Another important P-type ATPase family member that can affect Ca2+ concentration is the Na+/K+ ATPase11. Analyses have demonstrated altered expression levels of Na+/K+ ATPase subunits in lung cancer cells, specifically, overexpression of the α1 and α3 subunits12,13. Numerous investigations have shown that cardiac glycosides (inhibitors of Na+/K+ ATPase) could induce apoptosis in tumor cells14,15,16.
Traditional Chinese medicines (TCM) are a treasure trove of drugs that may be utilized for the treatment of different diseases. The clinical applications of Artemisinin in the treatment of malaria and Berberine in the treatment of type II diabetes aroused research interests regarding TCM17. In our previous studies, we screened a library of 800 natural compounds using MTT assays and identified Proscillaridin A (P.A) as having a relatively large anti-cancer effect in A549 and H1975 NSCLC cell lines18. In this study, we aimed to further investigate the mechanism of action of P.A, a constituent of squill-Drimia maritima, as a treatment for NSCLC. P.A has been previously investigated in several different kinds of cancer cells by other groups. It has been demonstrated that P.A can inhibit HIF-1α and reduce cell proliferation in prostate carcinoma and hepatocellular carcinoma19. Other researchers have demonstrated that the anti-cancer effect of P.A occurs through inhibition of DNA topoisomerase I and II in breast cancer20,21. P.A also induces apoptosis of cancer cells and suppresses tumor xenograft growth in a glioblastoma model22.
However, to our knowledge, there is currently no mechanistic study of P.A in NSCLC cells. Therefore, in this study, we aimed to first investigate the cytotoxicity of P.A in a panel of NSCLC cells by MTT assay. Second, we determined the functional effects of P.A by flow cytometry and colony formation assays. Then, the treatment mechanism of P.A was explored via sodium/potassium ATPase enzyme activity, flow cytometry, Western blotting and small interfering RNA transfection methods in NSCLC cells. Finally, we examined the in vivo efficacy of P.A in GFP-transformed H1975 stable cells in a xenograft mouse model.
Results
NSCLC cells are sensitive to P.A treatment
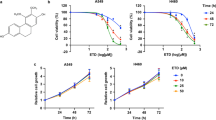
To measure the cytotoxic effects of P.A, we examined cytotoxicity in a panel of NSCLC cells. Among these NSCLC cell lines, we included six different NSCLC cell lines with common NSCLC driver mutations, including EGFR, ALK, KRAS, and ROS (see Supplementary Table 1). The cytotoxic effect of P.A on normal cells was also determined using a normal lung fibroblast cell line (CCD19-LU). As shown in Fig. 1a–h and Supplementary Table 1, P.A showed cytotoxic effects in NSCLC cells at a range of 5–30 nM. Interestingly, the cell viability of normal lung fibroblast cells (CCD19-LU) was the highest after P.A treatment. Even using a concentration 30-fold higher than the CC50 value for the EGFR mutant NSCLC cells (H1975 and HCC827) to treat CCD19-LU, there was still no significant inhibitory effect, implying low toxicity of P.A in normal lung fibroblast cells. P.A showed significant cytotoxicity in NSCLC cells and low cytotoxicity in normal cells, which may be advantageous for future pharmaceutical applications.

a The chemical structure of P.A; b–h P.A dose–response curves in NSCLC cell lines and a normal lung fibroblast cell line (CCD19-LU). The results are expressed as the mean ± S.E. (*p < 0.05, **p < 0.01, ***p < 0.001); i–k P.A specifically inhibited the phosphorylation of EGFR tyrosine residue 1173 in EGFR mutant NSCLC cells but had no effect on EGFR phosphorylation in EGFR wild-type NSCLC cells (A549)
P.A induces apoptosis in NSCLC cells and suppresses EGFR activation in EGFR mutant cell lines
Since P.A could inhibit cell proliferation in NSCLC cells harboring different common driver gene mutations, the most sensitive thrgee cell lines (A549, H1975, and HCC827) were chosen for treatment mechanism studies. As H1975 and HCC827 harbor an EGFR mutation and A549 is EGFR wild type, we compared the effects of P.A on EGFR activity among EGFR mutant and wild-type cells by measuring EGFR phosphorylation levels. The results showed that P.A inhibited the phosphorylation of EGFR at tyrosine residue 1173 in EGFR mutant cell lines (H1975 and HCC827) but not in the EGFR wild-type cell line (A549) (Fig. 1i–k). Next, we were interested in investigating whether the treatment of P.A affected the ability of cells to form colonies. The results demonstrated that, although EGFR was inhibited to different degrees in these cell lines, P.A could suppress clonogenic growth in all three NSCLC cell lines (Fig. 2a). Thus, the results demonstrated that P.A induced cell death through independent pathways.

a Data are shown as representative photomicrographs from colony formation assays after treatment of three NSCLC cell lines with different concentrations of P.A; b Morphological changes in three NSCLC cell lines after treatment with P.A; c Flow cytometric analysis of apoptosis levels after P.A treatment and statistical analysis results; d–f Statistical results of C (three independent experiments); g–i PARP, Caspase-3, Caspase-7, Caspase-9, and Bax were cleaved and activated by P.A, while Bcl-2 was downregulated and Akt activation was inhibited (*p < 0.05, **p < 0.01, ***p < 0.001)
To determine how P.A suppressed cell proliferation in NSCLC cells of different EGFR mutational statuses, we examined the apoptotic effect of P.A in A549, H1975 and HCC827 by microscopic and flow cytometric analyses. As shown in Fig. 2b, c, P.A significantly induced apoptosis in A549 and H1975 cells beginning at 12.5 and 25 nM, respectively, and induced apoptosis in approximately 70% of cells at 50 nM. In HCC827, P.A only induced apoptosis in ~30% of cells at 50 nM. Furthermore, as shown in Fig. 2g–i, PARP, Caspase-3, Caspase-7, and Caspase-9 were cleaved and activated by P.A treatment. The pro-survival regulator Akt was inhibited and the pro-apoptotic protein Bax was upregulated, while the anti-apoptotic protein Bcl-2 was downregulated, indicating that P.A-induced cell death was mediated through apoptosis. We further applied BAX immunofluorescence (IF) staining and mitochondrial extracted protein WB analysis to examine the localization of BAX after the treatment. From the results (supplementary Figure 5), we could observe the increased BAX fluorescence and upregulated BAX protein level in mitochondrial. To better understand the time-dependent cell death mechanisms, we tested P.A efficacy in different time points which exhibited the time-dependent manner of P.A (supplementary Figure 1-2). After comparing the P.A sensitivity results, the P.A-sensitive A549 and H1975 cells were chosen for subsequent functional assays.
P.A significantly inhibits Na+/K+ ATPase activity and increases Ca2+ levels, contributing to its cytotoxicity in cancer cells
To investigate whether P.A could inhibit the Na+/K+ ATPase, enzyme activity was assessed by in vitro Na+/K+ ATPase assay. As shown in Fig. 3a, the EC50 was 0.83 ± 0.3 nM, which is very potent, implying that P.A could directly inhibit the activity of Na+/K+ ATPase. As P.A inhibits Na+/K+ ATPase, it follows that ion levels would change23,24. Thus, we investigated whether inhibition of Na+/K+ ATPase would stimulate the Na+/Ca2+ exchanger, which would induce an increase in Ca2+ levels. As shown in Fig. 3b, P.A significantly increased intracellular Ca2+ levels at 12.5 nM in A549 and H1975 cells. We also tested the unfolded protein response after the treatment of P.A. In the results, the expression of BIP (GRP78) showed dosage-dependent decrease (supplementary Figure 6). However, in the normal lung fibroblast cell line, CCD19-LU, P.A treatment decreased Ca2+ levels, which may explain why CCD19-LU had the highest P.A CC50 among all the cell lines. The results also demonstrated that P.A treatment remarkably increased in vitro Ca2+ levels.

a P.A significantly inhibited Na+/K+ ATPase, and its IC50 was measured by in vitro Na+/K+ ATPase assay; b P.A elevated [Ca2+] levels after 6 h of treatment in A549 and H1975 cells; c, d Statistical results of b (three independent experiments); e P.A decreased [Ca2+] levels after 6 and 18 h treatment in CCD19-LU; f, g Statistical results of e (three independent experiments); h Increased intracellular [Ca2+] levels were required for P.A-induced apoptosis. A calcium chelator (BM) remarkably inhibited P.A-induced apoptosis; i, j Statistical results of e (three independent experiments) (*p < 0.05, **p < 0.01, ***p < 0.0001)
To ascertain whether Ca2+ elevation was important to P.A-induced apoptosis, the Ca2+ chelator BAPTA/AM (BM), which can decrease in vitro Ca2+ levels, was applied in conjunction with P.A. As shown by the flow cytometric results, the number of apoptotic cells significantly decreased following co-treatment with BM and P.A (Fig. 3h). The single treatment cytotoxicity of BM was shown in supplementary Figure 3. These results suggested that elevation of Ca2+ levels is a key mediator of P.A-induced apoptosis.
P.A activates AMPK and JNK phosphorylation and downregulates ACC phosphorylation
It is well known that the induction of calcium influx is one of the most important AMPK activation mechanisms25,26. Since P.A could elevate Ca2+ levels in vitro, we investigated whether P.A could induce activation of AMPK. As shown in Fig. 4a, b, P.A induced the phosphorylation of AMPK and down-regulated its direct downstream target acetyl-CoA carboxylase (ACC). To investigate whether the activation of AMPK was important for P.A-induced apoptosis, we applied compound C, a specific inhibitor of AMPK, to block the activation of AMPK. Our results demonstrated that compound C partially reduced the number of apoptotic cells in both the A549 and H1975 cell lines (Fig. 4c). Thus, AMPK is also an important modulator of P.A-induced apoptosis.

a, b P.A increased the phosphorylation of AMPK and its downstream target ACC; c Inhibition of AMPK by compound C partially rescued the cells from apoptosis; d, e Statistical results of C (three independent experiments); f The JNK inhibitor decreased apoptosis in both cell lines; g, h Statistical results of f (three independent experiments)
AMPK is a major upstream regulator of the mTOR pathway and AMPK activation can suppress the activity of the mTOR pathway27. Thus, we further investigated whether P.A could inhibit mTOR phosphorylation. As shown in Fig. 4a, P.A indeed inhibited the phosphorylation of mTOR. Moreover, c-Jun N-terminal kinase (JNK), which is closely associated with the mTOR pathway, was also shown to be involved in P.A-induced apoptosis. As shown in Fig. 4a, P.A significantly activated the phosphorylation of JNK. To ascertain whether the activation of JNK was required for P.A-induced apoptosis, we utilized the JNK specific inhibitor SP600125. As shown in Fig. 4f, JNK inhibition reduced the number of apoptotic cells when used in conjunction with P.A, indicating that the JNK pathway also contributes to P.A-induced apoptosis. The single treatment cytotoxicity of compound C and SP600125 was shown in supplementary Figure 3.
P.A upregulates DR4 expression and inhibits the DR4-suppressive NF-κB pathway
The Ca2+-regulated tumor necrosis factor (TNF) receptor is closely related to the apoptotic mechanism28,29. The TNF-related apoptosis-inducing ligand (TRAIL) can induce tumor cell death by interacting with TRAIL-receptor 1, also known as death receptor 4 (DR4)30,31. We investigated DR4 gene expression following treatment with P.A. As shown in Fig. 5a–c, both standard and quantitative PCR results demonstrated that the expression of DR4 was significantly elevated following P.A treatment. The protein level results were consistent with the gene expression results (Fig. 5d, e). In addition, when DR4 was knocked down, the percentage of apoptotic cells decreased (Fig. 5f). This may indicate that DR4 is an important mediator of P.A-induced apoptosis. WB was applied to confirm the efficacy of siRNA used to knockdown DR4 and the results supported it (supplementary Figure 4). Subsequently, NF-κB, a suppressor of DR432, has been shown to bind to the DR4 promoter at an NF-κB binding site (−1366/1356)33. NF-κB has also been shown to be downstream of mTOR pathway34. The results showed that the NF-κB pathway was significantly inhibited by P.A in a dose-dependent manner (Fig. 5h, i). This suggests that NF-κB signaling pathways could also be affected by P.A treatment.

a Standard and b, c quantitative RT-PCR were performed to determine the expression of DR4 after a 12-h P.A treatment; d, e DR4 and its downstream effector, Caspase-8, were upregulated by P.A; f A549 cells were transfected with si-DR4 and treated with or without P.A for the indicated amount of time. Cells were collected and analyzed by flow cytometry with PI and ANNEXIN V; g Statistical results of f (three independent experiments); h, i The activation of the NF-κB pathway was inhibited by P.A treatment
P.A suppresses tumor growth in H1975 xenografts
To further understand the pharmaceutical activity of P.A, we next investigated its in vivo efficacy. GFP-transformed H1975 stable cells were injected into nude mice, and the tumors were allowed to grow to ~100 mm3 in size before beginning 21 days of P.A treatment (7 mice in each group). As shown in Fig. 6a, c, tumor growth was significantly suppressed after 16 days in the P.A group compared to the vehicle group. On the last day of treatment, the net tumor mass was measured and the average tumor weight in the P.A treatment group was 35% less than the average tumor weight in the vehicle group (Fig. 6d, e). Mice in the P.A group displayed greater body weight compared to the vehicle group (the statistical analysis of body weight on their sacrificed day was shown in supplementary Figure 9). Compared to the Afatinib group, mice in the P.A group showed no apparent toxicity after treatment (Fig. 6b). We further examined the effect of P.A treatment on EGFR, AMPK, mTOR, Akt, and death receptor cascades in protein extracts derived from tumor tissues. Consistent with the in vitro data, we observed suppression of EGFR, Akt, and ACC phosphorylation and an increase of AMPK and DR4 (Fig. 6f). Moreover, as shown in Fig. 6g, immunohistochemistry analysis demonstrated that P.A treatment decreased EGFR phosphorylation (Tyr 1173) in tumor cells and notably reduced cell proliferation, as indicated by Ki-67 immunostaining. P.A also decreased cellularity, as measured by hematoxylin and eosin staining. Thus, these data demonstrate that P.A is detrimental to NSCLC tumor growth in preclinical lung xenograft models.

a Tumor volume was examined by caliper measurements from the beginning of the treatment period (when the tumor reached approximately 100 mm3 in size), n = 7 per group; b Quantification of body weight for each group; c Fluorescence intensity indicating the size of the tumor in vivo; d, e The weight of each tumor; f P.A decreased the phosphorylation of mTOR, EGFR (Tyr 1173 and Tyr 1068) and Akt activated AMPK and increased DR4 expression; g Immunohistochemistry analysis comparing P.A-treated tumor tissue to vehicle-treated tissue (*p < 0.05, **p < 0.01, ***p < 0.001)
Discussion
Recent reports have indicated that steroidal cardiac inhibitors could inhibit the in vivo growth of prostate and lung xenografts through significant up-regulation of caspase-3 activity35 and could induce apoptosis in glioblastoma cells through activation of the p38 and JNK pathways36. Therefore, Na+/K+ ATPase is an emerging target in anti-cancer therapy37 and is currently being investigated for the development of novel therapeutics38. The balance of cellular membrane potential is regulated by P-type ATPase family pumps, including Na+/K+ ATPase and Na+/Ca2+ exchanger39. These ion pumps are closely linked. For example, if Na+/K+ ATPase activity is interrupted, cellular Na+/K+ homeostasis and, subsequently, other ionic gradients such as Ca2+ will also be disrupted11. As a second messenger, Ca2+ is involved in various fundamental functions, including suppressing Bcl-2 expression and activating the caspase pathway24 to induce apoptosis40 and participating in Ca2+/calmodulin-dependent kinase kinase β (CaMKKβ)-mediated AMPK activation and other various metabolic pathways41,42,43,44.
In this study, we have found that P.A has anti-cancer effects through its ability to inhibit Na+/K+ ATPase. As shown in Fig. 7a, P.A inhibits Na+/K+ ATPase and elevates Ca2+ levels, resulting in the activation of the AMPK pathway. More interestingly, after an 18-hour P.A treatment in the normal lung fibroblast cell line CCD19-LU, Ca2+ levels decreased by almost 30%. Other investigators have demonstrated that cardiac glycosides have distinct effects on the regulation of Ca2+ levels and apoptosis in human fibroblasts and cancer cells, which is in line with the results of our study45. Compared with other cardiac glycosides, the CC50 of P.A is 50% of that of Digoxin in A549s, almost 10% of that of Digoxin in H1975s46, and less than the CC50 of Digitoxin in both EGFR-related cell lines18. However, P.A. demonstrated less toxicity in normal cells than both Digoxin and Digitoxin. Activation of AMPK exerts apoptotic effects through two main pathways: suppression of ACC phosphorylation and hence mTOR signaling or stimulation of the JNK pathway. Moreover, elevations in Ca2+ levels can also induce ER stress47, which will inhibit EGFR phosphorylation48,49, thus elucidating the connection between Ca2+ and EGFR. As shown in EGFR mutant cells (Fig. 7b), P.A induces the elevation of Ca2+ levels, which further inhibits EGFR phosphorylation. Inhibition of EGFR also affects other apoptotic signaling pathways, for example by inactivating phosphor-Akt and activating the JNK pathway, both of which will further induce apoptosis.

The proposed mechanism of action of P.A in NSCLC cells
High intracellular Ca2+ is not only related to inhibition of AMPK and EGFR but also inhibition of another important apoptotic cytokine, tumor necrosis factor (TNF). The TNF family is known to transduce death signals via cell membrane receptors. DR4, after binding to the apoptotic cytokine TRAIL, can recruit and activate the apoptosis-initiating protease caspase-8, which in turn activates ‘effector’ caspases, including caspase-3, -6, and -7, which execute the cell’s apoptotic demise50,51. Recent research shows that a combination of TRAIL and the herbal extraction apigenin (APG) or the chemotherapeutic drug cis-diaminedichloro- platinum (II) (CDDP) can induce apoptosis by upregulating the expression of DR452 and DR553 in different cancer cells. APG alone has low cytotoxicity in cancer cells. Compared to the reports above, P.A shows better treatment efficacy and enhances DR4 expression at a nanomolar level and the combination treatment with TRAIL showed significantly additive effects, increased nearly 20% apoptosis at 12.5 nM with 25 ng/ml TRAIL subtract both single treatment apoptotic percentage (supplementary Figure 7-8).
NF-κB, a suppressor of the DR4 pathway, is one of the most important signal transduction pathways and exhibits the ability to promote cell proliferation, suppress apoptosis, promote cell migration, and suppress differentiation54. It has been reported that a hemi-synthetic cardiac inhibitor, UNBS1450, can induce apoptotic cell death by inhibiting NF-κB transactivation. Recent studies have also demonstrated that TRAIL-mediated cell death is enhanced upon infection of neuroblastoma cells with a dominant-negative mutant of IκB kinase, a kinase essential for NF-κB activation32. P.A inhibits the activation of IKKβ, IκBα, and NF-κB through regulation of AMPK, the mTOR cascade and the EGFR and Akt pathways.
The significance and novelty of this study lie in the identification of an effective Na+/K+ ATPase inhibitor, P.A, which can significantly inhibit the growth of NSCLC cells via Ca2+-induced DR4 up-regulation at the nanomolar level. This is the first demonstration of the connection between Ca2+ gradients, DR4 and apoptotic signaling pathways in NSCLC cells following P.A treatment. This compound also shows low toxicity in normal lung fibroblast cells, demonstrating its promise for further development as a new anti-cancer drug. Though there have been studies using P.A in glioblastoma22, breast cancer20,21, and colon cancer55, the compound was less effective in these tumor types than in NSCLC cells. Further, the anti-cancer mechanisms demonstrated by other studies were shown through only one angle, such as the inhibition of HIF-1α19 or the inhibition of topoisomerase I and II20,21. We, for the first time, demonstrate how P.A can function through a combination of AMPK activation and EGFR inhibition, resulting in a potent anti-cancer effect.
In summary, we provide new evidence that P.A is useful in the treatment of EGFR-mutant NSCLC cell lines and displays a different treatment mechanism in these cell lines compared with non-EGFR-mutant NSCLC cell lines. P.A can elevate the expression of DR4 and inhibit its suppressor, NF-κB, which is a key mediator of tumor cell survival. P.A also has the ability to suppress tumor growth in vivo. We believe that P.A is a promising candidate for treating NSCLC in the future.
Materials and methods
Cell culture and reagents
All cell lines used in this study were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). Proscillaridin A was purchased from Sigma (St Louis, MO, USA). Primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The fluorescein-conjugated anti-rabbit secondary antibody was purchased from Odyssey (Belfast, ME, USA).
MTT assay
Cells were seeded in a 96-well microplate with 3000–5000 cells/well and incubated overnight to allow cells to adhere. Cells were treated with different concentrations of P.A or the vehicle control dimethyl sulfoxide (DMSO). Ten microliters of MTT (5 mg/ml) solution was added to each well for 4 h. After that, 100 μL of resolved solution (10% SDS and 0.1 mM HCL) was added to each well for another 4 h. Absorbance was measured at 570 nm (with a reference reading at 650 nm) on a microplate reader (Tecan, Morrisville, NC, USA).
Colony-formation assay
Colony-forming assays were performed as previously described18. Briefly, approximately 300 cells were plated into each well of a 6-well plate. After 48 h, cultures were rinsed with fresh medium and P.A (with different indicated dosages) was added to the medium. Forty-eight hours later, the cells were washed twice with PBS and then incubated in drug-free medium. The medium was changed every 5 days. After culturing for an additional 10–14 days, the cells were fixed with 3.7% paraformaldehyde for 20 min and incubated with 0.2% crystal violet solution in 10% ethanol for 10 min.
Apoptosis assay
A549, H1975, and HCC827 cells (1 × 105 cells/well) were seeded in a six-well plate for 24 h and then treated with the indicated concentrations of P.A for an additional 24 h at 37 °C. The cells were washed once with ice-cold 1× PBS and then harvested. Then, the cell pellets were re-suspended in 100 μl of 1× binding buffer. The cells were double-stained with 2 μl each of Annexin-V FITC and PI (100 μg/ml) for 15 min at room temperature (RT) in the dark. Apoptotic cells were quantified on a BD Aria III Flow Cytometer (BD Biosciences, San Jose, California, USA)
Western blot analysis
After incubating A549, H1975, and HCC827 cells with P.A for the indicated amount of time, cells were harvested and washed with cold 1× PBS. Then, cells were lysed with ice-cold RIPA lysis buffer containing protease and phosphatase inhibitors to extract the cellular proteins. Total extracted protein was quantified using the Bio-Rad DCTM protein assay kit (Bio-Rad, Philadelphia, PA, USA). Then, 35 μg of each protein sample was loaded and electrophoretically separated on an 8% SDS-PAGE gel and then transferred to a Nitrocellulose (NC) membrane. Membranes were blocked with 5% non-fat milk and PBS containing 0.1% Tween-20 (TBST) for 1 h at RT. After 1 h, membranes were incubated with primary antibodies (1:1000 dilution) at 4 °C overnight with gentle shaking. Membranes were washed three times with TBST (5 min/time) and incubated with a secondary fluorescent antibody (1:10,000 dilution) for 1 h at RT. After another three washes with TBST (15 min/time), bands were visualized on a LI-COR Odessy scanner (Belfast, ME, USA).
Na+/K+ ATPase enzyme activity assay
The enzymatic activity of Na+/K+ ATPase (purchased from Sigma as a lyophilized powder from porcine cerebral cortex) was measured by colorimetric quantification of Pi released during ATP hydrolysis. First, 10 μl of Na+/K+ ATPase (600 units/ml) was incubated with 2.5 μl of KCl/NaCl solution (45 mM KCl and 2 M NaCl) and either 5 μl of DMSO (control) or 5 μl of P.A in 67.5 μl of buffer (24 mM Tris HCl buffer with 0.68 mM ethylenediaminetetraacetic acid and 6.0 mM magnesium chloride, pH 7.8). ATP (5 μl of 80 mM solution) was then added. Trichloroacetic acid (30 μl of 100% w/v) was added to the reaction, then the mixture was centrifuged for 5 min. The supernatant (50 μl aliquot) was transferred to a 96-well plate containing 100 μl of Taussky–Shorr reagent. Following a 5 min incubation at RT, absorbance was read at 660 nm.
Measurement of intracellular calcium
Changes in intracellular free calcium were measured using a fluorescent dye, Fluo-3, as previously described56. Briefly, A549 and H1975 cells were washed twice with culture media after a 6-h P.A treatment (6.25–25 nM). Then, cell suspensions were incubated with 5 μM Fluo-3 at 37 °C for 30 min. The cells were then washed twice with HBSS and the re-suspended cell samples were subjected to FACS analysis. At least 10,000 events were analyzed
RNA extraction and quantitative real-time PCR
Cells were incubated with P.A for 12 h. Total RNA was then extracted from treated cells using a TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Quantitative real-time PCR was performed using High-productivity Real-Time quantitative PCR ViiATM7 (Life Technologies, Gaithersburg, MD, USA). The PCR primers used in our study were synthesized commercially, and the sequences are as follows: DR4: 5′- TTGTGTCCACCAGGATCTCA-3′ and 5′-GTCACTCCAGGGCGTACAAT-3′. The glyceraldehyde 3-phosphatase dehydrogenase (GAPDH) gene was used as the reference gene. GAPDH: 5′-AACGACCCCTTCATTGAC-3′; and 5′-TCCACGAC ATACTCAGCAC-3′. All data are represented as the mean fold change values normalized to GAPDH and obtained from triplicate analyses.
Transfection with small interfering RNA
A549 cells were seeded into six-well plates. After 24 h, cells were transfected with DR4 or control small interfering RNAs (siRNAs) using 2 μl of X-tremeGENE siRNA transfection reagent (Roche, Basal, Switzerland). All siRNAs were synthesized by GenePharma (Pudong New Area, Shanghai, China). The sequences of the siRNAs used are as follows (sense and antisense, respectively): siDR4 5′-r(CAAACUUCAUGAUCAAUCA)dTdT-3′ and 5′-r(UGAUUGAUCAUGAAGUUUG)dAdT-3′, and control siRNA 5′-r(UUCUCCGAACGUGUCACGU)dTdT-3′ and 5′-r(ACGUGACA CGUUCGGAGAA)dTdT-3′. Eight hours after transfection, cells were stimulated with 12.5 nM P.A. Cells were harvested 24 h after stimulation for determination of apoptosis by flow cytometry.
Xenograft study
All animals were acclimatized for 1 week. Six-week-old nude mice were injected with 5 × 106 GFP-transformed H1975 stable cells mixed with growth factor reduced Matrigel (Becton Dickinson, Franklin Lakes). Xenografts were allowed to grow to a size of ~100 mm3, then mice were randomized into the following three treatment groups: vehicle, Afatinib (10 mg/kg i.p.) and P.A (3 mg/kg i.p.). Mice were treated daily for 21 days and tumor volumes were measured every third day. In vivo tumor fluorescence intensity was measured using the In-Vivo XTREME Imager (Bruker BioSpin, Billerica, MA, USA). Histology and immunohistochemistry analyses were performed using the DAKO EnVisionTM + system (Dako, Glostrup, Denmark).
Statistical analysis
All the data are presented as the mean ± SD of three individual experiments. Differences were analyzed by one-way ANOVA using Graph Pad Prism 5.
References
Chen, W. et al. Cancer statistics in China, 2015. CA Cancer J. Clin. 66, 115–132 (2016).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 65, 5–29 (2015).
American Cancer Society. Cancer Facts & Figures 2015. Annual Publication (American Cancer Society Inc., Atlanta, 2015).
Nawaz, K. & Webster, R. M. The non-small-cell lung cancer drug market. Nat. Rev. Drug Discov. 15, 229–230 (2016).
Chen, Z., Fillmore, C. M., Hammerman, P. S., Kim, C. F. & Wong, K. K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer 14, 535–546 (2014).
Dubey, S. & Schiller, J. H. Three emerging new drugs for NSCLC: pemetrexed, bortezomib, and cetuximab. Oncologist 10, 282–291 (2005).
Marks, A. R. Intracellular calcium-release channels: regulators of cell life and death. Am. J. Physiol. 272(2 Pt 2), H597–H605 (1997).
Yang, S. L., Cao, Q., Zhou, K. C., Feng, Y. J. & Wang, Y. Z. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 28, 1320–1328 (2009).
Fixemer, T., Wissenbach, U., Flockerzi, V. & Bonkhoff, H. Expression of the Ca2+-selective cation channel TRPV6 in human prostate cancer: a novel prognostic marker for tumor progression. Oncogene 22, 7858 (2003).
McAndrew, D. et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 10, 448–460 (2011).
Sachs, G. & Munson, K. Mammalian phosphorylating ion-motive ATPases. Curr. Opin. Cell Biol. 3, 685 (1991).
Sreenivasan, Y., Raghavendra, P. B. & Manna, S. K. Oleandrin-mediated expression of Fas potentiates apoptosis in tumor cells. J. Clin. Immunol. 33, 1031 (2013).
Mijatovic, T. et al. The alpha1 subunit of the sodium pump could represent a novel target to combat non-small cell lung cancers. J. Pathol. 212, 170–179 (2007).
Calderónmontaño, J. M., Burgosmorón, E. & Lópezlázaro, M. The in vivo antitumor activity of cardiac glycosides in mice xenografted with human cancer cells is probably an experimental artifact. Oncogene 33, 2947–2948 (2014).
Kayali, F., Janjua, M. A., Laber, D. A., Miller, D. & Kloecker, G. H. Phase II trial of second-line erlotinib and digoxin for nonsmall cell lung cancer (NSCLC). Open Access J. Clin. Trials 3, 9–13 (2011).
Hallböök, H. et al. Ex vivo activity of cardiac glycosides in acute leukaemia. PloS ONE 6, e15718 (2011).
Leung, E. L. et al. Targeting tyrosine kinase inhibitor-resistant non-small cell lung cancer by inducing epidermal growth factor receptor degradation via methionine 790 oxidation. Antioxid. Redox Signal. 24, 263–279 (2016).
Zhang, Y. Z. et al. Compound library screening identified cardiac glycoside digitoxin as an effective growth inhibitor of gefitinib-resistant non-small cell lung cancer via downregulation of α-tubulin and inhibition of microtubule formation. Molecules 21, 374 (2016).
Zhang, H. et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc. Natl Acad. Sci. USA 105, 19579 (2008).
Winnicka, K., Bielawski, K., Bielawska, A. & Surażyński, A. Antiproliferative activity of derivatives of ouabain, digoxin and proscillaridin A in human MCF-7 and MDA-MB-231 breast cancer cells. Biol. Pharm. Bull. 31, 1131 (2008).
Bielawski, K., Winnicka, K. & Bielawska, A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol. Pharm. Bull. 29, 1493–1497 (2006).
Denicolaï, E. et al. Proscillaridin A is cytotoxic for glioblastoma cell lines and controls tumor xenograft growth in vivo. Oncotarget 5, 10934–10948 (2014).
Liu, J. et al. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 275, 27838–27844 (2000).
Mcconkey, D. J., Lin, Y., Nutt, L. K., Ozel, H. Z. & Newman, R. A. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 60, 3807–3812 (2000).
Newman, R. A. et al. Autophagic cell death of human pancreatic tumor cells mediated by oleandrin, a lipid-soluble cardiac glycoside. Integr. Cancer Ther. 6, 354–364 (2008).
Woods, A. et al. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 (2005).
Shi, W. Y. et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 3, e275 (2012).
Mollinedo, F. & Gajate, C. Fas/CD95 death receptor and lipid rafts: new targets for apoptosis-directed cancer therapy. Drug Resist. Updates 9, 51–73 (2006).
Tsai, C. T. et al. TNF-α down-regulates sarcoplasmic reticulum Ca2+ ATPase expression and leads to left ventricular diastolic dysfunction through binding of NF-κB to promoter response element. Cardiovasc. Res. 105, 318–329 (2015).
Chang, D. W. et al. c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 21, 3704–3714 (2002).
Wiley, S. R. et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 (1995).
Karacay, B., Sanlioglu, S., Griffith, T. S., Sandler, A. & Bonthius, D. J. Inhibition of the NF-κB pathway enhances TRAIL-mediated apoptosis in neuroblastoma cells. Cancer Gene Ther. 11, 681–690 (2004).
Mendoza, F. J., Ishdorj, G., Hu, X. & Gibson, S. B. Death receptor-4 (DR4) expression is regulated by transcription factor NF-kappaB in response to etoposide treatment. Apoptosis 13, 756–770 (2008).
Gao, Y. et al. Differential IKK/NF-κB activity is mediated by TSC2 through mTORC1 in PTEN-null prostate cancer and tuberous sclerosis complex tumor cells. Mol. Cancer Res. 13, 1602 (2015).
Honisch, S. et al. A steroidal Na+/K+ ATPase inhibitor triggers pro-apoptotic signaling and induces apoptosis in prostate and lung tumor cells. Anticancer Agents Med Chem. 14, 1161 (2014).
Garcia, D. G. et al. Na/K-ATPase as a target for anticancer drugs: studies with perillyl alcohol. Mol. Cancer 14, 105 (2015).
Dimas, K. et al. Steroidal cardiac Na+/K+ ATPase inhibitors exhibit strong anti-cancer potential in vitro and in prostate and lung cancer xenografts in vivo. Anti-Cancer Agents Med. Chem. 14, 762–770 (2014).
Prassas, I. & Diamandis, E. P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 7, 926 (2008).
Lingrel, J. B. & Kuntzweiler, T. Na+/K+-ATPase. J. Biol. Chem. 269, 19659–19662 (1994).
Mcconkey, D. J. & Orrenius, S. The role of calcium in the regulation of apoptosis. J. Leukoc. Biol. 59, 775 (1996).
Bagrov, A. Y., Shapiro, J. I. & Fedorova, O. V. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 61, 9–38 (2009).
Blaustein, M. P. Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and cell responsiveness. Am. J. Physiol. 264(6 Pt 1), C1367 (1993).
Jayaraman, T. & Marks, A. R. Calcineurin is downstream of the inositol 1,4,5-trisphosphate receptor in the apoptotic and cell growth pathways. J. Biol. Chem. 275, 6417–6420 (2000).
Gusarova, G. A. et al. Hypoxia leads to Na/K-ATPase downregulation via Ca2+ release-activated Ca2+ channels and AMPK activation. Mol. Cell. Biol. 31, 3546 (2011).
Winnicka, K., Bielawski, K., Bielawska, A. & Miltyk, W. Dual effects of ouabain, digoxin and proscillaridin A on the regulation of apoptosis in human fibroblasts. Nat. Product. Res. 24, 274–285 (2010).
Lin, S. Y. et al. Digoxin suppresses tumor malignancy through inhibiting multiple Src-related signaling pathways in non-small cell lung cancer. PloS ONE 10, e0123305 (2015).
Fujishita, T., Aoki, M. & Taketo, M. M. JNK signaling promotes intestinal tumorigenesis through activation of mTOR complex 1 in Apc(Δ716) mice. Gastroenterology 140, 1556–1563.e6 (2011).
Harr, M. W. & Distelhorst, C. W. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2, a005579-a (2010).
Fan, X. X. et al. (Z)3,4,5,4′-trans-tetramethoxystilbene, a new analogue of resveratrol, inhibits gefitinb-resistant non-small cell lung cancer via selectively elevating intracellular calcium level. Sci. Rep. 5, 16348 (2015).
Frese, S. et al. Cardiac glycosides initiate Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells by up-regulation of death receptors 4 and 5. Cancer Res. 66, 5867 (2006).
Kelley, S. K. & Ashkenazi, A. Targeting death receptors in cancer with Apo2L/TRAIL. Curr. Opin. Pharmacol. 4, 333 (2004).
Chen, M. et al. Apigenin potentiates TRAIL therapy of non-small cell lung cancer via upregulating DR4/DR5 expression in a p53-dependent manner. Sci. Rep. 6, 35468 (2016).
Nagane, M. et al. Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Res. 60, 847 (2000).
Dutta, J., Fan, Y. N., Fan, G. & Gelinas, C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene 25, 6800 (2006).
Felth, J. et al. Cytotoxic effects of cardiac glycosides in colon cancer cells, alone and in combination with standard chemotherapeutic drugs. J. Nat. Prod. 72, 1969–1974 (2009).
Liu, M. J., Wang, Z., Ju, Y., Wong, R. N. & Wu, Q. Y. Diosgenin induces cell cycle arrest and apoptosis in human leukemia K562 cells with the disruption of Ca2+ homeostasis. Cancer Chemother. Pharmacol. 55, 79–90 (2005).
Acknowledgements
This work was supported by the Macau Science Technology Development Fund (project code: 021/2013/A1, 005/2014/AMJ, and 010/2016/A1).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G-Q Chen
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, RZ., Fan, XX., Duan, FG. et al. Proscillaridin A induces apoptosis and suppresses non-small-cell lung cancer tumor growth via calcium-induced DR4 upregulation. Cell Death Dis 9, 696 (2018). https://doi.org/10.1038/s41419-018-0733-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0733-4
This article is cited by
-
Cardenolide glycosides sensitize gefitinib-induced apoptosis in non-small cell lung cancer: inhibition of Na+/K+-ATPase serving as a switch-on mechanism
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
The mechanistic role of cardiac glycosides in DNA damage response and repair signaling
Cellular and Molecular Life Sciences (2023)
-
Effects on the STAT3-shRNA in Non-Small-Cell Lung Cancer Therapy: Design, Induction of Apoptosis, and Conjugation with Chitosan-Based Gene Vectors
Journal of Ocean University of China (2021)
-
Microarray data analysis on gene and miRNA expression to identify biomarkers in non-small cell lung cancer
BMC Cancer (2020)
-
Ouabain impairs cancer metabolism and activates AMPK-Src signaling pathway in human cancer cell lines
Acta Pharmacologica Sinica (2020)
