
Abstract
Limb girdle muscular dystrophy type 2L (LGMD2L) and Miyoshi myopathy type 3 (MMD3) are autosomal recessive muscular dystrophy caused by mutations in the gene encoding anoctamin-5 (ANO5), which belongs to the anoctamin protein family. Two independent lines of mice with complete disruption of ANO5 transcripts did not exhibit overt muscular dystrophy phenotypes; instead, one of these mice was observed to present with some abnormality in sperm motility. In contrast, a third line of ANO5-knockout (KO) mice with residual expression of truncated ANO5 expression was reported to display defective membrane repair and very mild muscle pathology. Many of the ANO5-related patients carry point mutations or small insertions/deletions (indels) in the ANO5 gene. To more closely mimic the human ANO5 mutations, we engineered mutant ANO5 rabbits via co-injection of Cas9 mRNA and sgRNA into the zygotes. CRISPR-mediated small indels in the exon 12 and/or 13 in the mutant rabbits lead to the development of typical signs of muscular dystrophy with increased serum creatine kinase (CK), muscle necrosis, regeneration, fatty replacement and fibrosis. This novel ANO5 mutant rabbit model would be useful in studying the disease pathogenesis and therapeutic treatments for ANO5-deficient muscular dystrophy.
Similar content being viewed by others


Introduction
The limb girdle muscular dystrophies (LGMDs) are a diverse group of childhood and adult onset muscle diseases, characterized by progressive weakness of the hip and shoulder girdles and of the lower limbs, with muscle atrophy. The prevalence of LGMDs is about between 1 in 14,500 and 1 in 45,000 in the world1,2. To date, several genes have been identified in LGMDs including anoctamin-5 (ANO5), the causative gene for LGMD2L.
The ANO5 gene encodes ANO5, a member of the anoctamin/TMEM16 family of proteins with putative calcium-activated chloride channel and/or phospholipid scramblase activities3. The dominant mutations of ANO5 have been linked to gnathodiaphyseal dysplasia (GDD)4, while recessively inherited mutations cause LGMD2L and Miyoshi myopathy type 3 (MMD3)5,6,7. The phenotypical presentation of patients with ANO5 mutations varies remarkably, but symptoms typically begin in adulthood (age 20–50) with proximal lower limb weakness, high serum creatine kinase (CK) levels, asymmetric muscle atrophy and weakness, and sarcolemmal lesions, similar to dysferlinopathy8,9.
The molecular and cellular functions of ANO5 are not well understood. Due to the significant sequence homology with other anoctamin proteins, it is believed that ANO5 may function as either a calcium-activated chloride channel, or a phospholipid scramblase or both10,11. Similar to other anoctamin proteins, ANO5 is predicted to have 10 membrane-spanning helices and a hydrophilic cavity exposed to the lipid bilayer that likely represents the site of catalysis for phospholipid scrambling12. ANO5 is found in skeletal muscle, cardiac muscle, bone, testis and thyroid13, where it is primarily localized on the endoplasmic reticulum (ER) membrane14. Due to the phenotypical similarity between anoctaminopathy and dysferlinopathy, it is suggested that ANO5 may be involved in the membrane repair process. Moreover, the membrane repair efficiency was found to be defective in fibroblasts from a patient with ANO5 mutation and in skeletal muscle cells from an ANO5-knockout (KO) mouse model15. However, it remains to be determined how the ER-localized ANO5 plays a role in the cell plasma membrane repair.
At present, several ANO5-KO mice have been reported with different pathological outcomes15. In particular, two groups generated the ANO5-KO mice by disrupting the first or second exon of the gene, and the ANO5 transcripts were reported to be completely disrupted. These two ANO5-KO mice did not present with an overt myopathy. However, an ANO5 gene trapped mouse model with the insertion in intron 8 showed residual expression of mutant ANO5 transcript and presented with a mild muscular dystrophy phenotype. It is not very clear what exactly underlies the pathological variations in these ANO5-KO mice, however, the nature and localization of the mutations and the species difference are most likely responsible for the poor modeling of ANO5-deficient muscular dystrophy in mice. Of note, rabbits are considered better animal models than mice in recapitulating some human diseases because of the higher similarity in terms of physiology, anatomy and genetics with human beings than mice16.
Here we report the generation of ANO5 mutant rabbits, which recapitulate many features of LGMD2L patients by cytoplasmic microinjection of Cas9 mRNA and single guide RNA (sgRNA). We showed that engineered small insertions/deletions (indels) in the ANO5 gene of rabbits lead to muscle degeneration/regeneration, atrophy and elevation of serum CK levels. This novel ANO5-KO rabbit model could be used for pathogenesis studies and therapeutic development for ANO5-deficient muscular dystrophy.
Result
Design of CRISPR/Cas9 system and generation of ANO5-KO rabbits
To generate an ANO5-KO model, we designed a pair of sgRNAs targeting exon 12 and exon 13 to disrupt the open reading frame (ORF) of ANO5 in rabbit (Fig. 1a). To test the efficiency of gene targeting of ANO5 in zygotes, the mixed Cas9 mRNA and sgRNAs were microinjected into the zygotes, and cultured until the blastocyst stage. As shown in Table 1, 78.6% of injected embryos (N = 152) developed into the blastocyst stage, among which 75.4% carried mutations in ANO5 at the target sites. There were no significant differences in the developmental rate between the non-injected embryos and microinjected embryos (p > 0.05). These results showed that the dual sgRNA-directed CRISPR/Cas9 system is efficient for generation of mutations in the ANO5 gene in rabbit embryos.

a Schematic diagram shows the two different gRNAs targeting exon 12 and exon 13, respectively. b, c PCR analysis showing different mutations by the T7E1 assay using two different set of primers targeting exon 12 and exon 13, respectively. Gel images have been cropped. M, DL2000, has been used to indicate band size. Black line indicates the WT allele (492 and 488 bp). d Sanger sequencing results of the ANO5 RT-PCR product and the corresponding changes of amino acid sequences at the target site. Nonsense mutation was detected in puppies #7. ‘--’ indicates stop codon sites
To generate ANO5-KO rabbit, the 128 injected zygotes were transferred into the oviducts of four surrogate rabbits. Three of four surrogates were pregnant to term and produce 26 live pups (Table 2). The ear tissue of each animal was collected for genotyping. The mutations type was determined by the T7E1 assay and Sanger sequencing of the PCR products. As shown in Fig. 1b, c and Fig. S1, 13 of 26 (50%) newborn pups carried ANO5 mutations. We chose the rabbits with 7-bp deletion for further analysis. Sanger sequencing of the ANO5 RT-PCR product covering the exon 12 showed that the mutant transcript was expressed with a predicted premature stop codon at the deletion site (Fig. 1d). In order to examine the off-target effects in these ANO5-KO rabbits, the PCR products of top 12 potential off-target sites were subjected to Sanger sequencing and the T7E1 assay. No off-target mutations were detected at these potential sites in the ANO5-KO rabbits (Fig. S2).
Phenotype characterization of ANO5-KO rabbits
Due to the unavailability of a good anti-ANO5 antibody to detect rabbit ANO5 protein, we used quantitative reverse transcriptase-PCR (RT-PCR) to determine the effect of the engineered mutations in ANO5 on its expression in the KO rabbits using three sets of primers. The total RNA was extracted from gastrocnemius the of KO and WT rabbits. As shown in Fig. 2e, with the primer set2, where the reverse oligo is annealed to the gRNA targeting site in exon 12, the expression of ANO5 was significantly reduced in ANO5-KO rabbits compared with that of the heterozygous or WT controls. However, with the primer set 1 (annealed to the downstream region of ANO5 away from the gRNA target sites) and 3 (annealed to the upstream region of ANO5 away from the gRNA target sites), the expression levels of ANO5 transcripts were found to be similar between the KO and WT rabbits (Fig. 2e). These data suggest that the mutant ANO5 transcripts were expressed at the similar levels in the ANO5-KO rabbits as compared with those of the WT ANO5 transcripts in WT rabbits.

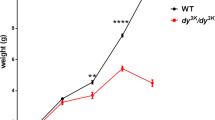
a, b The representative rabbit hind-limbs show no significant different among WT, ANO5+/- and ANO5-/- rabbit muscles at 8 weeks (n = 3). c Body weight, d percent survival rate, e ANO5 mRNA expression level by three different set of primers targeting upstream or downstream region of ANO5 away from the gRNA target sites and exon 12 region, and f serum CK levels of WT, ANO5+/- and ANO5-/- rabbit (n = 6). *p<0.05; **p<0.01;***p<0.001
To determine the gross physiological impact of the engineered ANO5 mutations, we collected the body weight data biweekly and calculated the mortality rate. As shown in Fig. 2a-c, the ANO5-KO rabbits showed no obvious difference as compared with their WT littermates. There were no significant changes in their body weights from 2 to 20 weeks of age compared with the age-matched WT or heterozygous littermates. There were also no significant changes in the mortality of these animals (Fig. 2d).
Muscular dystrophy presentation in ANO5-KO rabbits
Previous studies have reported that ANO5 mutant patients had marked elevation of serum CK levels and muscle weakness8,17. We evaluated clinical and histopathological features of ANO5-KO rabbits. As shown in Fig. 2f, the serum CK was significantly elevated in the ANO5-KO rabbits from 9 months of age compared with their age-matched littermate controls. To further examine the histopathology of the ANO5-KO rabbits, we performed hematoxylin and eosin (H&E) stainings of the gastrocnemius muscle sections from the rabbits at 6, 9, 12 and 15 months of age and Masson’s trichrome staining of the gastrocnemius at 15 months of age. As shown in Fig. 3a, the ANO5-/- rabbits displayed typical muscular dystrophy signs as evidenced by scattered necrotic muscle fibers with inflammatory infiltrates. There was no statistical difference in the average muscle fiber size between the wild-type and ANO5-/- rabbits (Fig. 3b). However, ANO5 rabbits displayed increased percentage of centrally nucleated fibers and necrotic area (Fig. 3c, d). The appearance of these phenotypes was typically observed starting at 12 months of age.

a The histomorphological analysis of WT, ANO5+/- and ANO5-/- rabbit gastrocnemius muscles by H&E at 6, 9, 12 and 15 months. Arrow indicates the presence of necrosis and regeneration area (H&E). b The analysis of muscle fiber area from WT, ANO5+/- and ANO5-/- rabbit . c The analysis of centrally nucleated fibers from WT, ANO5+/- and ANO5-/- rabbit. d The analysis of necrosis and regeneration area from WT, ANO5+/- and ANO5-/- rabbit. Scale bar: 50 µm. ***p<0.001; n = 3
Similarly, muscle necrosis was also observed in tibialis anterior muscle (Fig. 4a). Again, the fiber size in tibialis anterior muscle was not significantly different compared with their control counterparts (Fig. S4). Moreover, the pathological alterations were mostly evident in the tongue muscle (Fig. 4b), which showed extensive muscle degeneration, fibrosis and fatty replacement.

a The histomorphological characteristic of WT and ANO5-/- rabbit tibialis anterior muscles by H&E and Masson, at 15 months. b The histomorphological characteristic of WT and ANO5-/- rabbit tongue muscles by H&E and Masson, at 15 months. c The H&E staining shows the presence of muscle degeneration and irregular muscle fiber size in mutant rabbit diaphragm, whereas the Masson staining showed the collagen-rich tissue in fibrotic region (n = 3). Scale bar: 50 µm
The diaphragm muscles were also found to have extensive muscle degeneration and fibrosis (Fig. 4c), whereas the average fiber area was no significantly changed (Fig. S5). Interestingly, the pathological changes was also noted in smooth muscle of ANO5-/- rabbits. As shown in Fig. 5, the bladder of the ANO5-/- rabbits displayed extensive fibrosis. Therefore, these studies suggest that the loss-of-function mutations in the ANO5 gene lead to the pathological changes in both skeletal and smooth muscles.

a-c Representative images of H&E staining from WT, ANO5+/- and ANO5-/-rabbit. d-f Representative images of Masson's staining from WT, ANO5+/- and ANO5-/-rabbit. Scale bar: 50 µm
Finally, we studied whether ANO5 plays a role in muscle regeneration following cardiotoxin-induced injury in rabbits. At 14 days after cardiotoxin injection, the gastrocnemius muscle from the wild-type rabbits showed typical signs of regeneration as evidenced by the presence of central nuclei (Fig. 6a). However, the injured ANO5-/- muscles showed dramatic increase in fibrosis (Fig. 6a, b) and delayed regeneration with increased number of smaller muscle fibers (Fig. 6c). These results suggest that ANO5 plays a role in muscle regeneration.

a H&E (top) and Masson’s trichrome (bottom) stained muscle sections of WT and ANO5-/- rabbits at 14 days post-cardiotoxin injection. b Quantification of relative fibrotic area in WT and ANO5-/- rabbit muscles at 14 days after cardiotoxin injection. c Fiber size distribution of WT and ANO5-/- gastrocnemius muscles at 14 days post-cardiotoxin injection. Scale bar: 50 µm. ***p < 0.001; n = 3
Discussion
In the present study, we generated a novel rabbit model with ANO5 mutations via the zygote injection of Cas9 mRNA and a pair of sgRNA targeting the rabbit ANO5 gene. Our data demonstrate that CRISPR-induced indels within the exon 12 or 13 of the ANO5 gene lead to the development of pathological alterations in various muscles of the rabbit, resembling human patients with ANO5 mutations. Three independent laboratories including our own group reported the phenotypic results of the ANO5-KO mice, which were generated by traditional gene KO or gene-trapping strategy15,18,19. Two of these ANO5-KO mice, produced by disruption of either exon 118 or exon 219, do not exhibit any obvious muscle pathology; whereas the third line of ANO5-KO mice, produced by gene-trapping between exons 8 and 915, showed very mild myopathy as evidenced by a very small increase in serum CK and central nucleation. In contrast, the rabbits carrying small indels in exons 12 or 13 generated in the present study showed typical dystrophic features including muscle necrosis, regeneration, fatty replacement and fibrosis. Several possibilities may explain the different outcomes of muscle pathology in these ANO5 mutant models.
First, the species and genetic background may play an important role in muscle pathology associated with ANO5 mutations. There are many examples in the literature that the severity of disease could be affected by genetic backgrounds and/or species. For example, mdx mice, a widely used mouse model of Duchenne muscular dystrophy (DMD), have much milder pathology as compared with DMD patients20; the severity of muscular dystrophy in dysferlin-null mice depends on their genetic background21; a common disease-associated missense mutation in α-sarcoglycan fails to cause muscular dystrophy in mice22. Studies have shown that genetic modifiers may affect the disease outcomes in DMD and dysferlinopathy23,24. Thus, this raises a possibility that genetic modifiers may also contribute to the disease manifestation in muscular dystrophy associated with ANO5 mutations. In support of this, it has been shown that there is a wide variation of pathological outcomes among patients even with the same or similar ANO5 mutations25,26. Future investigations would be required to ascertain the involvement of genetic modifiers in the disease progression associated with ANO5 mutations and identify them. Elucidation of the genetic modifiers affecting muscular dystrophy associated with ANO5 mutations would not only shed light into the pathogenesis but may also offer us the opportunity to identify novel therapeutic targets to treat the disease.
Second, it is also plausible that the nature of different mutations is responsible for the different outcomes in these animal models. The complete disruption of ANO5 expression in the two lines of ANO5-KO mice does not cause muscle pathology, indicating that mice may have an efficient compensatory mechanism to maintain muscle function in the complete absence of ANO519. There are a total of 10 members of anoctamin protein family and some overlapping expression in skeletal muscle27. It is possible that these anoctamin homologs may compensate for the complete loss of ANO5 in mice. On the other hand, intragenic frame-terminating or shifting mutations in either the gene trapped mice or our ANO5 mutant rabbits result in muscle disease. Recently, Nigro and his colleagues screened a cohort of 786 undiagnosed patients with LGMD or nonspecific myopathic features using next-generation sequencing, and found that 33 out of 786 patients carry ANO5 mutations in both allele (either homozygous or compound heterozygous). Interestingly, the majority of these 33 patients carry at least one allele with a premature termination mutation (either nonsense or frame-shifting; only 9 carry both missense mutations)28. A common theme with the gene-trapping mouse, our CRISPR rabbit and human patients lies in that truncated ANO5 expression is expected, although the lack of a good antibody with high affinity and specificity to detect mouse or rabbit ANO5 makes it challenge to draw a firm conclusion. We recently showed that a LGMD2L patient carrying a frame-shifting mutation in ANO5 had truncated ANO5 expression, which is also prone to form intracellular aggregates, highlighting its potential contribution to the pathogenesis of muscular dystrophy. Thus, it appears that expression of truncated ANO5 peptides may have more deleterious effects on muscle health and function as compared with the complete absence of ANO5 protein, potentially via inhibiting the compensatory functions of other anoctamin proteins. Future studies with CRISPR gene-editing technology would allow us to unveil the compensatory mechanisms, as well as the mechanisms by which truncated ANO5 inhibits such compensatory pathways.
Taken together, the ANO5 mutant rabbit model generated by CRISPR gene-editing recapitulates many aspects of muscular dystrophy associated with ANO5 mutations in human patients. This new model would facilitate the basic research to understand the pathogenesis of ANO5-associated muscular dystrophy and the physiological function of ANO5, and the translational studies to develop novel therapeutic strategies for the treatment of this disease.
Materials and methods
Animals and ethics statement
The New Zealand rabbits used in this study were maintained at the Laboratory Animal Center of Jilin University. All rabbit experiments in this study were reviewed and approved by the Animal Care and Use Committee of Jilin University.
CRISPR/Cas9 sgRNA preparation, embryo microinjection and embryo transfer
The CRISPR/Cas9 sgRNA was designed and assembled as previously described29. The annealed sgRNA oligos were cloned into the BbsI sites of pUC57-T7-sgRNA cloning vector (Addgene ID 51306). The vector of pUC57-T7-sgRNA was PCR amplified using the T7 primers (T7-F: 5′-GAAATTAATACGACTCACTATA-3′ and T7-R: 5′-AAAAAAAGCACCGACTCGGTGCCAC-3′), and the PCR products were transcribed in vitro with MAXIscript T7 Kit (Ambion) and purified by miRNeasy Mini Kit (Qiagen) according to the manufacturer's instruction.
The 3xFLAG-NLS-SpCas9-NLS vector (Addgene ID 48137) was linearized with NotI and transcribed in vitro using the mMessage mMachine SP6 Kit (Ambion) and the RNeasy Mini Kit (Qiagen) according to the manufacturer's instruction. The microinjection procedure and embryo transfer was essentially the same as we previously described30.
Mutation detection in pups by PCR and sequencing
The genomic DNA from ANO5-KO and WT rabbits were extracted from a small piece of ear tissue using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) according to the manufacturer’s instruction. The sgRNA target sites were amplified by PCR using the primers (forward 1, 5′-CCCATATGCCTTGTTCTATT-3′; reverse 1, 5′-GCATGATTAGGAACCCTTT-3′; forward 2, 5′-CCCTCTGACTCACAAATAAA-3′; reverse 2, 5′-TCATAGCTTACCACCAAATC-3′). The PCR products were gel purified and cloned into pGM-T vector (Tiangen, Beijing, China). A minimum of 14 positive clones were sequenced and analyzed using DNAman.
T7EI cleavage assay
The T7EI cleavage assay was performed as described previously31. Briefly, the PCR products were purified, denatured and then re-annealed in NEBuffer 2 (NEB) using a thermocycler. Hybridized PCR products were digested with T7 endonuclease 1 (NEB, M0302L) for 30 min at 37 °C and subjected to 2% agarose gel electrophoresis.
Off-target analysis
The potential off-target sites were predicted by online CRISPR Design tool developed by Zhang’s group at MIT (http://crispr.mit.edu/). The PCR products for the top 12 off-target sites using the primers listed in Supplementary Table S1 were subjected to the T7EI assay and Sanger sequencing.
RNA isolation, RT-PCR and qRT-PCR
The total RNA was isolated from gastrocnemius muscle of WT and ANO5-KO rabbits using TRNzol-A+ reagent (Tiangen, Beijing, China), and treated with DNase I (Fermentas). The first-strand complementary DNA (cDNA) was synthesized using the cDNA first-strand synthesis kit (Tiangen, Beijing, China). The cDNA was used for RT-PCR and quantitative RT-PCR (qRT-PCR) analyses to examine the expression of ANO5. The primers used for RT-PCR and qRT-PCR were shown in Table S2. The qRT-PCR was performed using the BioEasy SYBR Green I Real Time PCR Kit (Bioer Technology, Hangzhou, China), and the 2-ΔΔCT formula was used to analyze the gene expression, Gapdh was used as a reference gene. All experiments were repeated three times. The data were expressed as the mean ± S.E.M.
Body weight, survival and statistical analysis
The body weight of age- and gender-matched WT and ANO5-KO rabbits were measure biweekly. All data were expressed as the mean ± S.E.M., and a minimum of three individual animals of each genotype were used in all experiments. The data were analyzed by the Student’s t-test using Graphpad Prism 7.0 software. A probability of p < 0.05 was considered statistically significant.
Serum biochemistry analysis
The blood samples were collected into heparinized tubes from the ear vein, and sera were prepared by precipitation and centrifugation. The serum CK was measured using the CK Test Kit (N-acetyl-l-cysteine method).
Histology analysis
Various tissues including gastrocnemius, tibial anterior, tongue, diaphragm and bladder were collected from ANO5-KO and WT rabbits (euthanized at 6, 9, 12 and 15 months of age). The tissues were fixed in 4% paraformaldehyde at 4 °C, dehydrated in increasing concentrations of ethanol (70% for 6 h, 80% for 1 h, 96% for 1 h and 100% for 3 h), cleared in xylene and embedded in paraffin for the histological examination. The 5-μm sections were cut for H&E32 and Masson’s trichrome33 as previously described. The stained sections were imaged with a Nikon TS100 microscope.
Morphometric analysis of myofibers
The H&E-stained cross-sections of gastrocnemius and tibial anterior muscles from the ANO5-KO and WT rabbits at the age of 15 months were analyzed for fiber size. A minimum of three different regions were counted per section. The fiber size was calculated using ImageProPlus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Cardiotoxin-induced muscle injury and regeneration
To induced muscle injury and regeneration, cardiotoxin (diluted to 30 μg/ml with sterile saline, 100 μl) was injected into the left gastrocnemius muscles of 12-month-old ANO5-/- and wild-type rabbits. An equal volume of sterile saline was injected into contralateral muscle as a vehicle control. The muscle biopsies were collected at 14 days post-cardiotoxin injection for histopathology analysis.
Change history
17 June 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Murphy, A. P. & Straub, V. The classification, natural history and treatment of the limb girdle muscular dystrophies. J. Neuromuscul. Dis. 2, S7–S19 (2015).
Blackburn, P. R. et al. Early-onset limb-girdle muscular dystrophy-2L in a female athlete. Muscle Nerve 55, E19–E21 (2017).
Suzuki, J., Umeda, M., Sims, P. J. & Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 (2010).
Tsutsumi, S. et al. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD). Am. J. Hum. Genet. 74, 1255–1261 (2004).
Bolduc, V. et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am. J. Hum. Genet. 86, 213–221 (2010).
Mahjneh, I. et al. A new distal myopathy with mutation in anoctamin 5. Neuromuscul. Disord. 20, 791–795 (2010).
Hicks, D. et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain 134, 171–182 (2011).
Bouquet, F. et al. Miyoshi-like distal myopathy with mutations in anoctamin 5 gene. Rev. Neurol. (Paris) 168, 135–141 (2012).
Penttila, S. et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 78, 897–903 (2012).
Schroeder, B. C., Cheng, T., Jan, Y. N. & Jan, L. Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–1029 (2008).
Almaca, J. et al. TMEM16 proteins produce volume-regulated chloride currents that are reduced in mice lacking TMEM16A. J. Biol. Chem. 284, 28571–28578 (2009).
Brunner, J. D., Lim, N. K., Schenck, S., Duerst, A. & Dutzler, R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212 (2014).
Mizuta, K. et al. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochem. Biophys. Res. Commun. 357, 126–132 (2007).
Pedemonte, N. & Galietta, L. J. Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 94, 419–459 (2014).
Griffin, D. A. et al. Defective membrane fusion and repair in Anoctamin 5-deficient muscular dystrophy. Hum. Mol. Genet. 25, 1900–1911 (2016).
Wang, Y. et al. Generation of knockout rabbits using transcription activator-like effector nucleases. Cell Regen. 3, 3 (2014).
Kadoya, M. et al. A Japanese male with a novel ANO5 mutation with minimal muscle weakness and muscle pain till his late fifties. Neuromuscul. Disord. 27, 477–480 (2017).
Gyobu, S. et al. A role of TMEM16E carrying a scrambling domain in sperm motility. Mol. Cell. Biol. 36, 645–659 (2015).
Xu, J. et al. Genetic disruption of Ano5 in mice does not recapitulate human ANO5-deficient muscular dystrophy. Skelet. Muscle 5, 43 (2015).
Bulfield, G., Siller, W. G., Wight, P. A. & Moore, K. J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 81, 1189–1192 (1984).
Han, R., Rader, E. P., Levy, J. R., Bansal, D. & Campbell, K. P. Dystrophin deficiency exacerbates skeletal muscle pathology in dysferlin-null mice. Skelet. Muscle 1, 35 (2011).
Collier, A. F. et al. Effect of ibuprofen on skeletal muscle of dysferlin-null mice. J. Pharmacol. Exp. Ther. 364, 409–419 (2018).
Xu, L. et al. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther.: J. Am. Soc. Gene Ther. 24, 564–569 (2016).
Fatehi, F. et al. Dysferlinopathy in Iran: clinical and genetic report. J. Neurol. Sci. 359, 256–259 (2015).
Little, A. A., McKeever, P. E. & Gruis, K. L. Novel mutations in the Anoctamin 5 gene (ANO5) associated with limb-girdle muscular dystrophy 2L. Muscle Nerve 47, 287–291 (2013).
Sarkozy, A. et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum. Mutat. 34, 1111–1118 (2013).
Duran, C. & Hartzell, H. C. Physiological roles and diseases of Tmem16/Anoctamin proteins: are they all chloride channels? Acta Pharmacol. Sin. 32, 685–692 (2011).
Savarese, M. et al. Next generation sequencing on patients with LGMD and nonspecific myopathies: findings associated with ANO5 mutations. Neuromuscul. Disord. 25, 533–541 (2015).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Sui, T. et al. CRISPR/Cas9-mediated mutation of PHEX in rabbit recapitulates human X-linked hypophosphatemia (XLH). Hum. Mol. Genet. 25, 2661–2671 (2016).
Guschin, D. Y. et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 649, 247–256 (2010).
Gutpell, K. M., Hrinivich, W. T. & Hoffman, L. M. Skeletal muscle fibrosis in the mdx/utrn+/- mouse validates its suitability as a murine model of Duchenne muscular dystrophy. PLoS. ONE. 10, e0117306 (2015).
Mu, X. et al. The role of Notch signaling in muscle progenitor cell depletion and the rapid onset of histopathology in muscular dystrophy. Hum. Mol. Genet. 24, 2923–2937 (2015).
Acknowledgements
We thank Peiran Hu for excellent technical assistance at the Embryo Engineering Center. This study was financially supported by the National Key Research and Development Program of China Stem Cell and Translational Research (2017YFA0105101) and GuangDong Province science and technology plan project (2014B020225003). R.H. is supported by US National Institutes of Health grants (R01HL116546 and R01 AR064241).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sui, T., Xu, L., Lau, Y.S. et al. Development of muscular dystrophy in a CRISPR-engineered mutant rabbit model with frame-disrupting ANO5 mutations. Cell Death Dis 9, 609 (2018). https://doi.org/10.1038/s41419-018-0674-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0674-y
This article is cited by
-
Regulation of phospholipid distribution in the lipid bilayer by flippases and scramblases
Nature Reviews Molecular Cell Biology (2023)
-
Ano5 modulates calcium signaling during bone homeostasis in gnathodiaphyseal dysplasia
npj Genomic Medicine (2022)
-
Dysregulated calcium homeostasis prevents plasma membrane repair in Anoctamin 5/TMEM16E-deficient patient muscle cells
Cell Death Discovery (2019)
-
Genetic Disruption of Anoctamin 5 in Mice Replicates Human Gnathodiaphyseal Dysplasia (GDD)
Calcified Tissue International (2019)
