
Abstract
Cancer-initiating cells (CICs) are responsible for tumor initiation, progression, and therapeutic resistance; moreover, redox homeostasis is important in regulating cancer stemness. Previously, we have identified that cancer cells containing low intracellular reactive oxygen species levels (ROSLow cells) display enhanced features of CICs. However, the specific metabolic signatures of CICs remain unclear and are required for further characterization by systemic screenings. Herein, we first showed CICs mainly relying on glycolysis that was important for the maintenance of stemness properties. Next, we revealed that NRF2, a master regulator of antioxidants, was able to maintain low intracellular ROS levels of CICs, even though in the absence of oxidative stress. We further characterized that NRF2 activation was required for the maintenance of CICs properties. Of ROSLow cells, NRF2 activation not only directly activates the transcription of genes encoding glycolytic enzymes but also inhibited the conversion of pyruvate to acetyl-CoA by directly activating pyruvate dehydrogenase kinase 1 (PDK1) to lead to inhibition of tricarboxylic acid (TCA) cycle; therefore, to promote Warburg effect. A positive regulatory ROS-independent ER stress pathway (GRP78/p-PERK/NRF2 signaling) was identified to mediate the metabolic shift (Warburg effect) and stemness of CICs. Lastly, co-expression of p-PERK and p-NRF2 was significantly associated with the clinical outcome. Our data show that NRF2 acting as a central node in the maintenance of low ROS levels and stemness associated properties of the CICs, which is significantly associated with the clinical outcome, but independent from ROS stress. Future treatments by inhibiting NRF2 activation may exhibit great potential in targeting CICs.
Similar content being viewed by others


Introduction
Cancer-initiating cells (CICs) exploit the characteristics of self-renewal and differentiation to drive tumor growth and progression1. Previously, we have enriched and identified head and neck CICs (HN-CICs) through sphere culture2. Our most recent study shows that a subset of HN-CICs contains lower ROS levels. Consequently, the sorted ROSLow cells possess enhanced stemness properties and tumorigenicity and acquire a quiescent state. Furthermore, compared with ROSLow cells, the other subset of HN-CICs with high ROS levels (the ROSHigh cells) are more proliferative but exhibit the less self-renewal capacity3. Given the importance of redox homeostasis in regulating the stemness of CICs, we need to understand the unique physiology to balance the ROS levels and stemness of CICs.
In various cancers, CICs are considered highly heterogeneous and harbor a distinct metabolic phenotype in terms of stemness features4. Of note, ROS is intimately tied to cellular metabolic phenotype5. Additionally, mitochondria are the major source of ROS production through oxidative phosphorylation (OXPHOS)5. Interestingly, CICs have been described as preferentially relying on the Warburg effect or OXPHOS in a cancer type-dependent manner6,7,8,9. Warburg effect not only provides sufficient energy demands but also minimizes ROS production in mitochondria8, 10. Furthermore, we recently have demonstrated that ROSLow cells highly express the high-affinity glucose transporter, GLUT33. Indeed, metabolic reprogramming of cancer cells tightly regulates defense against oxidative stress, thus promoting tumorigenesis and chemoresistance11.
From an initial screen of molecular mechanisms known to play a role in mediating CICs metabolism, we found a transcription factor NRF2 activity correlated with the Warburg effect (see the following contexts). NRF2 is a master regulator of ROS-scavenging enzymes12. Indeed, NRF2 has been considered to regulate the self-renewal of various kinds of normal stem cells. A recent study demonstrated that NRF2 is required for the switch to glycolysis by promoting HIFα activation in iPSC reprogramming13. Further, NRF2 has shown prognostic significance in many solid tumors14, 15. Nevertheless, the mechanisms by which NRF2 controls cell metabolism that maintain redox homeostasis, and therefore sustains CICs properties, remain to be elucidated. Furthermore, the molecular mechanisms by which NRF2 can be activated in CICs also remain elusive.
Our current study provides several insights into distinct subsets of cancer cells with different ROS levels, in which metabolic reprogramming and activation of NRF2 signaling are the main mechanisms regulating cancer stemness.
Results
Reprogrammed glucose metabolism in HN-CICs
Previously, we and others demonstrated that CICs, enriched within the sphere cells under serum-free culture conditions of cancer cells2, 16. To unravel the metabolic features of CICs, we first investigated possible pathways of glucose metabolism in HN-CICs. Initially, the expression profile of TCA cycle-related genes in sphere cells (SAS-S) and in parental cells (SAS-P) was analyzed by gene set enrichment analyses. Notably, TCA cycle-related genes were significantly downregulated in sphere cells (Figs. 1a, b). We further confirmed these results by measuring the mitochondrial membrane potentials of the parental and sphere cells with JC-1 staining. Red JC-1 aggregates are typical of healthy mitochondria17. Indeed, the sphere cells had fewer red JC-1 aggregates than the parental cells that indicate the occurrence of mitochondrial depolarization within the sphere cells (Fig. 1c). In addition, we found an approximately 2–3 folds reduction in the mitochondrial mass in sphere cells versus parental cells (Fig. 1d; SAS-P: 70.3% vs. SAS-S: 21.6%; OECM1-P: 80% vs. OECM1-S: 44.5%). Strikingly, the sphere cells displayed a higher expression of glycolytic enzymes in order to promote glycolysis (Figs. 1e, f and Figure S1a,S1b). Given that radiation-resistant cells have been reported to have characteristics of cancer stemness18, we set out to evaluate the correlation between glycolytic enzymes expression profile and radioresistance properties. Interestingly, expression of glycolytic enzymes was significantly higher in radiation-resistant cells than in parental cells (Figure S1a). Next, we wanted to address whether genetically or pharmacologically inhibition of glycolysis would abrogate the stemness properties of CICs. Genetic inhibition of HK2 or PFKFB3 by shRNAi, respectively, both reduced the expression of stemness markers (NANOG and OCT4) in SAS sphere cells (Fig. 1g). Furthermore, pharmacologic inhibition of glycolysis by addition of 2-deoxyglucose (2-DG) showed the most significant effect on increasing cytotoxic effects and decreasing expression of cancer stemness marker (OCT4 and CD44) in sphere cells (Figure S1c-S1e).

a Heatmap depicting expression of 24 genes encoding tricarboxylic acid (TCA) cycle pathway in parental (SAS-P) and sphere cells (SAS-S). b Gene Set Enrichment Analysis (GSEA) of a 24-gene set comprising (TCA) cycle pathway in SAS-P versus SAS-S (NES normalized enrichment score; P = nominal P-value; FDR false discovery rate). c The mitochondrial membrane potentials (Δψ, red/green ratios) of the parental and sphere cells were determined using JC-1 staining followed by FACS. (OECM1 parental (OECM1-P) and OECM1 sphere cells (OECM1-S)). d The mitochondrial masses of the parental and sphere cells were determined using MitoTracker Red staining followed by FACS. e Heatmap depicting expression of genes encoding glycolytic pathway in parental and sphere cells. f Immunoblots detecting the protein level of glycolytic enzymes in parental and sphere cells. g Immunoblots showing the expression level of HK2, PFKFB3, NANOG and OCT4 proteins in control, sh-HK2 or sh-PFKFB3 sphere cells, respectively. h Schematic depicting features of ROSLow, ROSHigh, and ROSMedi cells. i Top 10 toxicology pathway lists identified in ROSLow cells from RNA-Seq analysis. P-value, Fisher’s exact test. Threshold, the minimum significance level (−Log [P-value] as 1.25). Ratio, the molecules from the data set divided by the total molecules that map to the toxicology pathway from within the IPA software. j Heatmap depicting expression of genes encoding glycolytic pathway in ROSLow, ROSMedi, and ROSHigh cells (P < 0.001, Fisher’s exact test). k Immunoblots detecting the glycolytic enzymes and epithelial differentiation markers in sorted ROSLow, ROSMedi, and ROSHigh cells
We recently demonstrate that ROSLow cells were quiescent but exhibited the highest tumorigenicity; whereas ROSHigh cells showed higher proliferative activity and moderate tumorigenicity (Fig. 1h)3. To gain the mechanistic insight into distinct subpopulation cells, we performed RNA-Seq analyses with sorted ROSLow cells versus ROSHigh cells to examine associated changes in cellular pathways and functions. The transcriptome of the ROSLow cells displayed a very distinct expression profile compared with that of the ROSHigh and ROSMedi subpopulation cells (Figure S1f). Interestingly, the metabolic process was highlighted as having the first ranking among the significant differences (Figure S1g). Specifically, we also discovered the major mechanistic features, mitochondrial dysfunction, was up-regulated in the ROSLow cells (Fig. 1i and Figure S1h). Consistent with these results, the ROSLow cells exhibited reduced mitochondrial membrane potentials than the ROSHigh and ROSMedi cells (Figure S1i). In addition, the glycolysis-related genes were significantly up-regulated in the ROSLow cells (Fig. 1j,k). Finally, to further examine whether pharmacologic modulation of glycolysis contributed to cell death in ROSLow cells, ROSLow cells were treated with 2-DG. Treatment of 2-DG caused a significant increase in cell death in ROSLow cells (Figure S1j).Collectively, these results suggest that HN-CICs may rely on the glycolytic pathway and exhibit impaired OXPHOS, which is consistent with a “Warburg effect” profile.
Elevated NRF2 expression in HN-CICs
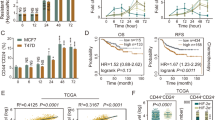
To further elucidate the underlying molecular mechanisms to mediate metabolic regulation of redox homeostasis, we performed a screening of the candidate transcription factors (NRF2, PGC1α, and p-MYC), which are known to correlate with the metabolic phenotypes and the stress/antioxidant response in cancer cells19,20,21,22. Interestingly, we found that only the protein level of NRF2 but not others were up-regulated in sphere cells compared with that of parental cells (Fig. 2a). These data were consistent with the IPA analyses identifying that the activation of NRF2 signaling pathway was the most significantly affected pathways in ROSLow cells (Fig. 1i). NRF2 activation in cells with different levels of ROS was further examined to validate the observation that low ROS levels were positively regulated by NRF2. It was shown that increased expression of the NRF2 protein in ROSLow was observed in comparison with that in ROSMedi or ROSHigh cells (Fig. 2b). Moreover, the expression profile of NRF2-mediated downstream targets was also increased in SAS sphere cells compared with those in SAS parental cells (Figures S2a and S2b). Phosphorylation of NRF2 (p-NRF2) is required for nuclear translocation and activation of NRF2 to increase drug resistance23. Consistently, sphere cells contained more p-NRF2 expression than the parental cells did (Fig. 2c). Subsequently, immunoblotting and immunofluorescence revealed an approximately 3 folds increase in the amount of nuclear NRF2 in the sphere cells (Figs. 2d, e; immunofluorescence: SAS-P:18.6% vs. SAS-S:73.6%; OECM1-P: 21.2% vs. OECM1-S:56.0%). On the basis of these results, we focused on further characterizing the effect of NRF2 activation on glucose metabolism of CICs. It has been demonstrated that NRF2 directly activates PPP and glutamine metabolism pathway in cancer cells24. However, a relationship between NRF2 and glycolysis in CICs remains elusive. We first investigated the relationship between the expression of NRF2 and glycolytic genes in clinical patients by analyzing the TCGA database. A positive correlation was observed NRF2 protein expression with the expression of glycolytic or PPP genes (Fig. 2f). Further, higher glycolytic genes expression predicted poor survival for HNSCC cancer (Fig. 2g). In addition, the increased protein level of NRF2 was positively correlated with worse survival in cancer patients (Fig. 2h). In summary, NRF2 was highly expressed in both sphere cells and ROSLow cells, which may be correlated with NRF2-mediated metabolic phenotypes. Therefore, we hypothesize that NRF2 activation is crucial for regulating the CIC metabolism.

a Immunoblots showing NRF2, PGC1α, and p-MYC protein levels in parental and sphere cells. b Immunoblots showing NRF2 protein level in ROSLow, ROSMedi, and ROSHigh cells. c FACS analyses showing the profiles of p-NRF2-positive parental and sphere cells (n = 3 independent experiments; t-test, *** indicates P < 0.001). d NRF2, actin and lamin B1 protein levels in the nuclear and cytosolic fractions prepared from parental or sphere cells. e Immunofluorescence staining showing the subcellular localization of the NRF2 protein (red) and nuclei stain DAPI (blue) in parental and sphere cells. Quantification of the colocalization efficiency between NRF2 and DAPI confirming that NRF2 was present in sphere cell nucleus. f Correlation analyses between the protein level of NRF2 and mRNA level of glycolytic genes in HNSCC, according to the TCGA database, respectively. Correlation coefficients (r) and P-values are displayed (n = 346). Data were obtained from the cBioPortal website. g Kaplan–Meier analysis showing the overall survival of patients with Head and neck cancers according to the expression of seven glycolytic transcripts. Data were obtained from the SurvExpress website. h Head and neck cancers (n = 346) data obtained from TCGA database (The Cancer Proteome Atlas (TCPA) website) were used for Kaplan-Meier analysis to predict the overall survival of patients according to the expression of the NRF2 protein
NRF2 directly activates glycolytic pathway and PPP in sphere cells
For a better understanding of the role of NRF2 activation in regulating CICs metabolic phenotypes, global mapping of NRF2 binding sites on genomic loci of the sphere and parental cells using ChIP-Seq analyses was performed. We identified 1258 NRF2 genomic binding sites in parental SAS cells and 5151 NRF2 genomic binding sites in SAS sphere cells within 5 kb of the gene (Figures S3a and S3b). Of note, sphere cells showed a constitutive increase nuclear accumulation of NRF2 (Figs. 2d, e), which may, at least in part, explained the larger number of NRF2 binding sites identified in sphere cells rather than in the parental cells. Strikingly, NRF2-targeted sites in sphere cells (Ranking 10th) occupied substantially more ontology categories in the metabolic process than in parental cells (Ranking 16th) (Figure S3c). By combining the NRF2 ChIP-Seq and the ROSLow and ROSHigh cells-upregulated transcriptome datasets, we identified the genes that were directly regulated by NRF2 in the ROSLow and ROSHigh cells (Fig. 3a). Further, through integrating the NRF2 ChIP-Seq data from the sphere cells with the RNA-Seq data from the ROSLow cells, we observed that NRF2 was able to directly activate the key enzymes in the glycolysis (HK2, and PFKFB3 et al.), the checkpoint in regulating OXPHOS (PDK1), an enzyme in the non-oxidative PPP pathway (TKT) in sphere cells and these genes indeed were upregulated in ROSLow cells (Fig. 3b). However, that bounding between NRF2 and genomic loci was lost in parental cells by ChIP-Seq analyses (Figure S3d). In contrast, the combined analysis of the NRF2 ChIP-Seq data from sphere cells and the RNA-Seq data from ROSHigh cells revealed enrichments in oxidative PPP and glutamine metabolic targets (G6PD and GLS) (Figure S3e).

a (Upper left panel) Venn diagram depicting the overlap genes (Group A; purple) between the NRF2-bound genes (ChIP-Seq analyses) and genes with significantly altered expression in ROSLow cells (blue). Otherwise, the Venn diagram depicts the overlap genes (Group B; orange) between NRF2-bound genes and genes with significantly altered expression in ROSHigh cells. (Right panel) NRF2 mediated metabolic genes, involved in glycolysis and the non-oxidative PPP pathway in ROSLow cells (Group A), or involved in the oxidative PPP and glutamine pathway in ROSHigh cells (Group B). (Lower left panel) Summary of NRF2 regulating distinct metabolic pathways in ROSLow cells (quiescent CICs) and ROSHigh cells (proliferating CICs). b ChIP-Seq tracks of NRF2 in SAS sphere cells, assessing the binding of glycolysis and PPP. Location of ChIP-Seq peaks that were identified by using Strand NGS software. RNA-Seq traces (UCSC genome browser) for each gene in ROSLow cells and ROSHigh cells
In summary, NRF2 activation not only directly activates the genes encoding glycolytic enzymes but also inhibits the TCA cycle by directly activating PDK1. These findings have led to the hypothesis that NRF2 may maintain low ROS levels within CICs by enhancing the Warburg effect.
NRF2 activation promotes the “Warburg effect”
We next to further determine whether NRF2 activation plays a crucial role in up-regulating the Warburg effect. Compared with the ControlGFP cells, the NRF2 overexpressing (NRF2Over) cells displayed the increased protein levels of glycolytic enzymes (Fig. 4a). In opposite, NRF2 knockdown impaired glycolysis, including inhibiting the expression of glycolytic enzymes, reducing the glucose consumption ability, inhibiting the lactate level, and decreasing the GLUT3 positive cells (Figs. 4b–e and Figure S4a). Conversely, NRF2 overexpression increased the lactate levels (Fig. 4f), which again supported our observation that NRF2 bound to the LDHA promoter of CICs by ChIP-Seq analyses (Fig. 3b). We next measured the glycolytic flux (extracellular acidification rate or ECAR) to further understand the effect of NRF2 activation on glycolytic activity. As shown in Figure S4b, the baseline ECAR was significantly increased in the NRF2Over cells. We also found that NRF2 directly induced PDK1 expression (Figs. 3b and 4a) thus to inactivate PDH activity, as a result, to suppress the influx of glycolytic metabolites into the mitochondria25, ultimately, to lead to a decrease of ROS production26. In opposite, the sh-NRF2 sphere cells showed more PDHA1 expression and red JC-1 aggregates than the Control cells (Fig. 4c and Figure S4c).

a Glycolytic enzyme protein levels in ControlGFP or NRF2Over cells cultivated in selection medium for 2 weeks. b Total transcripts were prepared from sh-NRF2 and Control sh-Luc sphere cells (SAS and OECM1 cells) and further analyzed by Real-time RT-PCR. c Total proteins, prepared from sh-NRF2 and control sphere cells (SAS-S, OECM1-S), were analyzed by immunoblotting with the indicated antibodies. d Glucose uptake levels in sh-NRF2 and Control sphere cells were measured by detecting the uptake of fluorescent 2-NBDG dye. e Intracellular lactate levels measured in sh-NRF2 and control sphere cells. f Intracellular lactate levels measured in ControlGFP or NRF2Over cells cultivated within selection medium. g LC-MS metabolic profiling showing the metabolic intermediates of glycolysis, PPP, TCA, and glutaminolysis. (n = 3 independent experiments; t-test, *P < 0.05; **P < 0.01; ***P < 0.001)
To further elucidate the mechanisms by which activated NRF2 reprogrammed glucose metabolism, the metabolomes of NRF2Over cells or Control cells were determined by liquid chromatography-mass spectrometry metabolic profiling. Consistent with our previous analyses, the NRF2Over cells showed increases in the levels of intermediate metabolites of glycolysis and non-oxidative PPP; however, the levels of the intermediate metabolites of TCA cycle were significantly decreased (Fig. 4g and Figure S4d). Based on these data, we further hypothesized that NRF2 activation enhancing the Warburg effect, which might be required to maintain the properties of CICs.
NRF2 expression is required for CICs maintenance
As shown in Figs. 3 and 4 that NRF2 mediated the CIC metabolism. Therefore, we further expected that NRF2 would regulate the stemness features of CICs. We first examined the endogenous NRF2 expression in HNSCC cancer cell lines. The endogenous NRF2 protein level was higher in SAS and OECM1 cells than in the other HNSCC cells (Figure S5a). Furthermore, human oral keratinocytes expressed the least amount of NRF2 protein among the examined cell lines (Figure S5a). As shown in Figure S5b, the sh-NRF2 sphere cells did not maintain floating spheres but showed more attached epithelial-like cells. Furthermore, NRF2 knockdown attenuated the expression of stemness markers (OCT4 and NANOG) (Fig. 5a). Since redox homeostasis could be mainly regulated by NRF2, we examined the ROS levels in sh-NRF2 sphere cells. As expected, the percentage of ROSLow cells was significantly reduced in sh-NRF2 sphere cells, supporting the importance of NRF2 activation for ROS control (Fig. 5b). We have showed that the memGrp78+ cells possess enhanced stemness properties and tumorigenicity18. Accordingly, silencing NRF2 expression in sphere cells markedly decreased their sphere-forming capacity (Fig. 5c and Figure S5c) and inhibited the amounts of cell surface Grp78 (memGRP78+) and CD44+ cells3, 27 (Fig. 5d and Figure S5d). In contrast, the sh-NRF2 sphere cells displayed enhanced differentiation and elevated apoptosis (Figures S5e and S5f). We also observed that the sh-NRF2 sphere cells showed decreased anchorage-independent growth ability and migration (Fig. 5e and Figure S5g, S5h). Strikingly, Control sphere cells generated tumors in mice, whereas NRF2-knockdown sphere cells failed to form tumors (Fig. 5f and Figure S5i).

a Immunoblots showing NRF2, OCT4, NANOG, and actin protein levels in the Control (sh-Luc) and sh-NRF2 sphere cells. b Expression profiles of ROSLow subpopulation cells in distinct lentivirus-infected sphere cells were determined via FACS. c Secondary spheres forming ability of sh-NRF2 sphere cells. d The percentage of memGRP78-positive cells in the sh-NRF2 sphere cells were determined via FACS. e Soft agar colony formation ability of sh-NRF2 sphere cells (SAS-S (right panel), OECM1-S (left panel)). f–g Tumor growth curves of the sh-Luc or sh-NRF2 SAS-S cells (f) and ControlGFP cells or NRF2Over cells (g). Error bars correspond to SD (***P < 0.001). h NRF2, OCT4 and NANOG protein expression levels in xenografted tumors derived from ControlGFP cells and NRF2Over cells or in NRF2Over OECM1 cells. i NRF2, OCT4 and NANOG protein levels in HNSCC cells expressing GFP or NRF2-GFP cultivated in selection medium for 2 weeks. j–k The percentages of ROSLow (j) and memGRP78 (k) cells in ControlGFP or NRF2Over cells, grown in selection medium for 2 weeks, were determined via FACS. l The percentage of each subpopulation of ROSLow, ROSMedi and ROSHigh cells from the ControlGFP cells or NRF2Over cells, grown in selection medium for 4 weeks, were determined via FACS. The results are presented as the mean ± SD of triplicate samples from three experiments (*P < 0.05; ***P < 0.001)
We further examined whether NRF2 overexpression could promote their cancer-initiating capacity. The NRF2Over (NRF2-overexpressing) cells displayed increased expression of stemness markers versus the control cells (ControlGFP) (Figures S5j-S5m). Notably, NRF2 overexpression also enhanced the tumorigenicity of OECM1 cells in vivo (Fig. 5g and Figure S5n). Empirically, we showed increased expression of stemness genes in xenograft-derived NRF2Over cells compared with xenograft-derived ControlGFP cells (Fig. 5h). Additionally, microscopic images of control tumor specimens displayed pleomorphism and keratin whorls, suggestive of a differentiated cell phenotype, while the NRF2Over specimens showed a more malignant cellular phenotype (Figure S5o).
We next sought to determine whether NRF2 overexpression would promote the CIC properties when the HNSCC cells were cultivated within defined selection medium. As expected, the NRF2Over cells showed an enhanced sphere-forming capacity (Figure S5p). Furthermore, more ROSLow cells and increased expression levels of stemness markers were detected in the NRF2Over cells than in the Control cells after cultivation in defined selection medium for 2 weeks (Fig. 5i–k and Figure S5q-S5s). Additionally, the NRF2Over cells gave rise to a large increase in the number of ROSLow cells but a decrease in the number of ROSHigh cells compared with the control cells after a four-week cultivation (Fig. 5l). Thus, NRF2 activation not only promotes CIC properties but also facilitates an increase in the amount of ROSLow cells.
NRF2 up-regulates the expression of glycolytic enzymes and stemness markers by activating GRP78/PERK signaling in HN-CICs
We next wanted to identify the molecular mechanism by which upregulated NRF2 activation to further reprogram the metabolism in HN-CICs. It has also been reported that NRF2 is directly activated by the Grp78/p-PERK signaling, which strongly correlates with chemotherapy resistance, tumor grade, and overall survival28,29,30,31. Therefore, we compared the expression profile of GRP78 and p-PERK proteins in sphere cells to that in parental cells. Accordingly, the protein levels of p-PERK were up-regulated in sphere or ROSLow cells (Figs. 6a, b). Although the protein level of the GRP78 in ROSLow, ROSHigh, and ROSMedi subpopulations of cells did not show a significant difference (Fig. 6b), the expression profile of memGRP78+ was significantly up-regulated in the ROSLow cells than that in the ROSHigh or ROSMedi cells (Fig. 6c). Furthermore, immunofluorescent staining revealed co-expression of NRF2 with memGRP78 in the sphere cells (Fig. 6d). Together, the levels of the KEAP1 was not down-regulated in ROSLow cells (Fig. 6b), suggesting that the NRF2 constitutive activation in ROSLow cells is mediated through GRP78/p-PERK signaling but not by KEAP1 down-regulation.

a–b GRP78, p-PERK, PERK and KEAP1 protein levels in parental and sphere cells (a) or ROSLow, ROSMedi and ROSHigh cells (b). c FACS analyses showing the expression profiles of memGRP78+ cells in ROSLow, ROSMedi, and ROSHigh cells, respectively. d Confocal IF staining of GRP78 and NRF2 in SAS-S cells. e Immunoblots showing the effects of GRP78 knockdown (sh-GRP78), PERK knockdown (sh-PERK) and NRF2 overexpression on GRP78/p-PERK/NRF2 signaling, NANOG, OCT4 and PFKFB3 expression in SAS-S cells. f–h Quantification of the ROSLow (f), CD44 (g) or memGRP78 (h) positive SAS-S cells with PERK knockdown and/or NRF2 overexpression. The data are presented as the mean ± SEM. i Overview of the transfection strategy. Effects of GRP78 overexpression and/or NRF2 knockdown on the activation of GRP78/p-PERK/NRF2 signaling, and expression of stemness proteins and glycolytic enzymes in SAS and OECM1 cells. j (Upper panel) IHC staining for p-NRF2 and p-PERK in HNSCC tumor tissues (p-NRF2-positive staining (black arrows: nucleus staining; yellow arrows: cytoplasmic staining)). (Lower panel) Fisher’s exact test showing the association according to p-NRF2 and p-PERK expression. k–l Kaplan–Meier analysis showing the overall survival of patients with HNSCC according to p-NRF2 (k) and p-PERK (l) protein expression
Next, we wanted to assess GRP78/p-PERK inactivation in sphere cells to determine whether GRP78/p-PERK signaling controls NRF2 activation in CICs. As shown in Fig. 6e and Figure S6a, knockdown of GRP78 or PERK diminished the protein levels of NRF2, stemness markers, and downstream targets. Conversely, NRF2 overexpression partially restored the inhibitory effects of GRP78 or PERK knockdown in sphere cells (Fig. 6e). As expected, PERK knockdown in sphere cells caused a significant reduction in the ROSLow subpopulation cells and the amounts of CD44+ and memGRP78+ cells; again, this inhibitory effect of PERK knockdown was significantly reversed by NRF2 overexpression (Figs. 6f–h and Figure S6b-S6d). Moreover, ectopic overexpression of GRP78 not only up-regulated p-NRF2 and p-PERK expression but also enhanced the protein levels of glycolytic enzymes and stemness markers in HNSCC cells grown in defined selection medium (Fig. 6i). However, the GRP78over cells which were co-transfected with sh-NRF2 to deplete NRF2 expression showed decreased expression of glycolytic enzymes and stemness markers (Fig. 6i).
We next established the ontogeny of p-NRF2 or p-PERK expression in tissue microarrays from 57 resected HNSCC specimens to assess whether co-expression of p-NRF2 and p-PERK was associated with clinical prognosis. High p-PERK expression was positively correlated with nuclear NRF2 accumulation and was associated with a poor outcome (Fig. 6j). Furthermore, the survival of patients with HNSCC was negatively correlated with the expression of p-NRF2 and p-PERK in the Kaplan-Meier survival analysis (Figs. 6k, l).
In summary, up-regulation of the GRP78/p-PERK pathway increases the expression of NRF2 and enables NRF2 to promote the glycolytic activity and antioxidant properties, which can promote stemness properties (Fig. 7).

Heterogeneity of CICs is demonstrated with respect to redox status. NRF2 activation directly increases glycolysis and non-oxidative PPP activity through GRP78-PERK signaling activation in ROSLow cells
Discussion
ROS status is associated with metabolic reprogramming of both stem cells and CICs, which results in adaptations to microenvironmental stress8. Activation of glycolysis and suppression of mitochondrial metabolite flux are an effective strategy for stem cell maintenance32. In fact, the promotion of “Warburg effect” results in several metabolic benefits for CICs, such as reduced oxygen dependence and ROS production, maintenance of ATP production in response to hypoxic stress and increased production of glycolytic intermediates for biosynthesis6, 33, 34. Because mitochondrial OXPHOS is a major source of ROS production and has been intimately linked to stem cell proliferation and differentiation35, 36, the quiescent ROSLow CICs may benefit from using glycolysis, as they successfully survive and maintain their quiescent state and stemness properties in response to the stress from the tumor microenvironment.
Distinct subsets of CICs with different ROS levels display different metabolic activities, which are associated with the NRF2-targeted genes (Fig. 3). Indeed, NRF2 activated the glycolysis-related genes in quiescent ROSLow cells, which was advantageous for maintaining low ROS levels within the CIC niche. In contrast, NRF2 activated the expression of enzymes required for oxidative PPP and glutathione synthesis in ROSHigh cells (Fig. 3a). Our observations are consistent with others’ study showing that NRF2 directly increases the expression of enzymes involved in the PPP and glutamine synthesis in cancer cells to facilitate increased proliferation24. Overall, NRF2 activation would affect various metabolic pathways within different tumor subtypes to generate tumor heterogeneity, consistent with the observation that NRF2 may play distinct roles in stem cells because it activates various target genes12, 37.
NRF2 may regulate over 100 genes within the promoter regions38. Intriguingly, we observed a decreasing trend in the expression levels of some NRF2-regulated canonical genes in sphere cells in the microarray. Additionally, our ChIP-Seq analyses revealed that sphere cells were NRF2 up-regulated but did not trans-activate typical NRF2 target gene NQO1. Accordingly, sphere cells displayed decreased expression of NQO1 (Figure S2c), suggesting that NRF2 may regulate genes other than classic cytoprotective genes in CICs.
ROS-independent NRF2 activation as a major effector of PERK-mediated cancer cell and stem cells survival has been recognized28, 39, 40. The potential contribution of global translation arrest control through p-PERK regulation of eIF2α cannot be excluded41. In addition, we also uncovered that the EIF2 signaling was upregulated in ROSLow and sphere cells (Figure S1h). These data lead us to hypothesize that active PERK-NRF2/EIF2 signaling may also play an additional role to maintain the quiescent state of ROSLow cells. Upon ER stress, GRP78 activates PERK/NRF2 signaling to promote cancer cell survival42. Stress may promote the re-localization of GRP78 to the cell membrane, which acts as a critical regulator of oncogenic signaling18, 43, 44. Here, overexpression of GRP78 in cells grown in defined selection medium promoted the re-localization of GRP78 to the membrane (Figure S6f), which activated both PERK and NRF2 and increased the stemness properties and glycolytic activity (Fig. 6 and Figure S6e). Consistent with our results, GRP78 is associated with modulation of the glycolysis pathway in stem cells31. Interestingly, GRP78 was one of the direct targets of NRF2 through NRF2 ChIP-Seq analysis of sphere cells (Figure S3f). NRF2 overexpression could restore GRP78 expression in GRP78-knockdown sphere cells (Fig. 6e and Figure S6a). Thus, the positive GRP78/p-PERK/NRF2 feedback loop in ROSLow cells appears to be a major mechanism for constitutively maintaining lower ROS levels.
In summary, this is the first finding that a positive regulatory loop of GRP78/p-PERK/NRF2 signaling mediates the Warburg effect and stemness properties of CICs in a ROS-independent manner. NRF2 signaling regulated the levels of different metabolic activities to adapt to the environment and to modulate cell behaviors in CICs containing different ROS levels. Therefore, the molecular mechanisms underlying these distinct metabolic pathways may reveal strategies for tumor-specific therapy.
Materials & Methods
Chromatin immunoprecipitation (ChIP) sequencing
For ChIP-Seq libraries of human SAS sphere cells or parental cells, input DNA has been used as the control. Full details are given in the in the Supplementary Material and Methods.
Fluorescent glucose uptake assay
Following 30 min of glucose starvation, stable NRF2-knockdown sphere cells were incubated in the presence of a fluorescent glucose analog, 2-[N-(7-nitrobenz-2-oxa-1,3-diaxol-4-yl)amino]-2-deoxyglucose (2-NBDG) for 30 min at 37 °C. The cells were then resuspended in PBS containing 2% FBS and stained with propidium iodide. The 2-NBDG fluorescence was measured using a FACScanto flow cytometer (Becton Dickinson).
Patient population and immunohistochemistry
Between 2004 and 2006, 57 patients with operable head and neck cancer, without histories of radiation therapy or chemotherapy, underwent surgical treatment at Taipei Veterans General Hospital (Taipei, Taiwan) (IRB No.1000075). This study was approved and accorded with the institutional review board and ethics committee of Taipei Veterans General Hospital and National Yang Ming University. The deparaffinization, rehydration, antigen retrieval, antibody hybridization, visualization, and grading were performed as described previously18, 45.
Statistical analysis
The data are presented as the mean ± SD of three independent experiments. Continuous variables between groups were compared with the independent Student’s t-test or ANOVA. We used the Statistical Package for the Social Sciences software (version 13.0) (SPSS, Inc., Chicago, IL) to generate the Kaplan-Meier survival curves and to determine statistical significance. P < 0.05 was considered a significant difference for all of the tests.
References
Baumann, M., Krause, M. & Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 8, 545–554 (2008).
Chiou, S. H. et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res. 14, 4085–4095 (2008).
Chang, C. W. et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 74, 6291–6305 (2014).
Yamashita, T. & Wang, X. W. Cancer stem cells in the development of liver cancer. J. Clin. Invest. 123, 1911–1918 (2013).
Yeo, H. et al. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. Embo. J. 32, 2589–2602 (2013).
Sancho, P., Barneda, D. & Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 114, 1305–1312 (2016).
Xie, H. et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell. Metab. 19, 795–809 (2014).
Schieber, M. S. & Chandel, N. S. ROS links glucose metabolism to breast cancer stem cell and EMT phenotype. Cancer Cell. 23, 265–267 (2013).
Ferreira, L. M., Hebrant, A. & Dumont, J. E. Metabolic reprogramming of the tumor. Oncogene 31, 3999–4011 (2012).
Dong, C. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 23, 316–331 (2013).
Costa, A., Scholer-Dahirel, A. & Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 25, 23–32 (2014).
Hochmuth, C. E., Biteau, B., Bohmann, D. & Jasper, H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell. Stem. Cell. 8, 188–199 (2011).
Hawkins, K. E. et al. NRF2 Orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep. 14, 1883–1891 (2016).
Yamadori, T. et al. Molecular mechanisms for the regulation of Nrf2-mediated cell proliferation in non-small-cell lung cancers. Oncogene 31, 4768–4777 (2012).
Frohlich, D. A., McCabe, M. T., Arnold, R. S. & Day, M. L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 27, 4353–4362 (2008).
Todaro, M. et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell. Stem. Cell. 14, 342–356 (2014).
Padanad, M. S. et al. Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 16, 1614–1628 (2016).
Wu, M. J. et al. Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Mol. Cancer 9, 283 (2010).
Mitsuishi, Y., Motohashi, H. & Yamamoto, M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2, 200 (2012).
Vazquez, F. et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 23, 287–301 (2013).
Koppenol, W. H., Bounds, P. L. & Dang, C. V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337 (2011).
Sancho, P. et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell. Metab. 22, 590–605 (2015).
Bu, Y. & Diehl, J. A. PERK Integrates Oncogenic Signaling and Cell Survival During Cancer Development. J. Cell. Physiol. 231, 2088–2096 (2016).
Mitsuishi, Y. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 22, 66–79 (2012).
Dupuy, F. et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell. Metab. 22, 577–589 (2015).
Kim, J. W., Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell. Metab. 3, 177–185 (2006).
Tamada, M. et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res. 72, 1438–1448 (2012).
Del Vecchio, C. A. et al. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 12, e1001945 (2014).
Bobrovnikova-Marjon, E. et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29, 3881–3895 (2010).
Kaira, K. et al. Expression of ER stress markers (GRP78/BiP and PERK) in adenoid cystic carcinoma. Acta Otolaryngol. 136, 1–7 (2015).
Miharada, K. et al. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell. Stem. Cell. 9, 330–344 (2011).
Khacho, M. et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell. Stem. Cell. 19, 232–247 (2016).
Dando, I. et al. The metabolic landscape of cancer stem cells. Iubmb. Life. 67, 687–693 (2015).
Liu, P. P. et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell. Death. Differ. 21, 124–135 (2014).
Le Belle, J. E. et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell. Stem. Cell. 8, 59–71 (2011).
Vega-Naredo, I. et al. Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells. Cell. Death. Differ. 21, 1560–1574 (2014).
Tsai, J. J. et al. Nrf2 regulates haematopoietic stem cell function. Nat. Cell. Biol. 15, 309–316 (2013).
Zhao, Y. et al. SoNar, a highly responsive NAD + /NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell. Metab. 21, 777–789 (2015).
Wang, L., Ryoo, H. D., Qi, Y. & Jasper, H. PERK limits Drosophila lifespan by promoting intestinal stem cell proliferation in response to ER stress. PLoS. Genet. 11, e1005220 (2015).
Pytel, D., Majsterek, I. & Diehl, J. A. Tumor progression and the different faces of the PERK kinase. Oncogene 35, 1207–1215 (2016).
Binet, F. & Sapieha, P. ER stress and angiogenesis. Cell. Metab. 22, 560–575 (2015).
Bellezza, I. et al. ROS-independent Nrf2 activation in prostate cancer. Oncotarget 8, 67506–67518 (2017).
Lee, A. S. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 14, 263–276 (2014).
Louessard, M. et al. Activation of cell surface GRP78 decreases endoplasmic reticulum stress and neuronal death. Cell. Death. Differ. 24, 1518–1529 (2017).
Lo, J. F. et al. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. Cancer Res. 71, 1912–1923 (2011).
Acknowledgements
We thank the Instrumentation Resource Center and Proteomics Research Center at National Yang-Ming University for technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, CW., Chen, YS., Tsay, YG. et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis 9, 194 (2018). https://doi.org/10.1038/s41419-017-0250-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-017-0250-x
This article is cited by
-
Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer
Experimental & Molecular Medicine (2024)
-
Angelica sinensis polysaccharides promote extramedullary stress erythropoiesis via ameliorating splenic glycolysis and EPO/STAT5 signaling-regulated macrophages
Journal of Molecular Histology (2024)
-
A novel HIF-2α targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPRER axis
Cell Death & Differentiation (2022)
-
The multifaceted role of NRF2 in cancer progression and cancer stem cells maintenance
Archives of Pharmacal Research (2021)
-
The role of redox system in metastasis formation
Angiogenesis (2021)
