
Abstract
Sterol regulatory element-binding proteins (SREBPs) belong to a family of transcription factors that regulate the expression of genes required for the synthesis of fatty acids and cholesterol. Three SREBP isoforms, SREBP1a, SREBP1c, and SREBP2, have been identified in mammalian cells. SREBP1a and SREBP1c are derived from a single gene through the use of alternative transcription start sites. Here we investigated the role of SREBP-mediated lipogenesis in regulating tumor growth and initiation in colon cancer. Knockdown of either SREBP1 or SREBP2 decreased levels of fatty acids as a result of decreased expression of SREBP target genes required for lipid biosynthesis in colon cancer cells. Bioenergetic analysis revealed that silencing SREBP1 or SREBP2 expression reduced the mitochondrial respiration, glycolysis, as well as fatty acid oxidation indicating an alteration in cellular metabolism. Consequently, the rate of cell proliferation and the ability of cancer cells to form tumor spheroids in suspension culture were significantly decreased. Similar results were obtained in colon cancer cells in which the proteolytic activation of SREBP was blocked. Importantly, knockdown of either SREBP1 or SREBP2 inhibited xenograft tumor growth in vivo and decreased the expression of genes associated with cancer stem cells. Taken together, our findings establish the molecular basis of SREBP-dependent metabolic regulation and provide a rationale for targeting lipid biosynthesis as a promising approach in colon cancer treatment.
Similar content being viewed by others


Introduction
Although diverse in type and underlying genetic alterations, cancers are fundamentally a disorder of cell growth and proliferation, which requires increased cellular building blocks, such as nucleic acids, proteins, and lipids1. To cope with these elevated requirements cancer cells undergo major metabolic modifications2,3. There has been increasing interest in cancer cell metabolism as a means to understand the functional distinction between transformed and normal cells and to provide critical mechanistic insights regarding cancer development and progression4. Among metabolic alterations, increased de novo lipid biosynthesis has been recognized as one of the important but not well-characterized hallmarks of cancer cells5. Relatively few studies have rigorously examined the role of lipogenesis in promoting colorectal cancer (CRC) and how lipogenic pathways are regulated.
Sterol regulatory element-binding proteins (SREBPs) is a small family of membrane-bound, basic helix-loop-helix leucine zipper (bHLH-LZ) transcription factors that regulate the expression of genes required for the synthesis of fatty acids, triglycerides and cholesterol6,7,8. Three SREBP isoforms, SREBP1a, SREBP1c, and SREBP2, have been identified in mammalian cells that control distinct but overlapping lipogenic transcriptional programs7,8,9. A rich body of research has demonstrated that SREBP1a activates fatty acid and cholesterol synthesis, SREBP1c fatty acid synthesis, and SREBP2 cholesterol synthesis in insulin-responsive tissues such as liver and adipose tissue. The activation process of SREBPs is known to be tightly controlled by the availability of sterols8,10. Specifically, the newly synthesized SREBPs are expressed as inactive precursors and reside as integral trans-membrane proteins within the endoplasmic reticulum (ER) membrane where they bind to the sterol-sensing SREBP cleavage-activating protein (SCAP). When intracellular sterol concentrations are low, the SREBP/SCAP complex trafficks to the Golgi where SREBP is cleaved by site-1 and site-2 proteases and the N-terminal bHLH-LZ domain of the protein is released and translocated to the nucleus where it binds to sterol regulatory element (SRE)-sequences in the promoters of its target genes, ultimately increases sterol levels8,9,10,11. As a feedback mechanism to regulate sterol synthesis, cholesterol and its hydroxylated derivatives, such as 25-hydroxycholesterol (25-HC), inhibit the proteolytic cleavage and prevent the activation of SREBPs12. Specifically, 25-HC binds to ER anchor protein Insig to promote the formation of SCAP–Insig complex and prevent trafficking of SREBP–SCAP complex to the Golgi13. Moreover, fatostatin, a non-sterol-like small molecule inhibitor of SREBP, has been developed to attenuate SREBP-dependent lipogenesis by binding to SCAP to block the ER–Golgi translocation of SREBPs14.
In addition to their role in maintaining the homeostasis of lipid metabolism8, emerging evidence suggests that increased activation of SREBPs is required to sustain cancer cell proliferation. For example, activation of SREBP1 and enhanced expression of its target genes have been observed in human glioblastoma multiforme carrying activating EGFR mutations and inhibition of lipid synthesis blocks the growth of xenograft tumors derived from glioblastoma cells expressing mutant EGFR15. In addition, it has been shown that the expression of SREBP1 is elevated in prostate cancer patients16. High SREBP1 expression is positively associated with tumor metastasis and predicts poor prognosis in breast cancer patients17. Moreover, activation of SREBP as a result of mTORC1 activation downstream of oncogenic PI3K and KRAS signaling has been shown to promote breast cancer cell growth and proliferation18. Consistently, the expression of SREBP target genes, such as fatty acid synthase (FASN) and steroyl-CoA desaturase (SCD), are also increased in a variety of human cancers1,19,20,21. However, the function of SREBPs in human CRC remains to be fully elucidated.
In this study, we investigated the functional importance of SREBP-mediated lipogenesis in regulating cellular metabolism and tumor growth in colon cancer. Inhibiting lipid biosynthesis by silencing SREBP1, SREBP2 or their regulator SCAP reduced the rate of cell proliferation as well as tumor-initiating potential in vitro. Moreover, downregulation of SREBP expression or inhibition of SREBP activation markedly altered cellular metabolic pathways. Importantly, knockdown of SREBPs inhibited xenograft tumor growth and initiation in vivo. Taken together, our findings demonstrate that SREBPs have an important role in promoting tumorigenesis in colon cancer.
Results
Knockdown of SREBP1 or SREBP2 decreases cell proliferation and spheroid formation in colon cancer cells
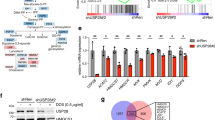
To begin to elucidate the role of SREBP-mediated lipid biosynthesis in colon cancer, we generated multiple SREBP1 and SREBP2 knockdown cell lines using two different lentivirus encoded shRNAs (Fig. 1a, b). Notably, both SREBP1-specific shRNAs target common regions in the SREBP1 gene shared by SREBP1a and SREBP1c isoforms. Downregulation of both the precursor and mature forms of SREBP1 and SREBP2 protein was confirmed by western blot analysis in corresponding knockdown cell lines (Fig. 1c, d). Consistent with their transcription factor function, silencing SREBPs led to a significant decrease in the expression of target genes in DLD1 and patient-derived Pt130 colon cancer cells (Fig. 1e, f). The target genes of SREBPs tested here are known to involved in cholesterol uptake and biosynthesis (including LDR receptor (LDLR), HMG-CoA reductase (HMGCR), and HMG-CoA synthase (HMGCS)) as well as fatty acid synthesis (including fatty acid synthase (FASN), acetyl-CoA carboxylase (ACACA), and stearoyl CoA desaturase (SCD)). Similar effects on decreasing target gene expression were observed in HCT116 cells (Supplementary Figure S1). As a consequence, intracellular levels of free fatty acids (FFA), cholesterol and triglycerides (TAG) were significantly decreased in SREBP knockdown cells (Fig. 1g).

a, b The real-time PCR analysis of SREBP1 and SREBP2 expression in control and knockdown DLD1 (a) and Pt130 (b) cells. Two different shRNA targeting sequences were used for knocking down SREBP1 or SREBP2. c, d The expression of SREBP1 and SREBP2 proteins were downregulated in stable DLD1 (c) and Pt130 (d) knockdown cells. Protein lysates from control and knockdown cells were analyzed by western blot. Both the precursor and mature forms of SREBP1 and SREBP2 proteins were detected. e, f Knockdown of SREBP1 and SREBP2 decreased the expression of downstream genes related to fatty acid synthesis and metabolism in DLD1 (e) and Pt130 (f) cells. Data represent the mean ± SD (*p < 0.001 and #p < 0.05 compared to sh-Control). g Knockdown of SREBP1 or SREBP2 decreased lipogenesis in colon cancer cells. Total cellular lipids were extracted from control and SREBP knockdown DLD1 and Pt130 cells and the levels of free fatty acids, cholesterol, and triglyceride were measured and normalized to the amount of total proteins. Data represent the mean ± SD (*p < 0.001, §p < 0.01, and #p < 0.05 compared to sh-Control)
To investigate the functional effect of silencing SREBPs, we monitored the rate of cell proliferation in control and SREBP1 or SREBP2 knockdown DLD1 and Pt130 cells. Silencing either SREBP1 or SREBP2 was sufficient to reduce cell growth in both cell lines when compared to control cells (Fig. 2a, b). Similar results were obtained in SREBP1 and SREBP2 knockdown HCT116 cells (Supplementary Figure S2a). Furthermore, we determined if SREBP-dependent lipogenesis is required for maintaining the tumor initiation potential of colon cancer cells. To this end, single cells of control and SREBP1 or SREBP2 knockdown DLD1 and Pt130 cells were cultured in suspension stem cell medium, a condition known to enrich tumor-initiating cells22, and numbers of spheroids (colonies) formed were counted after 6 days. While approximately 10% of control DLD1 and Pt130 cells formed tumor spheroids in suspension, the numbers of spheroids were significantly decreased in SREBP1 and SREBP2 knockdown cells suggesting inhibition of colony formation (Fig. 2c, d). In addition, knockdown of SREBP1 and SREBP2 resulted in a significant decrease in the number of tumor spheroids formed in HCT116 cells (Supplementary Figure S2b). Consistently, qPCR analysis revealed that the expression of genes associated with colon cancer stem cells, including CD44, CD133, LGR5, and AXIN2, was reduced in SREBP1 and SREBP2 knockdown DLD1 and Pt130 cells (Fig. 2e). Taken together, these data suggest that SREBP1-mediated and SREBP2-mediated lipid biosynthesis has an important role in supporting cell proliferation and tumor initiation potential of colon cancer cells.

a, b Knockdown of SREBP1 or SREBP2 decreased the rate of proliferation in DLD1 (a) and Pt130 (b) cells. Equal number of control and SREBP knockdown cells were allowed to grow for 4 days and the number of cells were counted each day. Data represent the mean ± SD (§p < 0.01 and #p < 0.05 compared to sh-Control). c, d Knockdown of SREBP1 or SRBEP-2 decreased the formation of tumor spheroids in the stem cell suspension medium. Control and SREBP knockdown DLD1 (c) and Pt130 (d) cells were seeded as single cells in the stem cell suspension medium and the number of colonies formed was determined after 6 days. Data represent the mean ± SD (¶p < 0.0001 and *p < 0.001 compared to sh-Control). e Knockdown of SREBP1 or SRBEP2 reduced the expression of genes associated with colon cancer stem cells. The relative expression of CD44, CD133, LGR5, and AXIN2 mRNA was determined using real-time PCR in control and SREBP knockdown DLD1 and Pt130 cells. Data represent the mean ± SD (§p < 0.01 and #p < 0.05 compared to sh-Control)
Inhibition of proteolytic processing of SREBPs decreases cell proliferation and spheroid formation in colon cancer cells
We next determined whether inhibition of SREBP activation by blocking proteolytic cleavage has similar effects as silencing SREBP directly. Consistent with previous reports, treating cells with 25-HC or fatostatin markedly decreased the levels of mature (transcriptionally active) form of SREBP1 and SREBP2 in DLD1 cells as shown by western blot analysis (Fig. 3a). Similar as those observed in SREBP1 and SREBP2 knockdown cells, 25-HC-induced and fatostatin-induced inhibition of SREBPs activation significantly reduced the expression of downstream target genes important for lipid biogenesis (Fig. 3b). To determine the functional effect of SREBP inhibition, DLD1 cells were cultured in the presence of 25-HC or fatostatin. We found that both fatostatin and 25-HC significantly inhibited cell growth, however, fatostatin showed a strong inhibitory effect (Fig. 3c). This is likely due to the fact that fatostatin binds and inhibits SCAP-mediated activation of SREBP in a sterol-independent manner, whereas 25-HC competes directly with sterols in cell culture media14. Furthermore, results from spheroid formation experiments showed that fatostatin treatment reduced the number of tumor spheroids formed in suspension (Fig. 3d). Interestingly, the 25-HC treatment was unable to inhibit tumor spheroid formation (data not shown). As the suspension medium used in this assay contains high levels of bovine serum albumin (BSA), BSA-bound lipids may attenuate the 25-HC-mediated inhibitory effect. In addition, 25-HC-induced and fatostatin-induced inhibition of SREBP activation decreased expression of genes associated with cancer stem cells (Fig. 3e).

a Treating cells with 25-HC (10 μg/ml) or fatostatin (30 μM) blocks the proteolytic processing of SREBPs in DLD1 cells. Protein lysates from control and treated cells were analyzed for the expression of SREPBs by western blot. b Inhibition of SREBPs decreased the cell survival. DLD1 cells were grown in the presence or absence of fatostatin and 25-HC for 72 h and numbers of cells was counted. The relative cell growth was calculated by normalizing the number of cells to the control condition. Data represent the mean ± SD (*p < 0.0001 and #p < 0.05 compared to control untreated cells). c Inhibition of SREBPs decreased the expression of downstream genes related to fatty acid synthesis and metabolism in DLD1 cells. Data represent the mean ± SD (*p < 0.001, §p < 0.01, and #p < 0.05 compared to control untreated cells). d Fatostatin treatment inhibited the formation of tumor spheroids in the stem cell suspension medium. DLD1 cells were seeded as single cells in the stem cell suspension medium in the presence of absence of fatostatin and the number of colonies formed was determined after 6 days. Data represent the mean ± SD (§p < 0.01 compared to untreated cells). e Inhibition of SREBPs reduced the expression of genes associated with colon cancer stem cells. The relative expression of CD44, CD133, LGR5, and AXIN2 mRNA was determined using real-time PCR in control and fatostatin or 25-HC treated DLD1 cells. Data represent the mean ± SD (§p < 0.01 and #p < 0.05 compared to control untreated cells)
To further confirm the functional importance of de novo lipid biosynthesis in colon cancer, we silenced the expression of SCAP, a sterol-sensing SCAP, in DLD1 and Pt130 cells using lentivirus-mediated RNAi. Consistent with its role in facilitating ER–Golgi translocation and proteolytic cleavage of SREBPs, downregulation of SCAP resulted in a similar decrease in the expression of genes required for lipid biosynthesis (Supplemental Figure S3a). In addition, knockdown of SCAP inhibited cell proliferation in both colon cancer cell lines (Supplemental Figure S3b). Furthermore, silencing SCAP significantly inhibited tumor spheroid formation and decreased expression of genes associated with cancer stem cells (Supplemental Figure S3c, d). Collectively, these results indicate that inhibition of lipid biosynthesis, via downregulation of SREBPs or inhibition of proteolytic cleavage of SREBPs, significantly inhibited cell growth and reduced cancer stem cell properties.
Downregulation of SREBP alters cellular metabolism
Accumulating evidence suggests that alterations in lipid metabolism contribute to overall metabolic reprogramming in cancer cells1,5,23. To better understand the mechanism underlying SREBP-mediated regulation of cell proliferation and stem cell properties, we determined the functional effect of SREBP-loss on modulating other metabolic pathways in colon cancer cells. To this end, we performed Mito Stress Tests using Seahorse XF96 Extracellular Flux Analyzer. Results showed that oxygen consumption rates (OCR) associated with both basal and maximal mitochondrial respiration was significantly decreased in SREBP1 and SREBP2 knockdown cells (Fig. 4a, b). In addition, mitochondrial respiration used for ATP production and reserved mitochondrial capacities were reduced in SREBP knockdown cells (Fig. 4a, b). Moreover, control, SREBP1 and SREBP2 knockdown cells were subjected to seahorse glycolysis stress tests to evaluate the glycolysis phenotype. Interestingly, silencing SREBP1 or SREBP2 significantly decreased the extracellular acidification rate (ECAR) associated with glycolysis, glycolytic capacity and glycolytic reserves in Pt130 cells (Fig. 4c, d). Similar results were obtained in SREBP1 and SREBP2 knockdown DLD1 cells (data not shown). Furthermore, bioenergetic measurements of relative utilization of mitochondrial respiration and glycolysis under basal conditions confirmed that knockdown of either SREBP reduced both the glycolytic potential and the mitochondria-mediated oxidative phosphorylation in colon cancer cells (Fig. 4e). These results suggest that SREBP-dependent lipid biogenesis has an important role in maintaining overall metabolic activity in cancer cells.

a Representative OCR measurements obtained from the Mito tress test performed in control (sh-Control) and SREBP knockdown (sh-SREBP1 and sh-SREBP2) Pt130 cells using the Seahorse XF96 Extracellular Flux analyzer. Oligomycin, FCCP, and antimycin A (Anti-A) were added at the indicated points. The shaded areas indicate the OCR levels associated with basal and maximal respiration, ATP turnover and reserved capacity, respectively. b Experiments as shown in a were quantified and the relative levels of OCR associated with basal and maximal respiration, ATP turnover and reserved capacity were calculated. Data represent the mean ± SEM (¶p < 0.0001 and #p < 0.05 compared to Sh-Control). c Representative ECAR measurements obtained from the glycolysis stress test performed in control and SREBP knockdown Pt130 cells using the Seahorse XF96 Extracellular Flux analyzer. Glucose, oligomycin, and 2-deoxyglucose (2-DG) were added at the indicated points. The shaded areas indicate the ECAR levels associated with glycolysis, glycolytic capacity, and glycolytic reserve, respectively. d Experiments as shown in c were quantified and ECAR associated with glycolysis, glycolytic capacity, and glycolytic reserve was calculated. Data represent the mean ± SEM (¶p < 0.0001 compared to Sh-Control). e Knockdown of SREBP resulted in metabolic shifts in Pt130 cells. The energetic map of control and SREBP knockdown Pt130 cells was based on the measurements obtained in a and c. The OCR and ECAR data shown in the map represent the OCR of basal respiration from the Mito stress test and the glycolysis-related ECAR from the glycolysis stress test, respectively (data represent the mean ± SEM)
To confirm the effects of SRBEP downregulation on modulating cellular metabolism, we assessed the levels of mitochondrial respiration and glycolysis in cells treated with fatostatin or 25-HC. Bioenergetic measurements obtained using the Seahorse XF Analyzer showed that inhibition of SREBP activation by treating cells with fatostatin or 25-HC decreased both basal and maximal mitochondrial respiration as well as mitochondrial respiration associated with ATP production and reserved capacities in DLD1 cells (Fig. 5a). In addition, fatostatin or 25-HC treatment significantly reduced levels of glycolysis, glycolytic capacity, and glycolytic reserve (Fig. 5b). The cell bioenergetic phenotype analysis revealed that inhibition of lipid biogenesis resulted in a decrease in both mitochondrial oxidative phosphorylation and glycolysis (Fig. 5c). Similar results were obtained in SCAP knockdown Pt130 cells (Fig. 5e, f). Furthermore, we determined whether SREBP expression is associated with cellular metabolic pathways by analyzing the Cancer Genome Atlas (TCGA) CRC gene expression data. Indeed, results from gene set enrichment analysis (GSEA) revealed that both SREBP1 (gene name: SREBF1) and SREBP2 (gene name: SREBF2) expression was positively associated with not only lipid biosynthesis pathways but also glucose metabolism and TCA cycle in CRC patients (Supplemental Figure S4). Taken together, our results indicate that the lipid biogenesis pathway is functionally connected with glycolysis and mitochondrial-dependent metabolic pathways in CRC.

Control and fatostatin or 25-HC treated DLD1 cells were subjected to metabolic analysis using the Seahorse XF96 Extracellular Flux analyzer as described in Fig. 4. a The relative levels of OCR associated with basal and maximal respiration, ATP turnover and reserved capacity were calculated based on the measurements obtained in the Mito stress test. Data represent the mean ± SEM (¶p < 0.0001 and §p < 0.01 compared to control untreated cells). b The levels of ECAR associated with glycolysis, glycolytic capacity, and glycolytic reserve were calculated based on the measurements obtained in the glycolysis stress test. Data represent the mean ± SEM (¶p < 0.0001 and §p < 0.01 compared to control untreated cells). c The energetic map of control and treated DLD1 cells was based on the measurements obtained in a and c. The OCR and ECAR data shown in the map represent the OCR of basal respiration from the Mito stress test and the glycolysis-related ECAR from the glycolysis stress test, respectively (data represent the mean ± SEM). d Control and SCAP knockdown Pt130 cells were subjected to metabolic analysis using the Seahorse XF96 Extracellular Flux analyzer. The relative levels of OCR associated with basal and maximal respiration, ATP turnover and reserved capacity were calculated based on the measurements obtained in the Mito stress test. Data represent the mean ± SEM (¶p < 0.0001 compared to Sh-Control). e The levels of ECAR associated with glycolysis, glycolytic capacity and glycolytic reserve was calculated based on the measurements obtained in the glycolysis stress test. Data represent the mean ± SEM (*p < 0.001 and §p < 0.01 compared to Sh-Control). f The energetic map of control and SCAP knockdown Pt130 cells was derived from the measurements obtained in d and e. The OCR and ECAR data shown in the map represent the OCR of basal respiration from the Mito stress test and the glycolysis-related ECAR from the glycolysis stress test, respectively (data represent the mean ± SEM)
Downregulation of lipid biogenesis decreases fatty acid oxidation
In normal insulin-responsive tissues, decreased fatty acid synthesis is often coupled with increased fatty acid oxidation (FAO). Here, we determined the effect of knocking down SREBPs on FAO in colon cancer cells. The FAO assays were performed to measure the OCR levels in the presence or absence of etomoxir (ETO), a carnitine palmitoyltransferase-1 (CPT-1) inhibitor, using Seahorse XF Analyzer. As no exogenous fatty acids were added in the assays, the differences in the OCR measurements were used to determine the utilization of endogenous fatty acids. Intriguingly, results obtained in control and SREBP1 and SREBP2 knockdown DLD1 and Pt130 cells showed that silencing either SREBP significantly reduced the levels of FAO in both cell lines (Fig. 6a, b). Similarly, knockdown of SCAP decreased the levels of FAO in both DLD1 and Pt130 cells (Fig. 6c). In supporting a functional connection between SREBP-mediated lipid biosynthesis and FAO in cancer cells, results from our GSEA study showed that the expression of SREBP1 and SREBP2 was positively correlated with the fatty acid metabolism pathway in CRC patient samples (Supplemental Figure S4). Taken together, these results suggest that inhibition of SREBP-mediated lipogenesis not only reduces the need for glycolysis and mitochondrial respiration but also decreases the rate of FAO in colon cancer cells.

a, b Knockdown of SREBP1 and SREBP2 reduced fatty acid oxidation (FAO) in DLD1 (a) and Pt130 (b) cells. The rate of cellular FAO was determined by subjecting control and SREBP knockdown cells to the FAO assays using the Seahorse XF96 Extracellular Flux analyzer. Data represent the mean ± SEM (§p < 0.01 compared to Sh-Control). c Knockdown of SCAP reduced FAO in DLD1 and Pt130 cells. The rate of cellular FAO was determined using Seahorse FAO assays. Data represent the mean ± SEM (#p < 0.05, compared to Sh-Control)
Silencing SREBP inhibits xenograft tumor growth in vivo
Given that inhibition of lipid biosynthesis by silencing SREBPs decreases cell proliferation and stemness in colon cancer cells, we next investigated the functional effects of SREBP-mediated lipid biosynthesis in controlling tumor growth in vivo. To this end, control and SREBP1- or SREBP2 knockdown Pt130 cells were injected subcutaneously into NSG mice and the tumorigenesis process was monitored over 28 days. We found that silencing SREBP1 or SREBP2 significantly reduced the rate of tumor growth over the follow-up period and decreased the average weight of tumors (Fig. 7a, b). In addition, tumor sections were stained with the Ki67 and cleaved caspase-3 antibodies to determine the rate of cell proliferation and apoptosis, respectively. IHC staining results showed that the numbers of Ki67-positive cells were significantly decreased in SREBP1 and SREBP2 knockdown tumors indicating decreased cell proliferation (Fig. 7c, d). In contrast, the numbers of cleaved caspase-3-positive cells were increased indicating elevated levels of apoptosis (Fig. 7e, f), Consistent with the results obtained in cultured cells, the expression of genes associated with cancer stem cells, including CD44, CD133, LGR5, and AXIN2, was significantly decreased in SREBP1 and SREBP2 knockdown tumors (Fig. 7g). Furthermore, to confirm the effect of SREBP downregulation on reducing the stemness of colon cancer cells, we performed tumor initiation experiments by injecting control and SREBP1 and SREBP2 knockdown Pt130 cells at 1000 cells per site in NSG mice. The number of tumors formed was determined after 3 months. While control cells were able to form tumors at 6 out of 8 injection site, the tumor forming ability of SREBP1 and SREBP2 knockdown cells was decreased (Fig. 7h), suggesting that silencing SREBPs inhibits tumor initiation. Collectively, our results demonstrate that SREBP-dependent lipid biosynthesis promotes colon cancer tumorigenesis.

a Control, SREBP1, and SREBP2 knockdown Pt130 cells were injected subcutaneously into NSG mice. The size of the tumors was measured every 3 days starting at day 14. Data represent the mean ± SEM (¶p < 0.0001 compared to Sh-Control group). b On day 29, tumors were excised and weighted. Data represent the mean ± SEM (¶p < 0.0001 and §p < 0.01 compared to Sh-Control group). c Detection of proliferating cells in tumors with IHC staining using the anti-Ki67 antibody. Scale bar, 25 μm. d Numbers of Ki67-positive cells were quantified in 500 randomly chosen tumor cells. Data in the graph represent the mean ± SD (¶p < 0.0001 compared to Sh-Control group). e Detection of apoptotic cells in tumors with IHC staining using the anti-cleaved caspase-3 antibody. Scale bar, 25 μm. f Numbers of cleaved caspase-3-positive cells were quantified in 500 randomly chosen tumor cells. Data in the graph represent the mean ± SD (¶p < 0.0001 compared to Sh-Control group). g Knockdown of SREBP reduced the expression of genes associated with colon cancer stem cells. The relative expression of CD44, CD133, LGR5, and AXIN2 mRNA was determined using real-time PCR in tumors derived from control, SREBP1 and SREBP2 knockdown Pt130 cells. Data represent the mean ± SD (*p < 0.001, §p < 0.01, and #p < 0.05 compared to Sh-Control group). h Tumor initiation experiments were performed using control, SREBP1, and SREBP2 knockdown Pt130 cells. The cells were mixed with Matrigel and subcutaneously inoculated into NSG mice at 1000 cells per site and total of eight injections were used for each cell line. The number of tumors formed was determined 3 months post inoculation.
Discussion
Cancer cells are rapidly dividing cells that have increased demands for energy and macromolecules. Emerging evidence suggests that the primary functions of activated oncogenes and inactivated tumor suppressors are to rewire cellular metabolic pathways in order to drive tumorigenesis24. Whereas most normal human cells utilize exogenous sources of fatty acids, tumors often exhibit a shift toward de novo lipid biosynthesis, despite the availability of extracellular lipids5,25,26. Here we show that inhibition of lipid biosynthesis by knocking down SREBP1 or SREBP2 significantly inhibits cell proliferation in vitro and tumorigenesis in vivo as a result of decreased fatty acid and cholesterol levels in colon cancer cells. In addition, silencing SREBP1 or SREBP2 expression in both established cancer cell lines and patient-derived cancer cells results in an overall shift in cellular metabolism as indicated by reduced levels of glycolysis, mitochondrial respiration as well as FAO. Importantly, knockdown of SREBPs inhibits the tumor initiation capacity of colon cancer cells and reduces the expression of cancer stem cell-associated genes. Furthermore, blocking the proteolytic processing of SREBP by treating cell with inhibitors or silencing SCAP, results in a similar inhibition of cell proliferation and alteration of cellular metabolism, thus confirming the functional importance of SREBP-dependent lipid biosynthesis. Taken together, results from our studies establish a role of SREBPs in promoting tumor growth and initiation in colon cancer.
Although it has been suggested that SREBP1 controls fatty acid and cholesterol synthesis whereas SREBP2 preferentially activates cholesterol synthesis9, genes related to fatty acid and cholesterol synthesis are similarly downregulated in SREBP1 and SREBP2 knockdown colon cancer cells and the functional outcomes are mostly comparable upon silencing either isoforms. One exception is that silencing SREBP1 has a stronger effect on reducing mitochondrial respiration compared to SREBP2. It is of particular interest to investigate if SREBP1 regulates expression of genes that are coupled directly with mitochondrial functions in future studies. Previous studies on defining the specificity of SREBP isoforms are generally conducted in the context of liver metabolism by overexpressing SREBP27,28. Our findings here suggest that SREBP1 and SREBP2 may have overlapping functions in colon cancer cells.
As a part of metabolic reprogramming, altered lipid metabolism has been increasingly recognized as an important hallmark of cancer cells1,2. In normal insulin-responsive tissues, such as liver and adipose tissue, decreased the cellular FAO can lead to increased production and accumulation of fatty acids29. Intriguingly, we find that inhibition of SREBP-mediated lipid biosynthesis reduces the mitochondrial activity associated with both glycolysis and FAO in colon cancer cells. These results suggest that levels of glucose metabolism are dictated by the need for lipid production in cancer cells, and fatty acids derived from de novo lipid biosynthesis are used to fuel tumor growth. Collectively, our findings highlight the significance of metabolic flexibility that adapted by cancer cells to fulfill the energy requirement for proliferation and survival. However, additional studies are needed to elucidate the molecular mechanism that allows the coupling of lipogenesis with FAO and other metabolic pathways in cancer cells. Recent studies using genome-wide ChIP-Seq approach have identified previously unknown target genes of SREBP1 and SREBP2 that are beyond lipid biosynthesis regulation30,31. For example, SREBP2 has been shown to directly activates autophagy related genes under sterol starvation condition31. In addition, it has been shown that inhibition of SREBP activation prevents cytokine-induced metabolic reprogramming in NK cells32. Thus, lipid biosynthesis-independent functions of SREBPs may also contribute to the regulation of cellular metabolism in colon cancer cells.
Given the notion that elevated lipid biosynthesis promotes tumorigenesis and tumor progression, targeting de novo lipogenesis represents an attractive option in developing new therapeutic agents in cancer treatment33. Indeed, it has been shown that depletion of SREBP1 and SREBP2 expression induces ER-stress, accumulation of reactive oxygen species and apoptosis in breast cancer and glioblastoma cells34. Downregulation of SREBP1 expression inhibits cell growth, migration and invasion and induces apoptosis in ovarian cancer cells35. Furthermore, fatostatin suppresses xenograft tumor growth of prostate and breast cancer cells36,37. In our effort to determine the levels of SREBP1 and SREBP2 expression in human CRC, we performed bioinformatics analysis using the Cancer Genome Atlas (TCGA) gene expression database. However, we did not find any significant increase in SREBP1 or SREBP2 expression in CRC patient samples compared to normal controls (data not shown). As the RAS/RAF and PI3K/AKT pathway are commonly activated in CRC, the activity of SREBPs may be increased via a post-translational mechanism downstream of Akt/mTOR rather than at the transcript level11,15,18.
Our study provides initial evidence that SREBP-mediated tumor promoting effects may be partially blocked by fatostatin. However, the overall anti-tumor effect of SREBP inhibition likely depends on the availability of exogenous lipids and the activation status of lipogenic signaling in cancer cells18,33,34. It remains an open question which types of cancer (or which cancer patients) are most likely to respond to therapies targeting de novo lipid biosynthesis pathways. Interestingly, increased lipid droplet formation has been observed in a subpopulation of colon cancer cells that express CD133, a putative marker of cancer stem cells, suggesting that cancer stem cells may maintain a higher level of lipid biosynthesis38. In addition, recent studies indicate that leukemia stem cells preferentially rely on FAO for the maintenance of stem cell properties23,39. We show here that silencing SREBP reduces FAO and the “stemness” of colon cancer cells as indicated by decreased colony formation ability in vitro and tumor initiation in vivo. Therefore, inhibition of lipid biosynthesis may provide an effective approach for targeting cancer stem cells.
In summary, results from our study demonstrate the functional importance of SREBP-mediated lipid biosynthesis in colon cancer. Future studies are needed to further explore the molecular mechanism by which lipogenesis regulates cancer stem cell properties and the potential application of therapeutic agents targeting lipid metabolism in cancer.
Materials and methods
Cells and reagents
Human colon cancer cell lines DLD1 and HCT116 cells were cultured in DMEM and McCoy’s 5A supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich, MO, USA) and 1% penicillin–streptomycin, respectively. These cells were purchased from ATCC and authenticated using short tandem repeat (STR) DNA profiling in March 2016 (Genetica, OH, USA). Primary colon cancer Pt130 cells were established from patient-derived xenografts (PDX) as described previously40. Briefly, this cell line, obtained from a 63-year old male with moderately differentiated colonic adenocarcinoma, contains BRAF (V600E) and TP53 mutations and was authenticated using short tandem repeat (STR) DNA profiling as a unique cancer cell line (Genetica). Stable SREBP1, SREBP2 and SCAP knockdown cells were generated using lentivirus-based RNAi40,41,42. The shRNA targeting sequences for SREBP1 (also named SREBF1) are as the following: 5′-GCCATCGACTACATTCGCTTT-3′ (a) and 5′-CCAGAAACTCAAGCAGGAGAA-3′ (b); for SREBP2 (also named SREBF2): 5′-CCTCAGATCATCAAGACAGAT-3′ (a) and 5′-GACCTGAAGATCGAGGACTTT-3′ (b); and for SCAP: 5′-GCTCAACGGTTCCCTTGATTT-3′ (a) and 5′-GCTCTGGTGTTCTTGGACAAA-3′ (b). The non-targeting control shRNA lentivirus plasmid (MISSION, SHC002) was obtained from Sigma-Aldrich (MO, USA). Fatostatin and 25-HC were purchased from Sigma-Aldrich.
Real-time PCR
Total RNA was isolated from human cancer cells or xenograft tumors using RNeasy kit (Qiagen, MD, USA). Equal amounts of RNA were used as templates for the synthesis of cDNA using High Capacity cDNA Reverse Transcription kit (Applied Biosysems, CA, USA). Real-time PCR reaction was performed using TaqMan probes specific for SREBP1, SREBP2, LDLR, FASN, SCD, ACACA, HMGCS, HMGCR, LGR5, CD44, CD133, LGR5, and AXIN2 using StepOne Real-Time PCR system (Applied Biosysems). All values were normalized to the level of β-actin.
Measurements of cellular fatty acid, cholesterol, and triglyceride levels
Equal numbers of colon cancer cells were seeded in 6-well plate in regular growth medium. After 16 h incubation, cells were lysed and extracted according to procedures specified by individual quantification kits. The Free Fatty Acid Quantification Kit, Cholesterol Quantification Kit (Sigma-Aldrich), and Triglyceride Colorimetric Assay Kit (Cayman, MI, USA) were used to quantify levels of cellular fatty acids, cholesterol, and triglyceride, respectively.
Cell proliferation assay
To determine the rate of proliferation, equal numbers of cells were seeded into 12-well plates (2 × 104 cells per well). The number of cells were counted daily for 4 days using a cell counter (Beckman-Coulter, Fullerton, CA, USA).
Spheroid formation assay
To determine the self-renewal capacity of colon cancer cells, 1,000 single cells were seeded in non-adherent 24-well plates in StemPro hESC SFM medium supplied with 1% GlutaMAX, 2% StemPro hESC supplement, 1.8% BSA, 8 ng/ml FGF-basic, and 0.1 mM 2-mercaptoethanol (Thermo Fisher Scientific, MA, USA). After 6 days in culture, numbers of spheroids were counted under a light microscope.
Seahorse extracellular flux analysis
The Seahorse XF96 Extracellular Flux Analyzer (Agilent, CA, USA) was used to measure the respiration activity of colon cancer cells. Cells were seeded at the density of 3 × 104 cells per well in a XF96 plate ~16 h before the measurement. The glycolysis and mitochondrial stress test were performed according to manufacturer’s protocol. All Seahorse measurements were normalized to the protein contents in each well. The relative levels of glycolysis, glycolytic capacity, and glycolytic reserve were calculated based on ECAR data obtained in the glycolysis stress tests, whereas the relative levels of basal, maximal, ATP production-related respiration, and reserved mitochondrial capacity were calculated based on OCR data obtained in the Mito stress tests using Seahorse Wave software for XF analyzers.
To measure the rate of cellular FAO, cells were incubated overnight in Substrate-Limited Medium (DMEM with 0.5 mM glucose, 1.0 mM glutamine, 0.5 mM carnitine, and 1% FBS) in XF96 cell culture plates. Forty-five minutes prior to the beginning of OCR measurement, the cells were switched to the FAO Assay Medium (111 mM NaCl, 4.7 mM KCl, 2.0 mM MgSO4, 1.2 mM Na2HPO4, 2.5 mM glucose, 0.5 mM carnitine, and 5 mM HEPES). Etomoxir (40 µM) was added to one set of cells to reveal the amount of FAO-associated OCR. All Seahorse measurements were normalized to the protein concentrations in each well. The FAO-dependent respiration was calculated by subtracting OCR levels in ETO-treated cells from those in untreated cells.
Western blot analysis
Colon cancer cells were harvested and detergent-solubilized cell lysates were obtained as described previously41,42,43,44. Equal amounts of cell lysates were resolved by SDS-PAGE and subjected to western blot analysis. The SREBP1 antibody was from BD Biosciences (CA, USA), whereas the SREBP2 antibody was from R&D Systems (MN, USA). Note that the SREBP antibodies recognize both the precursor and mature active forms of the respective proteins. The γ-tubulin antibody was from Sigma-Aldrich.
Xenograft tumor formation
All animal procedures were done using protocols approved by the University of Kentucky Animal Care and Use Committee. Six to eight week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG, The Jackson Laboratory) mice were used. Both male and female mice of equal numbers were included in each group. Control and SREBP knockdown Pt130 cells were collected in 50% Matrigel in DMEM and inoculated subcutaneously at 1 × 106 cells per injection site. After 2 weeks, the tumor size was measured every 3 days with a caliper, and the tumor volume was defined as (longest diameter) × (shortest diameter)2/2. At the end of experiments, tumors were harvested, weighted and fixed in 10% buffered formalin. The paraffin embedded samples were prepared, and 5 µm sections were used for IHC staining. For tumor initiation assay, Pt130 cells (1000 cells/injection site) were injected subcutaneously in NSG mice. The number of tumors formed was determined 3 months post injection.
Immunohistochemical (IHC) staining
Paraffin embedded xenograft tumor sections were deparaffinized, rehydrated, and treated with hydrogen peroxide. Antigen retrieval was performed using Dako Target Retrieval Solution (Agilent), and IHC staining was performed as previously described40,42. The anti-Ki67 antibody was from Dako (Agilent, CA, USA) and the cleaved caspase-3 antibody was from Cell Signaling. The stained sections were visualized using a Nikon Eclipse 80i upright microscope. Tumor sections from three different mice per group were stained and examined. In each stained section, numbers of Ki67 or cleaved caspase-3-positive cells were counted in seven randomly chosen images and the percentage of staining positive cells were quantified by normalizing to total numbers of cells. Results shown in Fig. 7 represent average results obtained in three mice.
Statistical analysis
In experiments to assess the rate of cell proliferation, mRNA expression, fatty acid and triacylglycerol (TAG) levels, bioenergetic measurements, and tumor weight were summarized using bar or line graphs and pairwise comparisons between different conditions were carried out using two-sample t-tests. A linear mixed model was employed to compare slope of tumor volume growth curves over time between groups. The relative mRNA expression results shown in Figs. 1–3 and 7 represent average of three separate qPCR experiments with four replicates for each gene in each experiment. For the Seahorse experiments, eight replicates were included for each cell line in each experiment and the experiments were repeated three times. Results shown in Figs. 4–6 were from one representative experiment. All other experiments were repeated three times and results shown represent the average of three experiments.
For the Gene Set Enrichment Analysis (GSEA), RNA sequencing data were obtained from the TCGA CRC study. Correlations between expressions of SREBP1 (gene name: SREBF1) and SREBP2 (gene name: SREBF2) and the other genes were quantified by Spearman’s correlation coefficient. The genes were then ordered from highest to lowest based on the correlation coefficient. This ranked list was inputted into the GSEA Desktop Application45 to identify pathways that are associated with SREBP expression.
References
Currie, E. et al. Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161 (2013).
Ward, P. S. & Thompson, C. B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308 (2012).
Cairns, R. A., Harris, I., McCracken, S. & Mak, T. W. Cancer cell metabolism. Cold Spring Harb. Symp. Quant. Biol. 76, 299–311 (2011).
Seyfried, T. N. & Shelton, L. M. Cancer as a metabolic disease. Nutr. Metab. 7, 7 (2010).
Menendez, J. A. & Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 (2007).
Bengoechea-Alonso, M. T. & Ericsson, J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr. Opin. Cell Biol. 19, 215–222 (2007).
Goldstein, J. L., DeBose-Boyd, R. A. & Brown, M. S. Protein sensors for membrane sterols. Cell 124, 35–46 (2006).
Shao, W. & Espenshade, P. J. Expanding roles for SREBP in metabolism. Cell Metab. 16, 414–419 (2012).
Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 (2002).
Osborne, T. F. & Espenshade, P. J. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been. Genes Dev. 23, 2578–2591 (2009).
Krycer, J. R., Sharpe, L. J., Luu, W. & Brown, A. J. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 21, 268–276 (2010).
Adams, C. M. et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 279, 52772–52780 (2004).
Radhakrishnan, A., Ikeda, Y., Kwon, H. J., Brown, M. S. & Goldstein, J. L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl Acad. Sci. USA 104, 6511–6518 (2007).
Kamisuki, S. et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 16, 882–892 (2009).
Guo, D. et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2, ra82 (2009).
Huang, W. C., Li, X., Liu, J., Lin, J. & Chung, L. W. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. 10, 133–142 (2012).
Bao, J. et al. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 12, 2409–2416 (2016).
Ricoult, S. J., Yecies, J. L., Ben-Sahra, I. & Manning, B. D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 35, 1250–1260 (2016).
Peck, B. et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. 4, 6 (2016).
Zaytseva, Y. Y. et al. Cancer cell-associated fatty acid synthase activates endothelial cells and promotes angiogenesis in colorectal cancer. Carcinogenesis 35, 1341–1351 (2014).
Zaytseva, Y. Y. et al. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget 6, 18891–18904 (2015).
Kreso, A. et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 20, 29–36 (2014).
Carracedo, A., Cantley, L. C. & Pandolfi, P. P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer 13, 227–232 (2013).
Nagarajan, A., Malvi, P. & Wajapeyee, N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2, 365–377 (2016).
Swinnen, J. V., Brusselmans, K. & Verhoeven, G. Increased lipogenesis in cancer cells: new players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 9, 358–365 (2006).
Zaidi, N. et al. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 52, 585–589 (2013).
Horton, J. D. et al. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Invest. 101, 2331–2339 (1998).
Pai, J. T., Guryev, O., Brown, M. S. & Goldstein, J. L. Differential stimulation of cholesterol and unsaturated fatty acid biosynthesis in cells expressing individual nuclear sterol regulatory element-binding proteins. J. Biol. Chem. 273, 26138–26148 (1998).
Fucho, R., Casals, N., Serra, D. & Herrero, L. Ceramides and mitochondrial fatty acid oxidation in obesity. FASEB J. 31, 1263–1272 (2017).
Seo, Y. K. et al. Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc. Natl Acad. Sci. USA 106, 13765–13769 (2009).
Seo, Y. K. et al. Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 13, 367–375 (2011).
Assmann, N. et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 18, 1197–1206 (2017).
Guo, D., Bell, E. H., Mischel, P. & Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 20, 2619–2626 (2014).
Griffiths, B. et al. Sterol regulatory element-binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 1, 3 (2013).
Nie, L. Y. et al. Sterol regulatory element-binding protein 1 is required for ovarian tumor growth. Oncol. Rep. 30, 1346–1354 (2013).
Li, X., Chen, Y. T., Hu, P. & Huang, W. C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 13, 855–866 (2014).
Freed-Pastor, W. A. et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258 (2012).
Tirinato, L. et al. Lipid droplets: a new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 33, 35–44 (2015).
Ye, H. et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19, 23–37 (2016).
Wen, Y. A. et al. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 8, e2593 (2017).
Li, X. et al. PHLPP is a negative regulator of RAF1, which reduces colorectal cancer cell motility and prevents tumor progression in mice. Gastroenterology 146, 1301–1312 (2014).
Liu, J. et al. Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene 28, 994–1004 (2009).
Wen, Y. A. et al. Loss of PHLPP protects against colitis by inhibiting intestinal epithelial cell apoptosis. Biochim. Biophys. Acta 1852, 2013–2023 (2015).
Wen, Y. A., Stevens, P. D., Gasser, M. L., Andrei, R. & Gao, T. Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol. Cell Biol. 33, 4594–4605 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Acknowledgements
This work was supported by R01CA133429 (T.G.) and R01CA208343 (B.M.E. and T.G.). The studies were conducted with support provided by the Redox Metabolism, Biospecimen Procurement and Translational Pathology, and Biostatistics and Bioinformatics Shared Resource Facilities of the University of Kentucky Markey Cancer Center (P30CA177558).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wen, YA., Xiong, X., Zaytseva, Y.Y. et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis 9, 265 (2018). https://doi.org/10.1038/s41419-018-0330-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0330-6
This article is cited by
-
SF3B3-regulated mTOR alternative splicing promotes colorectal cancer progression and metastasis
Journal of Experimental & Clinical Cancer Research (2024)
-
The effects of metabolism on the immune microenvironment in colorectal cancer
Cell Death Discovery (2024)
-
The RORɣ/SREBP2 pathway is a master regulator of cholesterol metabolism and serves as potential therapeutic target in t(4;11) leukemia
Oncogene (2024)
-
Metabolic reprogramming in colorectal cancer: regulatory networks and therapy
Cell & Bioscience (2023)
-
Synthetic lethality in large-scale integrated metabolic and regulatory network models of human cells
npj Systems Biology and Applications (2023)
