
Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the deadliest cancers, and it requires novel treatment approaches and effective drugs. In the present study, we found that treatment with plumbagin, a natural compound, reduced proliferation and survival of the KYSE150 and KYSE450 ESCC cell lines in a dose-dependent manner in vitro. The drug also effectively inhibited the viability of primary ESCC cells from fresh biopsy specimens. Furthermore, plumbagin-induced mitotic arrest and massive apoptosis in ESCC cells. Notably, the drug significantly suppressed the colony formation capacity of ESCC cells in vitro and the growth of KYSE150 xenograft tumors in vivo. At the molecular level, we found that exposure to plumbagin decreased both polo-like kinase 1 (PLK1) and phosphorylated protein kinase B (p-AKT) expression in both ESCC cell lines. Enforced PLK1 expression in ESCC cells not only markedly rescued cells from plumbagin-induced apoptosis and proliferation inhibition but also restored the impaired AKT activity. Furthermore, signal transducer and activator of transcription 3 (STAT3), a transcription factor of PLK1, was also inactivated in plumbagin-treated ESCC cells; however, the overexpression of a constitutively activated STAT3 mutant, STAT3C, reinstated the plumbagin-elicited blockade of PLK1-AKT signaling in ESCC cells. Taken together, these findings indicate that plumbagin inhibits proliferation and potentiates apoptosis in human ESCC cells in vitro and in vivo. Plumbagin may exert these antitumor effects by abrogating STAT3-PLK1-AKT signaling, which suggests that plumbagin may be a novel, promising anticancer agent for the treatment of ESCC.
Similar content being viewed by others


Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the most common malignancies worldwide and the fourth leading cause of cancer-related deaths in China1,2. ESCC is an aggressive malignant disease with a poor prognosis, and its treatment remains a significant challenge3. Few currently available drugs are effective in patients with ESCC; thus, potent anticancer agents are urgently needed.
Plumbagin (5-hydroxy-2-methyl-1,4-napthoquinone, PL), a natural quinoid constituent isolated from the roots of the medicinal plant Plumbago zeylanica L., exhibits striking anticancer activity in various human cancers, including lung, breast, ovarian, prostate, and colon cancer4,5,6,7,8,9,10,11. It has been shown that plumbagin can suppress malignant activity of cancer cells through multiple mechanisms, such as the inhibition of growth, invasion, and metastasis; induction of apoptosis; and anti-angiogenesis4,5,7,8,9,10. However, the antitumoral efficacy of plumbagin in ESCC remains to be determined.
In this work, we evaluated the potential antitumor effects of plumbagin against ESCC cells in vitro and in vivo. Furthermore, we explored the underlying mechanisms of these effects in ESCC cells.
Results
Plumbagin inhibits the proliferation of ESCC cells
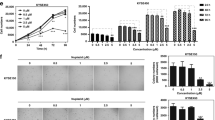
To explore the potential cytotoxicity of plumbagin to ESCC cells, we first assessed the effect of plumbagin on cell proliferation. The treatment of two independent ESCC cell lines, KYSE150 and KYSE450, with various concentrations of plumbagin significantly inhibited cell proliferation in a concentration-dependent and time-dependent manner (Fig. 1a). The IC50 values of plumbagin in KYSE150 and KYSE450 cells were 6.4 ± 0.2 and 8.0 ± 0.3 μM, respectively (Fig. 1b).

a,b ESCC cell line KYSE150 and KYSE450 were treated with increasing concentrations of plumbagin for 24–72 h. Cell viability was determined with a CCK-8 assay. a The effects of plumbagin on cell viability of ESCC cells. Data are presented as mean ± SEM (n = 3). **P < 0.01, ***P < 0.001. b ESCC cells were treated with plumbagin for 24 h, and the IC50 was calculated. c,d Primary ESCC cells cultured in vitro were treated with plumbagin for 72 h, and cell viability were assessed using ATP-TCA. c Primary ESCC cells from five patients were treated with increasing doses of plumbagin. d Primary ESCC cells from nine patients were treated with 10 μM plumbagin. e Comparison of drug sensitivity in primary ESCC cells from two patients
To further examine the antitumor efficacy of plumbagin in clinical specimens, we performed a chemosensitivity assay using primary ESCC cells isolated from fresh biopsy tissues. Exposure to plumbagin for 72 h inhibited the proliferation of ESCC cells in a concentration-dependent manner, and the average IC50 and IC90 values of plumbagin in tested samples were 1.8 ± 0.4 and 7.5 ± 0.9 μM (n = 5), respectively (Fig. 1c). In addition, 10 μM of plumbagin reduced proliferation by ∼90% in 89% (8/9) of biopsy specimens (Fig. 1d), indicating that most of the ESCC cells responded well to the drug. Moreover, we compared the efficacy of plumbagin with two drugs currently used in ESCC chemotherapy, 5-Fu and Cisplatin (DDP). Notably, in the samples from two ESCC patients, the IC50 and IC90 of plumbagin were markedly lower than those of 5-Fu and DDP (Fig. 1e), indicating that plumbagin is more effective compared to either 5-Fu or DDP in these cases.
Plumbagin induces cell cycle arrest and apoptosis in ESCC cells
To decipher the mechanisms underlying the plumbagin-mediated inhibition of proliferation, we analyzed the cell cycle distribution using flow cytometry. Following 12 h of plumbagin treatment, the number of KYSE150 and KYSE450 cells in the G2/M phase significantly increased, which was accompanied by a marked decrease of cells in the G0/G1 phase (Fig. 2a and b, Supplementary Table 1). In addition, obvious changes in apoptotic morphology were observed in KYSE150 and KYSE450 cells 24 h after exposure to plumbagin (Fig. 3a), and this plumbagin-induced apoptosis was dose-dependent in both cell lines (Fig. 3b).

KYSE150 and KYSE450 cells were treated with 5 or 7.5 μM of plumbagin for 12 h, and the cell cycle distribution was then assessed using flow cytometry. a Representative results of the cell cycle analysis. b Histogram of cell cycle distribution. Data are represented as mean ± SEM (n = 3). **P < 0.01, ***P < 0.001

KYSE150 and KYSE450 cells were incubated with 0, 5, 7.5, or 10 μM plumbagin for 24 h. a Representative images of cells treated with plumbagin obtained with a phase contrast microscope. Scale bar = 100 μm. b Cell apoptosis was assessed based on Annexin V-FITC/PI double staining and flow cytometry. Representative results are shown and percentage of apoptotic cells was plotted. Data are represented as mean ± SEM (n = 3). *P<0.05, ***P < 0.001
Plumbagin suppresses the tumorigenicity of ESCC cells
Subsequently, we investigated the effects of plumbagin on the tumorigenicity of ESCC cells. First, we observed that plumbagin markedly and dose-dependently suppressed the colony formation capacity of KYSE150 and KYSE450 cells (Fig. 4a). Then, xenograft tumors derived from KYSE150 cells were utilized to evaluate the antitumor efficacy of plumbagin in vivo, since the xenograft tumors derived from KYSE450 cells were mainly comprised of necrotic tissues. The intraperitoneal (i.p.) administration of plumbagin (2 mg/kg/day, 5 days per week for 3 weeks) drastically attenuated the growth of subcutaneous (s.c.) KYSE150 xenograft tumors in nude mice, as determined by marked decreases in tumor volume compared with vehicle-treated controls (Fig. 4b). However, plumbagin treatment did not result in observable weight loss during the experimental period (Fig. 4c), indicating that the drug lacks apparent systemic toxicity in vivo. The weights of plumbagin-treated xenograft tumors were also significantly lower than those of control tumors (Fig. 4d, e). In accordance with the strong antitumor effect of plumbagin observed in vivo, treatment with this drug also evidently inhibited proliferation and enhanced apoptosis in xenograft tumors, as evidenced by Ki67 immunostaining and TUNEL assays performed on s.c. tumor sections (Fig. 4f, g).

a Colony formation assays with ESCC cells exposed to the indicated concentrations of plumbagin (PL) for 6 h. Representative results are shown, and the number of colonies is plotted. Data are presented as mean ± SEM (n = 3). ***P < 0.001. b–g Nude mice bearing established KYSE150 xenograft tumors (~50 mm3) were i.p. injected with 2 mg/kg plumbagin or vehicle control 5 days per week for 3 weeks. The tumor volumes b and body weight of mice c were measured at the indicated time after plumbagin administration. Data are presented as mean ± SEM (n = 5). Body weight is expressed as a percentage relative the initial weight at the beginning of treatment. *P < 0.05, **P < 0.01. d Xenograft tumors. Scale bar = 1 cm. e Comparison of tumor weights. f Intratumoral proliferation was assessed based on Ki67 immunostaining. Bar = 50 μm. g Apoptosis was measured with a TUNEL assay. Bar = 50 μm
PLK1 is a pivotal cellular target of plumbagin in ESCC cells
Our previous studies have demonstrated that the overexpression of PLK1 plays an essential role in the proliferation and apoptosis resistance of ESCC cells12,13. Specifically, depletion of PLK1 with a specific siRNA resulted in marked G2/M arrest and drastic apoptosis in KYSE150 and KYSE450 cells (Supplementary Fig. 1); plumbagin induced similar effects. Hence, we speculated that plumbagin may suppress cell proliferation and survival by suppressing PLK1 expression in ESCC cells. To test this hypothesis, we examined changes in PLK1 expression in plumbagin-treated ESCC cells. Indeed, exposure to plumbagin for 24 h reduced the PLK1 protein levels in KYSE150 and KYSE450 cells in a dose-dependent manner, indicating that plumbagin is a potent inhibitor of PLK1 (Fig. 5a).

a KYSE150 and KYSE450 cells were treated with increasing concentrations of plumbagin (PL) for 24 h. b KYSE150 and KYSE450 cells were transiently transfected with PLK1 siRNA or control non-silencing siRNA for 48 h. c KYSE150 and KYSE450 cells were treated with 7.5 μM plumbagin for the indicated times. Cell lysates were immunoblotted for the indicated proteins. β-actin was used as a loading control
PLK1 is a key regulator of cell division14. It has been reported that PLK1 inhibition efficiently arrested cells in mitosis by preventing cyclin B1 degradation at anaphase15. The present study demonstrated that PLK1 depletion resulted in a mitotic block with a concomitant increase in the levels of cyclin B1 and phospho-Histone H3 (Ser10), a mitosis marker, in both ESCC cell lines (Supplementary Fig. 1a and b, Fig. 5b). In parallel, the levels of cyclin B1 and p-Histone H3 (Ser10) were also elevated in plumbagin-treated ESCC cells (Fig. 5c), implying that plumbagin-mediated PLK1 downregulation prevents cyclin B1 degradation, which contributes to drug-induced mitotic arrest.
In most cancer cells, mitotic arrest induced by PLK1 inhibition triggers apoptosis14,16,17,18. Our earlier work showed that PLK1 depletion lead to apoptosis by activating the mitochondrial apoptotic pathway in ESCC cells13. As with PLK1 knockdown, treatment of ESCC cells with plumbagin induced the cleavage of pro-caspase-9, but not pro-caspase-8 in KYSE150 and KYSE450 cells. Meanwhile, the levels of pro-caspase-3 and full-length poly (ADP-ribose) polymerase (PARP), a cleavage target of caspase-3, were reduced upon drug treatment (Fig. 6a). Moreover, enforced PLK1 expression partially restored the plumbagin-induced cleavage of pro-caspase-9, pro-caspase-3, and full-length PARP in both cell lines (Fig. 6b). Collectively, the data indicated that plumbagin-elicited PLK1 downregulation potentiates apoptosis by activating the intrinsic mitochondrial apoptotic pathway in ESCC cells.

a KYSE150 and KYSE450 cells were treated with plumbagin for 24 h, and the levels of indicated proteins were assessed by Western blotting. β-actin was used as a loading control. b-e The inducible stable clone strains KYSE150-TO-PLK1-V5 and KYSE450-TO-PLK1-V5 cells were pre-treated with 1 μg/ml doxycyclin for 24 h to induce exogenous PLK1 expression, and then treated with the indicated dose of plumbagin (PL) for 24 h. Cells stably transfected with Empty vector (EV) were used as controls. b Cell lysates were immunoblotted for the indicated proteins. c Cell viability was assessed using a CCK-8 assay. d Apoptosis were analyzed with Annexin V-FITC double-staining and flow cytometry. The representative results are shown and percentages of apoptotic cells were plotted. e Colony formation assays of cells exposed to the indicated concentrations of plumbagin. Representative results are shown and colony number was quantified. c-e. Data are represented as mean ± SEM (n = 3). **P < 0.01; *** P < 0.001
Remarkably, ectopically expressed PLK1 attenuated the plumbagin-mediated inhibition of proliferation, survival, and colony formation in both ESCC cell lines compared to control cells. This further demonstrated that plumbagin-induced PLK1 downregulation largely imparts antitumor activity in ESCC cells (Fig. 6c–e).
Plumbagin inactivates AKT by downregulating PLK1
Next, we identified critical downstream signaling pathways modulated by plumbagin in ESCC cells. Among the pathways that are closely associated with cell proliferation and survival, plumbagin treatment markedly suppressed PI3K-AKT signaling in KYSE150 and KYSE450 cells, as demonstrated by a reduction in the p-AKT levels (Fig. 7a). Likewise, PLK1 knockdown abrogated the activation of AKT (Fig. 7b). Moreover, exogenous PLK1 overexpression effectively reversed this reduction in the AKT levels (Fig. 7c).

a ESCC cells were treated with 0, 7.5 or 10 μM plumbagin (PL) for 24 h. b ESCC cells were transiently transfected with PLK1 siRNA or non-silencing control siRNA for 48 h. c PLK1 overexpressed stable clone cells or empty vector (EV) control cells described in Fig. 6 were treated with 7.5 μM plumbagin (PL) for 24 h. Cell lysates were immunoblotted for the indicated proteins. β-actin was used as a loading control.
STAT3 is involved in the plumbagin-mediated blockade of PLK1-AKT cascade
Finally, we explored the molecular mechanism underlying the plumbagin-induced downregulation of PLK1. Exposure to plumbagin drastically decreased the PLK1 mRNA levels, implying that plumbagin influences PLK1 transcription (Fig. 8a). Accordingly, earlier studies have shown that plumbagin can inhibit STAT3 activity4,19,20,21. In addition, we previously demonstrated that STAT3 can transcriptionally activate PLK1 expression in ESCC cells. Therefore, we herein examined changes in STAT3 activity in plumbagin-treated ESCC cells. As expected, plumbagin treatment decreased the p-STAT3 levels (Fig. 8b). In addition, blocking JAK2/STAT3 signaling with a highly selective inhibitor JSI-124 (cucurbitacin I) distinctly downregulated PLK1 at both the mRNA and protein levels, and these changes were accompanied by diminished AKT activity (Fig. 8c, d). Moreover, the overexpression of a constitutively activated STAT3 mutant, STAT3C, markedly restored the reduction of PLK1 expression mediated by plumbagin (Fig. 8e, f). These data suggest that plumbagin may inhibit ESCC tumor growth by abrogating the STAT3-PLK1-AKT cascade.

a,b KYSE150 and KYSE450 cells were treated with plumbagin (PL) for 24 h. a The PLK1 mRNA levels were examined by qRT-PCR. GAPDH was used as an internal reference. b The activity of STAT3 were assessed by Western blotting. β-actin was used as a loading control. c,d Cells were treated with 10 μM JSI-124 for 24 h, and the PLK1 mRNA and indicated protein levels were then measured with qRT-PCR c and Western blotting d. e,f ESCC cells stably transfected with pBabe-Stat3C or empty vector were treated with 7.5 μM plumbagin (PL) for 24 h, and the mRNA or protein level of PLK1 was measured with qRT-PCR e and Western blotting d. a,c and e Data are represented as mean ± SEM (n = 3). **P < 0.01; ***P < 0.001
Discussion
ESCC is a common cancer with poor prognosis, and is unresponsive to most conventional chemotherapeutic agents. We show here that plumbagin effectively suppresses the proliferation and survival of ESCC cells in vitro and in vivo. Furthermore, our data indicate that the plumbagin-mediated tumor-inhibitory effects are closely associated with the inhibition of STAT3-PLK1-AKT pathway in ESCC cells.
Although plumbagin has exhibited potent antitumor activity in several cancer types10, its impact on ESCC cells remains unclear. Our present work revealed that plumbagin has strong antitumor efficacy against ESCC cells in culture and in mice, which is consistent with its effects on some other cancer types4,7,22,23,24. In addition, our xenograft assay showed that the effective dose of plumbagin (2 mg/kg, i.p.) had no apparent systemic toxicity in vivo, reminiscent of an earlier finding that plumbagin did not markedly influence the viability of normal cells, i.e., peripheral blood mononuclear cells, even at 100 µM6. Particularly, we confirmed the growth-inhibitory effect of plumbagin in primary ESCC cells from biopsy tissues. Cisplatin (DDP) and 5-Fu are currently used in patients with unresectable locally advanced or metastatic ESCC; however, their antitumor efficacy and response rate are not satisfactory3. Notably, ∼90% of ESCC biopsy specimens were sensitive to plumbagin, even in DDP and 5-Fu insensitive cases. Altogether, our data suggest that plumbagin is a novel promising agent for ESCC therapy.
To date, the mechanisms underlying the anticancer action of plumbagin are not fully understood10, and the cellular targets of plumbagin in ESCC cells remain unidentified. In the present study, we found that plumbagin treatment resulted in pronounced G2/M arrest and apoptosis in ESCC cells, which contributes to its antitumor effects. PLK1 is a member of the highly conserved serine/threonine protein kinase family, and it is a key regulator of cell division25,26,27. We previously reported that PLK1 was overexpressed in ∼70% of ESCC tissues. Furthermore, suppression of PLK1 expression or activity significantly inhibited the proliferation and survival of ESCC cells in culture and in mice12,13,28. PLK1 depletion and plumbagin treatment resulted in identical phenotypes (i.e. G2/M arrest and apoptosis), which raises the possibility that plumbagin suppresses cell proliferation and survival by regulating PLK1 expression in ESCC cells. As expected, plumbagin treatment decreased PLK1 expression. Moreover, overexpression of exogenous PLK1 prominently decreased the inhibitory potency of plumbagin. Collectively, these data reveal that PLK1 is an essential target of plumbagin, and the cytotoxicity of plumbagin to ESCC cells is at least partially due to downregulation of PLK1.
PLK1 is a cell cycle-related kinase required for proper M-phase progression. PLK1 inhibition leads to pronounced mitotic arrest followed by apoptosis14,16,17,18. Plumbagin treatment also arrested ESCC cells in the mitosis phase, as indicated by the elevated cyclin B1 and p-Histone H3 (Ser10) levels. Cyclin B1 is an important regulator of mitosis. At the end of mitosis (anaphase), cyclin B1 is degraded by the anaphase-promoting complex (APC), thereby permitting cells to exit mitosis. PLK1 has been shown to activate the APC by phosphorylating several of its components. The activated APC catalyzes the polyubiquitination of cyclin B1 and causes its degradation15. Altogether, our results imply that plumbagin-mediated PLK1 downregulation lead to aberrant cyclin B1 accumulation at anaphase, thereby arresting the cells in mitosis. In contrast, plumbagin-induced earlier G2/M arrest in some cancer types, which was associated with decreased amounts of cyclin B129,30,31. These different findings imply that the effects of the drug may be cell type-specific or cell context-specific, which may be largely dependent on its cellular targets and associated signaling pathway. Furthermore, our work demonstrated that plumbagin activated the intrinsic mitochondrial apoptotic pathway by impairing PLK1 expression in ESCC cells.
Previous reports have shown that plumbagin can inhibit cell proliferation and survival by modulating multiple signaling pathways4,9,10,21,32. However, the impact of plumbagin on the PLK1-related network is not clear. PLK1 is known to be an upstream kinase of the AKT33,34, and we found that both plumbagin treatment and PLK1 depletion inactivated AKT in ESCC cells. More importantly, enforced PLK1 expression markedly restored AKT activity in plumbagin-treated ESCC cells, suggesting that PLK1 is a critical mediator of plumbagin-induced AKT inactivation. Moreover, our data also suggest that plumbagin directly downregulates PLK1 mRNA expression by abolishing the activity of STAT3, a transcriptional factor of PLK1. Intriguingly, a bioinformatics analysis predicted that plumbagin can inactivate STAT3 by directly binding to the DNA-binding domain of STAT3 and packing against its residues, such as Gly-421, Gly-419, Cys-418, Arg-417, Glu-415, and Arg-38235, implying that STAT3 is a direct cellular target of plumbagin. Taken together, this work provides evidence that plumbagin can exert its antitumor effects by abrogating STAT3-PLK1-AKT signaling, thus revealing a novel anticancer mechanism of the drug.
Conclusions
Here, we report that plumbagin elicits potent antitumor activity against ESCC by reducing cell proliferation and survival in vitro and in vivo. We further identify the STAT3-PLK1-AKT signaling pathway as a pivotal molecular target of plumbagin in ESCC cells. Overall, our results suggest that plumbagin is a promising anticancer drug for the treatment of ESCC, which build a foundation for the clinical trials of this novel therapeutic agent.
Materials and methods
Tumor tissues
ESCC biopsy tissues form nine patients were collected at Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS & PUMC), Beijing, China. The clinicopathological features of biopsy specimens is described in Supplementary Table 1. None of the patients had received chemotherapy of radiotherapy. All patients signed separate informed consent forms for sampling and research. This study was approved by the Ethics Committee/Institutional Review Board of National Cancer Center/Cancer Hospital, CAMS and PUMC (No. NCC2013RE-025).
Cell culture and treatments
The human ESCC cell lines KYSE 150 and KYSE 450 were gifts from Dr. Y. Shimada (Kyoto University, Kyoto, Japan). The background information of both cell lines is provided in Supplementary Table 2. The cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (Invitrogen, San Diego, CA), penicillin (100 U/ml), and streptomycin (100 mg/ml) at 37 °C under 5% CO2 in a humidified incubator. ESCC cells were incubated with plumbagin (Sigma, St. Louis, MO) at the indicated concentrations and for the indicated times. Vehicle (DMSO)-treated cells were used as controls.
Cell viability assay
Cells were seeded in triplicate in 96-well plates at a density of 8000 cells per well. After an overnight incubation, the cells were exposed to different concentrations of plumbagin for the indicated times. Cell viability was assessed with a Cell Counting kit-8 (CCK-8, Dojindo, Japan) according to the manufacturer’s protocol. The absorbance at 450 nm was measured using an ELX808 microplate spectrophotometer (BioTek Instruments, Winooski, VT, USA).
Colony formation assay
To assess the clonogenic capacity of cells in vitro, 500 cells were plated in six-well culture plates in triplicated. Following a 24 h incubation, the cells were treated with plumbagin for 6 h. After being washed with PBS, the cells were allowed to grow for 10–14 days to form colonies, and then fixed with methanol and stained with crystal violet. The number of colonies was counted.
Cell cycle analysis
Cells were seeded at a density of 5 × 105 per well in six-well plates and incubated at 37 °C overnight. The cells were treated with plumbagin (0, 5 and 7.5 μM) for 12 h and then collected by trypsinization and fixed in ice-cold 70% ethanol overnight at 4 °C. The cell cycle distribution was analyzed using a Cell cycle detection kit (Nanjing KeyGen Biotech, Nanjing, China) and BDTM LSRΙΙ flow cytometer (BD Biosciences, San Jose, CA, USA). The resultant cell cycle profiles were analyzed using the ModFit software (Verity Software House, Topshem, ME, USA).
Apoptosis detection
The morphology of ESCC cells treated with plumbagin was observed under a phase-contrast microscope (Leica DMI4000B, Leica Microsystems Wetzlar GmbH, Wetzlar, Germany). Apoptotic cells were double-labeled with AnnexinV-FITC and PI using the Annexin V/FITC kit (Neo Bioscience, Beijing, China) and analyzed with a BDTM LSRΙΙ flow cytometer (BD Biosciences). Annexin V-positive cells were calculated and defined as apoptotic cells. A TUNEL assay using the Dead-End Fluorometric TUNEL System (Promega, Madison, WI) was performed to detect intratumoral apoptosis as described previously[12].
Chemosensitivity assay
Fresh ESCC tissue specimens were digested and separated to generate single-cell suspensions, which were seeded in 96-well plates at a density of 2–4 × 104 cells per well. The primary cultured ESCC cells were then treated with plumbagin, 5-fluorouracil (5-Fu, Tianjin Kingyork Group Co., Ltd., Tianjin, China), or Cisplatin (DDP, Qilu Pharmaceutical Co., Ltd., Jinan, China) for 72 h. The viability of cells was assessed using an ATP bioluminescence tumor chemosensitivity assay (ATP-TCA). All essential materials and reagents, including the tissue dissociation enzyme, red blood cell lysis buffer, culture medium, ATP standard, ATP maximal inhibitor, luciferase–luciferin mix, and various buffers, were provided by the tumor chemosensitivity assay kit (Beijing Gold Amethyst Pharm & Biotech Co. Ltd., Beijing, China). Light units were measured using a microplate luminometer (BHP9504, Hamamatsu Photo Medical Techniques Inc., Beijing, China). The growth inhibition rate was calculated using the following formula: (1-absorption of experimental group/absorption of vehicle control) ×100%.
Small interfering RNA and plasmid constructs
The following siRNA target sequence was used to silence human PLK1: 5′-AGATCACCCTCCTTAAATATT-3′13. A scrambled siRNA sequence, 5′-TTCTCCGAACGTGTCACGT-3′, was used as a negative control. The oligonucleotides were chemically synthesized by GenePharma (Shanghai, China). All plasmid constructs generation are described in the Supplementary materials.
Transfection and lentiviral transduction
Cells were transfected with siRNA or plasmid vectors using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. To generate the lentivirus, 293FT cells (Invitrogen) were cotransfected with psPAX2, pMD2.G, and either pLenti6.3-TO-PLK1-V5 or pLenti6.3-TO-EV. Forty-eight hours after transfection, the lentiviral supernatants were harvested and passed through a 0.45 μM filter. The lentiviruses were added to media supplemented with 8 µg/ml polybrene (Sigma) to transduce ESCC cells following the manufacturer’s instructions.
Stable clone selection and induction of PLK1 expression
To establish inducible PLK1-overexpressing ESCC cell strains, KYSE150 or KYSE450 cells were infected with pLenti6.3-TO-PLK1-V5 lentivirus and further selected for 10 days with 1.5 and 0.7 μg/ml blasticidin (Invitrogen), respectively, to generate stable pools named KYSE150-TO-PLK1 or KYSE450-TO-PLK1. Concurrently, KYSE150 or KYSE450 stable clones infected with pLenti6.3-TO-V5-EV, named KYSE150-TO-EV and KYSE450-TO-EV, were used as controls. To induce exogenous PLK1 expression in KYSE150-TO-PLK1 and KYSE450-TO-PLK1 cells in vitro, 1 μg/ml doxcycline (Dox, Selleckchem, Houston, TX, USA) was added to the culture medium. The expression of PLK1-V5 in stable clone cells was verified by Western blotting using V5 antibody.
Western blot analysis
SDS-PAGE and Western blotting were performed according to standard protocols. Briefly, the protein lysates were separated on SDS-PAGE gels and then transferred onto PVDF membranes (Millipore, Bedford, MA, USA). The blots were blocked and probed with primary antibodies against PLK1 (Abcam, Cambridge, UK), V5 (Invitrogen), pro-caspase-3, full-length PARP (FL-PARP) (Proteintech, Wuhan, China), p-Histone-H3(p-HH3)(Ser10)(Immunoway Biotechnology, TX, USA), pro-caspase-8 (Atlas Antibody, Stockholm, Sweden), cyclin B1, pro-caspase-9, Histon-H3, p-ERK (Thr202/Tyr204), ERK1/2, p-AKT (Ser473), AKT, Stat3, and p-Stat3 (Tyr705) (Cell Signaling Technology, Danvers, MA, USA). β-actin (Amart, Shanghai, China) was used as a loading control. The signals were visualized with a super-enhanced chemiluminescence detection reagent (Applygen).
Xenograft model antitumor assay
All animal procedures were approved by the Institutional Animal Care and Use Committee at the National Cancer Center/Cancer Hospital, CAMS & PUMC (No. NCC2013A014), and followed the National Institutes of Health guide for the care and use of Laboratory animals. KYSE150 cells were subcutaneously inoculated into the flanks of 4-week-old female nude mice (BALB/c-nu, HFK Bioscience, Beijing, China). A total of 1×106 cells were injected per animal. When the tumor volume reached ∼50 mm3, the mice were randomly divided into two groups (n = 8) and treated daily for 5 days per week with plumbagin (2 mg/kg body weight, i.p.) prepared in 25% PEG or vehicle control. The mice were weighed and tumor size was measured twice weekly. The tumor volume (V) was calculated with the following formula: V = 0.524×L×W2, where L is the length and W is the width of the xenograft tumor. The mice were euthanized, and the average weight of tumor tissues was obtained after 3 weeks of treatment.
Immunohistochemistry
Xenograft tumors from vehicle-treated and plumbagin-treated mice were fixed in 10% neutral buffered formalin, embedded in paraffin, and used to prepare 4 μM-thick sections. These slides were immunohistochemically stained with anti-Ki67 antibody (Cell Signaling Technology) as described previously12.
Real-time quantitative RT-PCR
Total RNA isolation, cDNA synthesis and real-time quantitative RT-PCR (qRT-PCR) was performed as previously described [12]. GAPDH was used as an internal reference.
Statistical analysis
Statistical analysis was performed using the SPSS 19.0 program (SPSS Inc., Chicago, IL). Experimental results were statistically evaluated using the Student’s t-test, a one-way ANOVA, or the nonparametric Kruskal–Wallis test. A P-value < 0.05 was considered significant.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 66, 7–30 (2016).
Chen, W. Cancer statistics: updated cancer burden in China. Chin. J. Cancer Res.=Chung-kuo yen cheng yen chiu. 27, 1 (2015).
Ohashi, S. et al. Recent advances from basic and clinical studies of esophageal squamous cell carcinoma. Gastroenterology 149, 1700–1715 (2015).
Aziz, M. H., Dreckschmidt, N. E. & Verma, A. K. Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer. Res. 68, 9024–9032 (2008).
Manu, K. A. et al. Plumbagin inhibits invasion and migration of breast and gastric cancer cells by downregulating the expression of chemokine receptor CXCR4. Mol. Cancer 10, 107 (2011).
Subramaniya, B. R. et al. Apoptosis inducing effect of plumbagin on colonic cancer cells depends on expression of COX-2. PLoS One 6, e18695 (2011).
Lai, L. et al. Plumbagin inhibits tumour angiogenesis and tumour growth through the Ras signalling pathway following activation of the VEGF receptor-2. Br. J. Pharmacol. 165(4b), 1084–1096 (2012).
Sinha, S. et al. Plumbagin inhibits tumorigenesis and angiogenesis of ovarian cancer cells in vivo. Int. J. Cancer 132, 1201–1212 (2013).
Li, Y. C. et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 344, 239–259 (2014).
Liu, Y., Cai, Y., He, C., Chen, M., Li, H. Anticancer properties and pharmaceutical applications of plumbagin: a review. Am. J. Chin. Med. 45, 423–441 (2017).
Niu, M. et al. Plumbagin inhibits growth of gliomas in vivo via suppression of FOXM1 expression. J. Pharmacol. Sci. 128, 131–136 (2015).
Zhang, Y. et al. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology 142, 521–30 e3 (2012).
Feng, Y. B. et al. Overexpression of PLK1 is associated with poor survival by inhibiting apoptosis via enhancement of survivin level in esophageal squamous cell carcinoma. Int. J. Cancer 124, 578–588 (2009).
Gutteridge, R. E., Ndiaye, M. A., Liu, X. & Ahmad, N. Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol. Cancer Ther. 15, 1427–1435 (2016).
Liu, X. & Erikson, R. L. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc. Natl. Acad. Sci. USA 99, 8672–8676 (2002).
Chopra, P., Sethi, G., Dastidar, S. G. & Ray, A. Polo-like kinase inhibitors: an emerging opportunity for cancer therapeutics. Expert Opin. Investig. Drugs 19, 27–43 (2010).
Rudolph, D. et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 15, 3094–3102 (2009).
Steegmaier, M. et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 17, 316–322 (2007).
Powolny, A. A. & Singh, S. V. Plumbagin-induced apoptosis in human prostate cancer cells is associated with modulation of cellular redox status and generation of reactive oxygen species. Pharm. Res. 25, 2171–2180 (2008).
Hafeez, B. B. et al. Plumbagin inhibits prostate carcinogenesis in intact and castrated PTEN knockout mice via targeting PKCepsilon, Stat3, and epithelial-to-mesenchymal transition markers. Cancer Prev. Res. 8, 375–386 (2015).
Hafeez, B. B., Jamal, M. S., Fischer, J. W., Mustafa, A. & Verma, A. K. Plumbagin, a plant derived natural agent inhibits the growth of pancreatic cancer cells in in vitro and in vivo via targeting EGFR, Stat3 and NF-kappaB signaling pathways. Int. J. Cancer 131, 2175–2186 (2012).
Yan, W. et al. Suppressive effects of plumbagin on invasion and migration of breast cancer cells via the inhibition of STAT3 signaling and down-regulation of inflammatory cytokine expressions. Bone Res. 1, 362–370 (2013).
Hafeez, B. B. et al. Plumbagin, a medicinal plant (Plumbago zeylanica)-derived 1,4-naphthoquinone, inhibits growth and metastasis of human prostate cancer PC-3M-luciferase cells in an orthotopic xenograft mouse model. Mol. Oncol. 7, 428–439 (2013).
Sung, B., Oyajobi, B. & Aggarwal, B. B. Plumbagin inhibits osteoclastogenesis and reduces human breast cancer-induced osteolytic bone metastasis in mice through suppression of RANKL signaling. Mol. Cancer Ther. 11, 350–359 (2012).
Combes, G., Alharbi, I., Braga, L.G., Elowe, S. Playing polo during mitosis: PLK1 takes the lead. Oncogene 36, 4819-4827 (2017)
Strebhardt, K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 9, 643–660 (2010).
Archambault, V. & Glover, D. M. Polo-like kinases: conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 10, 265–275 (2009).
Lin, Y. et al. Epidemiology of esophageal cancer in Japan and China. J. Epidemiol. 23, 233–242 (2013).
Pan, S. T. et al. Plumbagin induces G2/M arrest, apoptosis, and autophagy via p38 MAPK- and PI3K/Akt/mTOR-mediated pathways in human tongue squamous cell carcinoma cells. Drug Des. Dev. Ther. 9, 1601–1626 (2015).
Kuo, P. L., Hsu, Y. L. & Cho, C. Y. Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol. Cancer Ther. 5, 3209–3221 (2006).
Hsu, Y. L., Cho, C. Y., Kuo, P. L., Huang, Y. T. & Lin, C. C. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induces apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation at serine 15 in vitro and in vivo. J. Pharmacol. Exp. Ther. 318, 484–494 (2006).
Zhou, Z. W. et al. Induction of apoptosis and autophagy via sirtuin1- and PI3K/Akt/mTOR-mediated pathways by plumbagin in human prostate cancer cells. Drug Des. Dev. Ther. 9, 1511–1554 (2015).
Zhang, Z. et al. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res. 74, 6635–6647 (2014).
Li, Z. et al. Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol. Cell Biol. 34, 3642–3661 (2014).
Jamal, M. S. et al. Anticancer compound plumbagin and its molecular targets: a structural insight into the inhibitory mechanisms using computational approaches. PLoS One 9, e87309 (2014).
Acknowledgements
This work was supported by the Beijing Natural Science Foundation (7151008), the National Natural Science Foundation (81330052, 81520108023, 81572841), and the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1-001, 2016-I2M-3-007). We thank Dr. Jacqueline F. Bromberg (Memorial Sloan-Kettering Cancer Center, New York, NY) for providing the plasmid vectors of pBabe-Stat3C and pBabe, Dr. Andrew S. Chi (Cancer center, Massachusetts General Hospital, Boston, MA) for his kind help in constructing the pLenti6.3-TO-PLK1-V5 plasmid. We also thank Prof. Wei Zhang and Ms. Yan-Fen Li (Detection Center of Tumor Biology, CAMS & PUMC, Beijing, China) for expert technical assistance in chemosensitivity asssay.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, YY., Yu, J., Liu, TT. et al. Plumbagin inhibits the proliferation and survival of esophageal cancer cells by blocking STAT3-PLK1-AKT signaling. Cell Death Dis 9, 17 (2018). https://doi.org/10.1038/s41419-017-0068-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-017-0068-6
This article is cited by
-
The phytochemical plumbagin: mechanism behind its “pleiotropic” nature and potential as an anticancer treatment
Archives of Toxicology (2024)
-
Molecular structure and bioactivities of 2, 4-Ditert butyl phenol extracted from Plumbago zeylanica, investigated using HPLC and NMR
Biomass Conversion and Biorefinery (2024)
-
Erianin Impedes the Proliferation and Metastatic Migration Through Suppression of STAT-3 Phosphorylation in Human Esophageal Cancer Cells
Applied Biochemistry and Biotechnology (2024)
-
Epigenetics and the role of nutraceuticals in health and disease
Environmental Science and Pollution Research (2023)
-
Pharmacogenomics of in vitro response of the NCI-60 cancer cell line panel to Indian natural products
BMC Cancer (2022)
