
Abstract
Any therapy that aims at eradicating a cancerous growth will have at its core a cell death-inducing component. Here we argue that paediatric oncology presents with its unique set of considerations and problems, which—while taking the lead from oncological research experiences obtained from the adult population—need to be clinically evaluated independently. This is particularly true when considering long-term side effects. Precision medicine offers a promising new approach in therapy, but given as a monotherapy and in a limited combination, as found in an apoptosis inducer/sensitiser combination, it will most likely lead to mutation escape of the target cell population and the emergence of resistance. However, using the increasing amount of the molecular data as the basis for a complex combination therapy combining several key components such as cell death-inducing agents, kinase inhibitors and BH3 mimetics, holds great promise.
Similar content being viewed by others


Facts
-
Paediatric, adolescent and adult cancers are different groups of diseases occurring in distinct environments.
-
Survival rates of patients afflicted with different paediatric malignancies have increased significantly over the last decades.
-
Long-term complications in childhood cancers are complex and need to be carefully evaluated.
-
More clinical trials are needed that focus exclusively on paediatric tumours.
-
Precision medicine has so far not been shown to be more effective than “doctor’s choice”.
Open questions
-
How best to collect, identify and analyse the relevant data on patients and disease?
-
How best to minimise and react to emerging long-term complications of novel therapeutic approaches?
-
How best to design a precision + approach that further increase treatment efficacy while reducing negative side effects?
Introduction
Modern cancer therapy is built on three pillars, surgery, radio- and chemotherapy, with a fourth—which actually precedes the latter two—currently gathering momentum: immunotherapy1,2,3. Surgery aside, all of these medical approaches are based on the induction of cell death, and they work. For example, the 10 years or more survival rate in the UK has more than doubled in the last 40 years and is now ~50%, although with large tumour-specific variations, ranging from 98% survival for prostate to 1% for pancreatic cancer4.
There are currently more than a dozen forms of cell death recognised. However, the preferred mode by which cell death is therapeutically induced remains a mechanism termed ‘apoptosis’, which also plays a crucial role in normal physiology and was first described more than 40 years ago1,5. Other forms of cell death that are of therapeutic interest are 'mitotic catastrophe', which is induced by some subgroups of chemotherapeutic reagents, 'autophagic cell death', which is closely related to 'autophagy', a cellular survival mechanism, and the unregulated cell death 'necrosis', which is immunogenic, i.e., causes inflammation and can elicit an immune response of potentially therapeutic value1,6,7.
The primary aim of cancer therapy has hardly changed since the first recorded cases of treating tumours of the breast 3500 years ago1, caedite eos - eradicating all tumour cells and thereby curing the patient. Only if the primary aim is unachievable, would we consider minimising tumour spread and rate of growth as to achieving maximal quality and quantity of life for the patients. However, both the means of reaching that goal and our understanding of what we are fighting have changed drastically in the last two and a half decades. From a dose-escalating arms race until the maximal tolerated doses (MTDs) are reached, and, thus, accepting severe and, in extremis, lethal side effects, the focus has shifted to a metronomic chemotherapy protocol, i.e., the continuous or more frequent administration of lower therapeutic doses8. In addition, the increasing acceptance of the cancer stem cell hypothesis, although at best still a large simplification of biological reality9, has led to a shift in our understanding of tumour organisation. We no longer view cancer as an egalitarian collective of malignant cells, but understand that a hierarchical order exists, leading to a complex ecosystem of supporting and competing populations, not all of which are necessarily comprised of malignant cells10.
The treatment of paediatric malignancies has been one of the few great success stories in oncology. The survival rates for a wide range of childhood cancers has drastically improved over the last 40 years (Fig. 1). Childhood leukaemia 10-year survival went from 27 to 81% in a period of 30 years, while during the same time span sympathetic nervous system tumour survival went from 15 to 60%. Even already good survival rates could be further improved, for example, going from 87 to 99% for retinoblastoma. Looking at 10 common childhood malignancies the average survival rate almost doubled (a 1.9-fold increase) from the early 1970s to the early 2000s (Fig. 1). There are, nevertheless, subgroups of paediatric malignancies that we cannot control as well, for example, 2–3% of childhood acute lymphoblastic leukaemia (ALL) are refractory at presentation, while 15% relapse reducing long-term survival to 40–50%, thus, making recurrent ALL one of the leading causes of cancer-related death in children and highlighting the need for novel therapeutic approaches11. Similarly, Ewing’s Sarcoma has a long-term survival rate of ~75%, which is reduced to <30% when the disease has metastasised, which is the case in 20–30% of all children at clinical presentation12, while other paediatric cancers generally present with a dismal diagnosis such as diffuse intrinsic pontine glioma, which is associated with an overall survival of just 8–10 months13. While a better fundamental understanding of these treatment-resistant malignancies is needed to improve treatment options, it is also the increasing awareness of the side effects of existing (and successful) therapeutic interventions that drives current research.

Shown is the percentage of patients suffering from ten common childhood cancers either in 1971–1975 or 2001–2005 who survived for 10 years or more, i.e., are considered cured. The data was obtained from Cancer Research UK (www.cancerresearchuk.org/health-professional/cancer-statistics) and based on National Cancer Intelligence Network. National Registry of Childhood Tumours Progress Report, 2012. Oxford: NRCT; 2013 and 10-year actuarial survival, children aged 0–14 years, Great Britain, the 1971–2005 data were provided by Charles Stiller at the National Registry of Childhood Tumours on request in 2013
A different group of diseases?
Ralph Waldo Emerson once put forward that “[a]ll diseases run into one, old age”. This probably holds true for no other illness more so than for cancer: our current understanding of this malignancy is that it is a stochastic disease, the accumulation and selection of unrepaired mutations over time, i.e., somatic mutation14. Generally, it is assumed that six mutations in key factors are necessary to initiate full blown cancer15,16,17, while, at least for some cancers, the need for only three driver mutations has been postulated18. However, cancer development is not necessarily a linear progression. Knudson, continuing from his seminal 1971 work suggests that two mutations are necessary to overcome the initial rate limiting steps19,20. While certain activated oncogenes can increase DNA double strand breaks and therefore the rate of mutations—the so-called mutator phenotype21—mathematical modelling suggests that selective pressure is sufficient to explain observed carcinogenesis14. Whether the increase of cancer with age (Fig. 2) can be merely reduced to cumulative acquisition of oncogenic mutations over time has been increasingly discussed and additional factors such as age-related changes of DNA methylation22 have also been suggested23. There seems little doubt that a general relationship exists between the total number of (stem) cells within a tissue, the number of cell divisions and the overall risk of developing cancer in that tissue24,25. Additional risk factors can significantly increase the risk of cancer by altering cell proliferation (i.e., increase the number of cell divisions), increasing the chances of mutation, or doing both, but they do not alter the underlying mechanisms at work. These risk factors can be both genetic, for example, Li-Fraumeni Syndrome, a germline mutation in the TP53 gene, which leads to an early onset of multiple types of cancer, or environmental like cigarette smoke, which drastically increases the risk of lung cancer, still one of the most common causes of cancer-related death26,27.

Shown are the cancer-related deaths (blue line), new diagnoses of cancer (red line) and the deaths per newly diagnosed cancer (grey line) for various age groups within the UK population.The scale to the left refers to the red and blue line, the scale on the right corresponds to the grey line. The area boxed red in the upper panel is blown up in the lower panel.Of note, new diagnoses exclude non-melanoma skin cancers as there are no sufficient records.Data were obtained from Cancer Research UK (www.cancerresearchuk.org/health-professional/cancer-statistics)
But what does it mean when a disease closely associated with ageing occurs in children? While around 5% of all childhood cancers are due to an inherited mutation, it was long assumed that most paediatric malignancies develop due to chance28. With more than 1 million individual molecular lesions per cell per day29 and a large enough population, chance would suggest that ‘bad luck’ might lead to cancer in some younger individuals. This is supported by the fact that cancer incidences are lowest in children (Fig. 2). However, it would also mean that—ignoring cancer associated with extrinsic risk factors, e.g. lung cancer and smoking—the cancer risk for individual cancers, like ALL, should be equal in childhood and total cancer. This is not the case (Table 1). Furthermore, we know that some cancers are linked to the different stages of development: for example, early puberty (the change between a child’s and an adolescent’s body) is a known risk factor for breast cancer, while at the other end of development, aromatase blockers only work in post-menopausal breast cancer patients30,31. Also, looking at two consecutive age groups, children (0–14 years of age) and adolescents (15–19 years), where the additional external factors are of minimal difference, one can observe stark variations in the age-adjusted incidences of various cancers (examples are shown in Table 2). Looking at mortality, one can further observe that paediatric cancer is actually a more lethal disease (in relation to incidences) than cancer in the working-age population (Fig. 2). If this were due to children being unable to cope with doses of radio- and chemotherapy applied to adults, it would be sufficient to show that paediatric cancer and adult cancer need different treatment approaches. However, although a recent ex vivo study has suggested that paediatric organs are more primed for apoptosis than those of adults32, this does not reflect clinical reality. Children younger than 3–5 years of age are generally not treated with radiotherapy, but can tolerate higher doses of chemotherapy than adults. Interestingly, both treatment modalities function similarly, by the induction of apoptosis.
An alternative model for oncogenesis is chromothripsis, the shattering and rearrangement of chromosomes, i.e., the replacement of the gradualism of acquiring individual mutations over time with a single catastrophic event33. While this hypothesis is not without controversy34,35, it might help to explain the development of childhood cancers. There is evidence that chromothripsis strongly contributes to Sonic-Hedgehog subtype of paediatric medulloblastoma development if a somatic TP53 mutation is present, while evidence for chromothripsis is only detected in 13% of all medulloblastoma36. For neuroblastoma estimates range from 0 to 10%, and for ALL cancer initiated by chromothripsis it has been shown again to be mainly associated with a particular predisposition, in this case, the Robertsonian chromosome37,38,39. The current data therefore suggest that chromothripsis is not a mechanism that distinguishes paediatric from adult cancers. Indeed, genetic analyses show that childhood tumours exhibit very few genetic alterations compared to adult cancers40. While this appears initially to fit to the model of accumulating mutations over time as cause of cancer, as an explanation it is insufficient, as cell divisions do not occur uniformly over time. Indeed, most, including telomere shortening as a surrogate readout for cell division, occur mainly prior to birth41. Experimentally, it was shown that maximal accumulation of mutations occurs during ontogeny, the development to maturity23, suggesting that the few additional events, which occur later in life are needed for tipping the scale. Taken together, the data suggest that the predisposition for paediatric cancers are caused early during development, often by de novo germline mutations42, and that tumours arise when a combination of a few particularly aggressive mutations occurs, i.e., fewer mutations are needed than in an adult tumour, and therefore the contribution of an individual mutation must be bigger, ceteris paribus. This has several therapeutic implications, as follows:
(A) Children where the mutations occur early during development, either in the form of germline mutations or affecting whole tissues, are genetically distinct from children who have not developed these mutations. This might affect systemic responses to therapy.
(B) That only few mutations have occurred might also point to the possibility that paediatric tumours are genetically more stable and therefore might not as easily develop resistances to therapy as adult tumours. For some paediatric malignancies, this might contribute to the increased therapeutic successes when compared to adult tumours.
(C) The reduced number of mutations unfortunately also means there is a reduced number of tumour-specific features, which can be targeted. However, it should also be pointed out that the focus on mutations tends to ignore the fact that dysregulation, such as overexpression and deregulated activity, are important factors contributing to the oncogenic phenotype3. It is particularly this latter area of interest where coordinated studies are lacking.
To summarise, in paediatric oncology, both the patient, as host for the disease, and the malignancy are distinct from adults and their tumours. Therefore, any therapeutic approach trying to improve the health of childhood cancer patients must take these specificities into account. Essentially, this has already been formulated more than a century ago by another German paediatrician: “Pediatrics does not deal with miniature men and women, with reduced doses and the same class of diseases in smaller bodies, but… it has its own independent range and horizon and gives as much to general medicine as it receives from it.”43.
One of the key reasons why cell death-inducing treatment has been a major success story in the treatment of many childhood cancers, particularly ALL40,44, is that a coordinated effort was made in the 1970s by the clinics to share information and experiences, helping to standardise and further develop treatment options3. This success further highlights the need to treat children and childhood cancers as distinct from adults. Yet, most clinical trials are not designed specifically for children and even thosen trials that look at children do not do so exclusively, but in most cases combine them with adolescents or all age groups (Fig. 3). Partially, this is understandable, as the incidences of cancer are much higher in the adult population. However, in terms of years of life lost and common causes of death, cancer can be very well viewed as a major problem affecting children (Table 3). Currently, there seem to be two fundamentally opposite philosophical approaches that need to be resolved: Protecting children from clinical trials (by extrapolating data from adult trials) vs. Protecting children with clinical trials (by finding the optimal therapeutic approach). The authors, both clinicians/researchers and parents/grandparents, are well aware that resolving this conflict is neither easy nor can it ever be fully satisfying or without considerable emotional burden.

.Interventional cancer-related clinical studies registered at ClinicalTrials.gov were categorised into four distinct groups, indicating whether the subjects were children, children and adolescents, adults or mixed (each study was allocated only one group). In a, absolute numbers are shown, thus indicating the development of the number of oncology studies over time, while in b, the distribution of the target age group of the oncology studies over time (in % of total) is depicted.Of note, the time period 1993–1998 precedes the establishment of the database and therefore only contains few appended entries, and studies without identifying starting date or with future starting date were excluded (cutoff date: 22 May 2017)
Long-term considerations
Caedite eos! Novit enim Dominus qui sunt eius already indicates that destroying a population is not possible without collateral damage. In adult cancer, we see this predominantly in the short-term complications of treatment, such as fatigue, diarrhoea, nausea, vomiting and hair loss, as well as neuropathy, anaemia and thrombocytopenia. In paediatric oncology, one also has to consider the long-term implications, as successful treatment hopefully means the patient has another three score and 10 years to live. In a recent review discussing radiation and brain tumours we identify the following complications, which might arise from using cell death-inducing treatment in children: A link has been postulated between the exposure of children to radiotherapy for leukaemia and the overall risk of developing glioblastoma45. Even computed tomography (CT) scans, needed to optimise targeting and monitoring therapy success, are associated with increased risk of brain cancer46. Childhood cancer survivors have increased risks for several malignancies later in life such as Acute Myeloid Leukaemia (AML), Non-Hodgkin lymphoma and colorectal cancer47,48. Overall, the risk of developing a second cancer is 3- to 6-fold higher in survivors of childhood cancer than in the general population49. Importantly, not only radiation, but also various forms of chemotherapy are known risk factors48,49.
Already in 1969, Bloom and co-workers demonstrated cognitive dementia among survivors of childhood medulloblastoma50, and while some earlier studies suggested that the tumour might be the causative agent for reduction in IQ, most large-scale studies agree that radiotherapy is an independent risk factor. Importantly, younger age of patient at treatment, radiation dose and time since treatment enhance the neurological deficits51. This might suggest that radiotherapy damages the pool of neurological stem cells, replenishment becoming more and more inefficient with increasing age, and thus neurological deficits increasing. This hypothesis is supported by the observation that conformal avoidance of the hippocampal neural stem cell compartment reduces neurological side effects52, and hyperfractionated radiotherapy, at doses of 1 Gy, can also prevent neurological decline, without the loss of therapeutic efficacy53. However, radiotherapy is also associated with additional side effects in childhood brain cancer survivors, such as hypothalamic-pituitory dysfunction and hearing loss54,55.
While radiation, by the nature of localised application, has rather specific side effects, often relating to the tumour-harbouring organs, chemotherapy is given systemically and therefore a wide range of side effects is to be expected. Indeed, here we find growth, intellectual development and entry into puberty being delayed, as well as reduced fertility and organ-specific damage, mainly to heart and lungs56. Importantly, the original damage caused by toxicity is often unrecognised or subclinical at the time and only during development, when the tissue is exposed to further stress, do the long-term side effects become apparent56.
In the recent decade, the large data sets on long-term survivors of several paediatric cancers became available, as a direct result of the successes in the 1970s. Previous data were mainly collected from predominantly benign tumours, like medulloblastoma, but now children also survive more aggressive tumours, partially due to more aggressive treatment. It is in the very nature of long-term side effects that our knowledge will always be lagging behind by a few decades and as long as we have to consider undetectable organ damage, we need to remain vigilant.
The aim of cell death-inducing therapies in paediatric oncology must be two-fold: In the large group of cancers, which we can successfully treat, we must minimise the severe, long-term side effects that occur, while for those tumour entities that remain therapy-resistant, we must improve our therapeutic choices. Precision medicine potentially addresses both those aims.
Precision medicine: chances and perspectives
Personalised medicine, i.e., the tailoring of the therapeutic intervention towards the individual cancer (and not actually the person), is—as such—not a new approach in paediatric oncology: already the early protocols were stratified according to risk groups, originally according to patient’s age and gender, but also cells of origin: for example, it has long been known that a T-cell ALL has a worse prognosis than a pre-B-cell ALL3. This was later augmented by cytogenetic analyses, looking at different chromosome content and composition; in ALL the presence of the so-called Philadelphia Chromosome (resulting from a t(9;22) translocation) calls for a particularly aggressive treatment. With the advent of high throughput and deep sequencing, we now have increasing data on the molecular composition of individual cell populations in the malignancies and the challenge has shifted to extracting therapeutically relevant information. This will mean both effective data mining techniques and the mapping of in silico information onto cellular processes. The conflict of how best to achieve this is reflected in the famous debate between Weinberg and Golub57,58. The INFORM study and the NCI-MATCH trial only identified targetable genetic abnormalities in around 10% of paediatric patients59,60. The equivalent studies looking at adult cancer have further shown that personalised medicine leads to no improvement over “doctor’s choice”61,62.
The current approach is implicitly linked to our understanding of how tumours arise, accumulating gain-of-function mutations (proto-oncogene to oncogene) and detrimental mutations (tumour suppressor inactivation) to acquire the hallmarks of cancer, which allow the tumours to grow independently of external control63,64. This is mainly mediated by kinases, predominantly tyrosine kinases (Fig. 4a). Identifying the mutations in the individual tumours indicates which pathways are abnormally activated and targeting these with pharmacological inhibitors should to a certain extent restore ‘normality’, e.g., reduce apoptosis resistance and make the tumour more amenable to cell death-inducing therapies. While the logic of this strategy is sound, there are several key factors, which might explain why the clinical application of this strategy has so far not been overly successful:
-
1.
The complexity of the signalling networks is far greater than usually admitted. While signalling pathways are generally described in linear terms with defined functions, in reality they are more akin to a circuit board with almost infinite intricacy64. The PI3K/Akt/mTOR cascade, for example, is highly interwoven with the MEK/Erk signalling pathway, interacting, both in an inhibitory, as well as an activating function, at multiple points and influencing cellular behaviour with respect to survival, cell cycle progression, metabolism, motility and DNA repair10,65. Our own research has highlighted the complexity of finding the optimal combination of inhibitor and cell death-inducing agent. Combing a dual inhibitor of PI3K and mTOR with the chemotherapeutic agent doxorubicin can have very distinct effects on neuroblastoma cells depending on the sequence of application, where inhibiting PI3K signalling after inducing apoptosis leads to a higher cell death rate than blocking it prior to the addition of doxorubicin, i.e., maximal inhibition of a survival cascade does not necessarily lead to maximal therapeutic benefits66. This was also confirmed by looking at the RIST therapy, a complex combination therapy that combines two chemotherapeutic agents with two pharmacological inhibitors in a compassionate use setting67. In an attempt to improve efficacy, When replacing rapamycin, an mTOR inhibitor, with GDC-0941, which blocks PI3K, i.e. acts more upstream in a glioblastoma cell culture system, this alternative RIST approach showed higher potency, but upon transferring it to a murine system all potency was lost. It appears that this was due to GDC-0941 reducing tumour vascularisation and, thus, minimising repeated delivery of the chemotherapeutic agents67. In essence, the inhibitor worked less well in combination because it worked better as a single agent. Translating these emerging pharmacokinetic and -dynamic interactions into the human system is going to be one of the major challenges of modern combination therapy.
-
2.
In personalised medicine the target is always selected by frequency, i.e. mutations found in the most common populations of cancer cells are detected and selected as targets. However, at initiation of treatment the tumour might contain 200,000 to 2,000,000 undetected cells that are distinct from the dominant subclone1. The most common population within a cancer is obviously the best adapted one (to the current microenvironment), but, of course, by initiating treatment we change the environment and create new selective forces. We have discussed the complex Darwinian mechanisms occurring in a tumour elsewhere1,10,65,68, but if one transfers Luria and Dellbrück’s seminal work from 1943 to the evolution of cancer, it would suggest that any precision therapy will lead to the emergence of a treatment-resistant disease69. There are computer models that devise a treatment strategy for bacteria so as to avoid antibiotic resistance70. On the basis of a similar sequential approach, we showed enhanced treatment efficacy in neuroblastoma66. We need to understand which tumour cells exist that might already be resistant to the treatment and which preferred routes of acquired resistance might be taken. This is also important as targeting a tumour-specific aberration can lead to emergence of rapid resistance via several possible routes (Fig. 4b).
In essence, a precision + approach is needed (Fig. 5), specific enough to minimise damage to the host, and broad enough also to target both minor and likely future cell populations. To quote Emerson again, “[w]hen you strike at a king, you must kill him”. This certainly holds true for the emperor of all maladies - once the decision is made to cure the cancer and not chronify it preventing the emergence of a recessive disease should be our primary aim1,10,65.
Fig. 5: Precision+Therapy 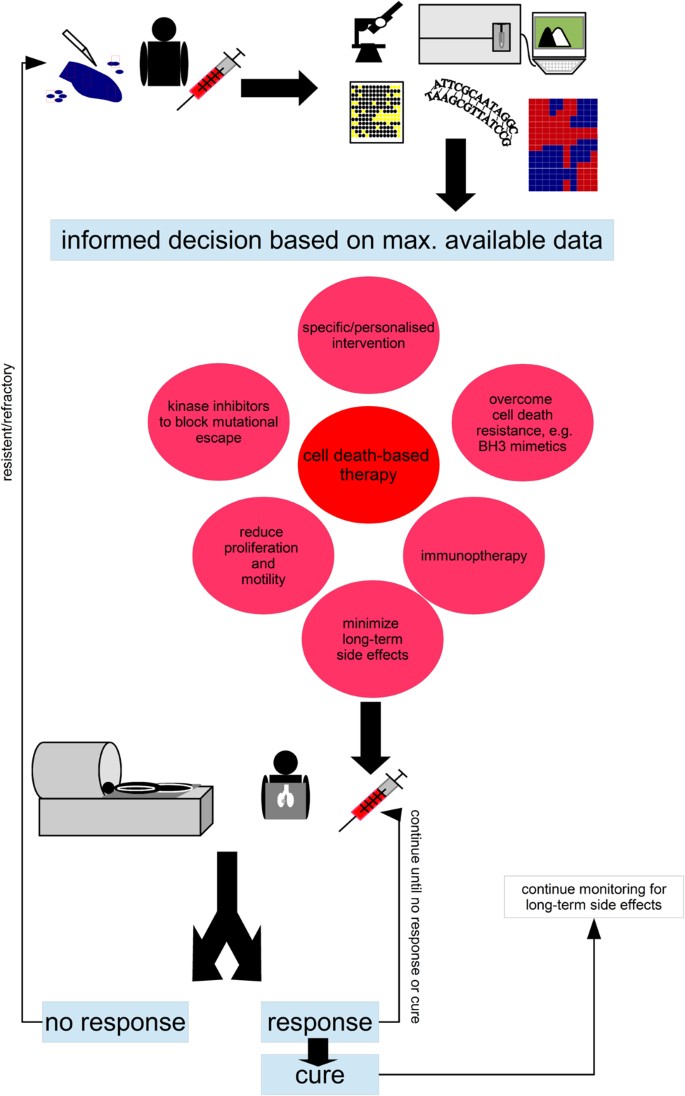
Collect as much data on the (epi)genetic make-up of the malignancy, its potential satellites and the patent. On the basis of somatic mutations, expression patterns, phosphorylation and levels of proteins as well as clinical knowledge devise a therapeutic intervention.Important questions that need answering: How much sampling is needed to get a comprehensive picture of the ecological niche and the various subclonal populations that make up the cancer? How best to identify the data that is of therapeutic relevance, tumours in adults can present >10,000 genetic alterations?93 The centre piece of any precision therapy will be cell death-based treatment, to reduce tumour burden, combine this pharmacological molecules that target tumour-specific alterations, drugs that overcome apoptosis resistance such as BH3 mimetics (as discussed in Fig. 6) and substances that minimise cell proliferation (no full blockage as many therapeutic interventions are dependent on proliferation) and block invasion, as to avoid ‘the cost of migration’ or ‘go or grow’: it has been shown that the lack of oxygen can induce genes that are responsible for motility, which, in turn, reduces proliferation73,74. Similarly, epithelial–to–mesenchymal transition which is most linked to invasion and several surface molecules, which also mediate adhesion should be prevented10,94. In addition, add additional inhibitors to prevent the emergence of resistance mechanisms as discussed in Fig. 4b. The patient’s immune system should also be trained to further sponge up cells which have escaped the initial therapeutic intervention, here modern immunotherapy holds several promises3. Finally, known side effects can also be countered at this stage already. For example, neurocognitive impairment, i.e., cognitive dementia, associated with radiotherapy can be reduced by neurological and pharmacological interventions95,96,97. Important questions that need answering: How predictable are the emerging resistance patterns, will blocking certain routes just provoke evolutionary escape via other mechanisms without greatly improving therapeutic outcome? Which sequence of administration should be chosen and how to further optimise this? The patients need to be closely monitored and any changes that suggest mutational escape of the tumour will need to fully restart the precision + therapeutic protocol. Even upon cure monitoring needs to be continued to identify long-term side effects, such as the emergence of secondary cancers, as early as possible. Important questions to ask: How to best monitor patients, as many radiation-based screening methods are themselves associated with increased cancer risks?46 Which screening methods should be employed for cured patients, as the long-term side effects have proven to be hard to detect and will be extremely varied for a multi-modular therapy. Importantly, the free and frank exchange between the paediatric oncology community which was the basis for the successes of the 1970s must be sustained throughout every step
-
3.
The choice of target also requires some consideration. Obviously, an early initiating mutation will be present in most, if not all, cancer cells, but there is no reason to assume that a genetic alteration necessary for oncogenesis is a good target to sensitise cells for treatment. Indeed, several tumour-specific alterations seem to have multiple contradictory functions in a tumour, for example in Ewing Sarcoma the fusion protein EWS/ETS and other proteins overexpressed in this tumour, such as DKK2, contribute negatively to proliferation, but enhance metastasis71,72. A similar relationship has also been observed in glioblastoma, where it is called ‘go or growth’ or ‘the cost of migration’: increased tumour invasion is linked to reduced proliferation73,74. In glioblastoma, we find a high incidence of MGMT promoter methylation, which is assumed to allow for higher tumour flexibility by increasing the rate of (unrepaired) mutations. However, while it also sensitises the tumour to treatment with temozolomide (a radical change in environment), promoter methylation strongly correlates with overall survival (independent of treatment) in paediatric glioblastoma75. Therefore, treating glioblastoma with temozolomide, while improving survival rates by a few months76, also selects for tumour cells with worse prognosis. Therefore, while mutated gene products or fusion proteins are an obvious and highly specific target, they could have several, competing functions that do not make them ideal targets. Using components within combination therapy that are closely and (as far as we know) fairly specifically associated with apoptosis resistance might be a promising alternative such as targeting the Bcl-2 family or IAPs (Fig. 6).
Fig. 6: The roles of the Bcl-2 family and IAPs in apoptosis regulation 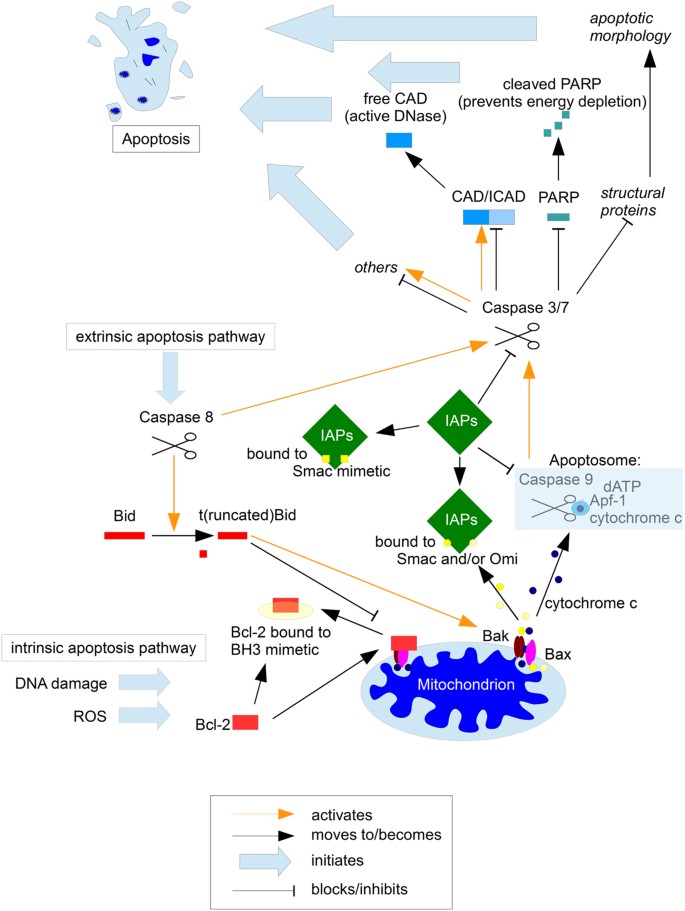
Apoptosis is a highly complex and tightly regulated cell death program10. In recent years two classes of proteins have come increasingly into the focus of potential therapeutic interventions, the Bcl-2 family and IAPs. The Bcl-2 family (red) comprises proteins that contain at least one Bcl-2 homology (BH) domain and fall into three subgroups: the BH3-only, the pro-survival and the proapoptotic proteins. Cellular stress stimulates BH3-only proteins which then alter the balance between pro- and anti-apoptotic Bcl-2 family proteins leading to BAX and BAK-forming pores and thus initiating mitochondrial apoptosis65. The most promising approach with regards to this protein family are BH3 mimetics that have a function similar to that of BH3-only proteins. While BH3 mimetics on their own can be sufficient to induce apoptosis, generally some form of additional stress is needed for optimal efficacy65. Interestingly, in a preclinical setting, successes with BH3 mimetics could be achieved particularly in the high-risk groups of ALL and neuroblastoma3. It is tempting to speculate that within particularly aggressive tumours, be it due to the high proliferation rate or the fact that invasive cells more frequently encounter a hostile environment, the intrinsic cellular stress is already at a higher level, so that less extrinsic force is needs to be combined with the mimetics. While the Bcl-2 family contribute to apoptosis resistance directly prior to/at the mitochondria, there is a second class of proteins (green) that mediate cell death resistance at the stages following mitochondrial involvement. Of the eight mammalian Inhibitor of Apoptosis Proteins (IAPs), three are ubiquitously expressed, XIAP, c-IAP 1 and 265. XIAP binds and thereby inhibits Caspases-3, -7 and -9, while the role of the c-IAPs seems slightly more complex; while there is considerable functional overlap with XIAP, they also have a crucial role in RIP1-dependent necrosis or necroptosis98,99. Therapeutically, mimetics of the naturally occurring IAP inhibitor, Smac/DIABOLO have shown great promise. Unlike BH3 mimetics which can either sensitise for, or induce cell death in a tumour-dependent manner65, Smac mimetics are generally considered as sensitisers and not inducers of apoptosis99. Their potency was first demonstrated using an orthotopic human glioblastoma model, where the combination of Smac mimetic and death ligand Apo2-L/TRAIL de facto cured the mice, while the individual substances had no or little effect on overall survival100



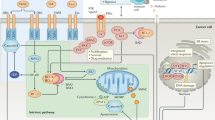
a Aberrant activation of tyrosine kinases as a mechanism for malignant transformation. Cancer cells are defined by overactive signalling cascades, often mediated by tyrosine (tyr) kinases. Common therapeutic strategies are either blocking of the tyr kinase receptor by inhibiting antibody/pharmacological inhibitor (which does not work for ligand-independent signals and has reduced potency if the target is overexpressed), or utilising pharmacological inhibitors that block kinase activity (dependent/independent of mutational status). Modified from a previous version3. b Three common escape mechanisms from kinase inhibition. The three common mechanism by which tumour cells escape the blocking effect of small molecule inhibitors are (from left to right): (I) Upregulation of the target structure, for example the tyr kinase receptor. This can occur independently of any genetic alterations and is potentially reversible, i.e., after discontinuation of treatment the tumour can become re-sensitised (as seen, for example, in AML89). (II) Mutation of the target structure, so that the inhibitor can no longer bind. This is frequently observed during monotherapy, where only one new selective pressure is applied. For example, initial reports on the novel tyrosine kinase inhibitor imatinib indicated that treatment restored the life expectancy of chronic myelogenous leukaemia patients to that of the general population90. While imatinib is a potent drug that greatly improves patients’ quantity and quality of life, treatment-resistant cancer cells frequently emerged and prevented a complete cure91. (III) Due to the interconnectedness of signalling cascades, inhibition of specific survival molecules can lead to a re-routing or re-wiring of the signals via alternative intermediates, which can result in the formulation of the Nile Distributary hypothesis, that postulates that some cancers are particularly aggressive as they use “multiple cross-covering growth enhancing pathways to grow and avoid cytotoxic interventions”92
An enhanced focus on these two groups might also reduce a risk associated with the use of kinase inhibitors. Many of these kinases associated with cancer also play a critical role in development and therefore will cause a distinct set of side effects in children compared to adults77. Currently, only a very limited number of targeted drugs are approved for the treatment. As this will have to change in future more work is needed to understand and counter these potential side effects.
It had long been hoped that in addition to the common cell death-inducing therapies, the induction of apoptosis via triggering death receptors, such as Apo1/CD95 and Apo2/TRAIL-R, might prove a feasible approach with less severe side effects78. However, two findings have rather impeded the development of death rector triggering as treatment: (1) the discovery of pro-tumourigenic properties of these receptors and their ligands79,80,81 and (2) the fact that paediatric cancers, such as neuroblastoma and primitive neuroectodermal brain tumours, often exhibit resistance to death receptor-triggered apoptosis via downregulation of Caspase-882,83. Importantly, the clinical evaluation of Apo2-L/TRAIL has also been disappointing, while for CD95 an antagonist, APG101, has been shown to be clinically beneficial for glioblastoma patients84,85. Death receptors remain an interesting potential target for cancer therapy; however, there is still considerable work needed to successfully integrate their triggering into a complex therapeutic regiment84.
Conclusions
If we want further to improve the cure rate of patients with paediatric malignancies several factors have to be considered. Neither host/patient nor disease are identical to post-pubescent individuals and their disease. While adult oncology will still be needed to inform and initiate novel therapeutic approaches, we will need to further our understanding of the nature of paediatric cancers and how they react to treatment.
Essentially, any therapeutic approach that aims to cure the patient will need to be built around cell death induction. However, in paediatrics, we have to be much more aware of the long-term consequences, be they developmental or the risk of treatment induced secondary malignancies. The introduction of metronomic chemotherapy protocols a decade ago, implementing the continuous or more frequent administration of lower therapeutic doses, has already lowered the risks of acute side effects. Moving away from monotherapies to increasingly complex combination therapies, an approach that was also pioneered in paediatric oncology before it was taken up for the treatment of adults, will further prevent mutational escape. We already have a considerable arsenal of kinase inhibitors for use in paediatric oncology (Table 4), and combining several of these into a cocktail with cell death inducing therapy and BH3 mimetics will open new avenues of intervention. Here, the challenge lies in understanding the complex interactions of signalling networks in malignant cells to allow the use of the optimal sequence of the optimal combination, personalised for both patient and disease.
Another treatment approach which should be combined with this complex combination therapy is immunotherapy, using both the patient’s innate immune system and adaptive immunity to kill cancer cells. Here several forms of medication are also already available (Table 4).
It has been estimated that in 1980 0.1% of all 20-year-old US Americans was a childhood cancer survivor86, yet paediatric malignancies are still a leading cause of death among children. We, therefore, need a coordinated effort both to understand long-term consequences of our treatments and to improve therapeutic efficacy. This information will have to come from a frank and free exchange of clinical information, i.e., the continuation of a successful approach initiated in the 1970s, and clinical trials. Here, allowing for the lower overall number of cases, we must rethink our trial designs and incorporate rolling six designs and continuous recruitment87,88.
References
Nonnenmacher, L. et al. Cell death induction in cancer therapy: past, present, and future. Crit. Rev. Oncog. 21, 253–267 (2016).
Coley, W. B. II Contribution to the knowledge of sarcoma. Ann. Surg. 14, 199–220 (1891).
Debatin K. M., Burdach S. Präzisionsmedizin in der pädiatrischen Onkologie, Chancen und Perspektiven. Schwerpunkt2017: Therapeutische/Medizinische Fortschritte. Berufsverband der Kinder- und Jugendärzte e.V. (BVKJ).http://www.bvkj-shop.de/infomaterial/broschueren/schwerpunktbroschueren.html
Quaresma, M., Coleman, M. P. & Rachet, B. 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971-2011: a population-based study. Lancet 385, 1206–1218 (2015).
Kerr, J. F. R., Wyllie, A. H. & Currie, A. R. Apoptosis: A Basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Marino, G., Niso-Santano, M., Baehrecke, E. H. & Kroemer, G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15, 81–94 (2014).
Melcher, A., Gough, M., Todryk, S. & Vile, R. Apoptosis or necrosis for tumor immunotherapy: what’s in a name? J Mol. Med. 77, 824–833 (1999).
Romiti, A. et al. Metronomic chemotherapy for cancer treatment: a decade of clinical studies. Cancer Chemother. Pharmacol. 72, 13–33 (2013).
Schneider, M. et al. A paired comparison between glioblastoma “stem cells” and differentiated cells. Int. J. Cancer 138, 1709–1718 (2016).
Westhoff, M. A., Bruhl, O., Nonnenmacher, L., Karpel-Massler, G. & Debatin, K. M. Killing me softly-future challenges in apoptosis research. Int. J. Mol. Sci. 15, 3746–3767 (2014).
Ceppi, F. et al. Improvement of the outcome of relapsed or refractory acute lymphoblastic leukemia in children using a risk-based treatment strategy. PLoS ONE 11, e0160310 (2016).
Hendershot, E. Treatment approaches for metastatic ewing’s sarcoma: A review of the literature. J. Pediatr. Oncol. Nurs. 22, 339–352 (2005).
Jansen, M. H. A., van Vuurden, D. G., Vandertop, W. P. & Kaspers, G. J. L. Diffuse intrinsic pontine gliomas: A systematic update on clinical trials and biology. Cancer Treat. Rev. 38, 27–35 (2010).
Tomlinson, I. P., Novelli, M. R. & Bodmer, W. F. The mutation rate and cancer. Proc. Natl Acad. Sci. USA 93, 14800–14803 (1996).
Armitage, P. & Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 8, 1–12 (1954).
Armitage, P. & Doll, R. A two-stage theory of carcinogenesis in relation to the age distribution of human cancer. Br. J. Cancer 11, 161–169 (1957).
Weinberg, R. A. How cancer arises. Sci. Am. 275, 62–70 (1996).
Tomasetti, C., Marchionni, L., Nowak, M. A., Parmigiani, G. & Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl Acad. Sci. USA 112, 118–123 (2015).
Knudson, A. G. Jr. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA 68, 820–823 (1971).
Knudson, A. G. Hereditary cancer: two hits revisited. J. Cancer Res. Clin. Oncol. 122, 135–140 (1996).
Hicks, W. M., Kim, M. & Haber, J. E. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329, 82–85 (2010).
Xu, Z. & Taylor, J. A. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis 35, 356–364 (2014).
DeGregori, J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene 32, 1869–1875 (2013).
Tomasetti, C. & Vogelstein, B. Cancer risk: role of environment-response. Science 347, 729–731 (2015).
Wu, S., Powers, S., Zhu, W. & Hannun, Y. A. Substantial contribution of extrinsic risk factors to cancer development. Nature 529, 43–47 (2016).
Hisada, M., Garber, J. E., Fung, C. Y., Fraumeni, J. F. Jr. & Li, F. P. Multiple primary cancers in families with Li-Fraumeni syndrome. J. Natl Cancer Inst. 90, 606–611 (1998).
Schwartz, A. G. & Cote, M. L. Epidemiology of lung cancer. Adv. Exp. Med. Biol. 893, 21–41 (2016).
Moore, S. W. Developmental genes and cancer in children. Pediatr. Blood Cancer 52, 755–760 (2009).
Lodish, H. et al. Molecular Biology of the Cell. 5th edn, (WH Freeman, New York, USA, 2004).
Apter D. A. N,Vihko R, Early menarche, a risk factor for breast cancer, indicates early onset of ovulatory cycles*. J. Clin. Endocrinol. Metab.57:82–86 (1983).
Chumsri, S., Howes, T., Bao, T., Sabnis, G. & Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J. Steroid Biochem. Mol. Biol. 125, 13–22 (2011).
Sarosiek, K. A. et al. Developmental regulation of mitochondrial apoptosis by c-myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell 31, 142–156 (2017).
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011).
Righolt, C. & Mai, S. Shattered and stitched chromosomes—chromothripsis and chromoanasynthesis—manifestations of a new chromosome crisis? Genes Chromosomes Cancer 51, 975–981 (2012).
Solorzano, C. O. S., Pascual-Montano, A., de Diego, A. S., Martinez-A, C. & van Wely, K. H. M. Chromothripsis: breakage-fusion-bridge over and over again. Cell Cycle 12, 2016–2023 (2013).
Rausch, T. et al. Genome sequencing of pediatric medulloblastoma links catastrophic dna rearrangements with tp53 mutations. Cell 148, 59–71 (2012).
Sausen, M. et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 45, 12–17 (2013).
Molenaar, J. J. et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593 (2012).
Li, Y. et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508, 98–102 (2014).
Mody, R. J., Prensner, J. R., Everett, J., Parsons, D. W. & Chinnaiyan, A. M. Precision medicine in pediatric oncology: Lessons learned and next steps. Pediatr. Blood Cancer 64, 3 (2017).
Weng, N. P., Hathcock, K. S. & Hodes, R. J. Regulation of telomere length and telomerase in T and B cells: a mechanism for maintaining replicative potential. Immunity 9, 151–157 (1998).
Zhang, J. et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346 (2015).
Jacobi, A., Robinso, W. J. (ed). Collected Essays Addresses, Scientific Papers and Miscellaneous Writings,. (Critic and Guide co., New York, USA, 1909).
Hunger, S. P. & Mullighan, C. G. Redefining all classification: toward detecting high-risk all and implementing precision medicine. Blood 125, 3977–3987 (2015).
Neglia, J. P. et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the childhood cancer survivor study. J. Natl Cancer Inst. 98, 1528–1537 (2006).
Pearce MS et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet. 380, 499–505 (2012).
Rihani, R., Bazzeh, F., Faqih, N. & Sultan, I. Secondary hematopoietic malignancies in survivors of childhood cancer. Cancer 116, 4385–4394 (2010).
Henderson, T. O., Oeffinger, K. C. & Whitton, J. et al. Secondary gastrointestinal cancer in childhood cancer survivors: A cohort study. Ann. Intern. Med. 156, 757–766 (2012).
Bhatia, S. & Sklar, C. Second cancers in survivors of childhood cancer. Nat. Rev. Cancer 2, 124–132 (2002).
Bloom, H. J., Wallace, E. N. & Henk, J. M. The treatment and prognosis of medulloblastoma in children. A study of 82 verified cases. Am. J. Roentgenol. Radium Ther. Nucl. Med. 105, 43–62 (1969).
Palmer, S. L. Neurodevelopmental impact on children treated for medulloblastoma: a review and proposed conceptual model. Dev. Disabil. Res. Rev. 14, 203–210 (2008).
Gondi, V. et al. Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): A phase ii multi-institutional trial. J. Clin. Oncol. 32, 3810–3816 (2014).
Carrie, C. et al. Conformal radiotherapy, reduced boost volume, hyperfractionated radiotherapy, and online quality control in standard-risk medulloblastoma without chemotherapy: Results of the French M-SFOP 98 protocol. Int. J. Radiat. Oncol. Biol. .Phys. 63, 711–716 (2005).
Clement, S. C. et al. Prevalence and risk factors of early endocrine disorders in childhood brain tumor survivors: A Nationwide, multicenter study. J. Clin. Oncol. 34, 4362–4370 (2016).
Merchant, T. E., Conklin, H. M., Wu, S., Lustig, R. H. & Xiong, X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J. Clin. Oncol. 27, 3691–3697 (2009).
Schwartz, C. L. Long-term survivors of childhood cancer: the late effects of therapy. Oncologist 4, 45–54 (1999).
Weinberg, R. Point: Hypotheses first. Nature 464, 678–678 (2010).
Golub, T. Counterpoint: Data first. Nature 464, 679–679 (2010).
Worst, B. C. et al. Next-generation personalised medicine for high-risk paediatric cancer patients - The INFORM pilot study. Eur. J. Cancer 65, 91–101 (2016).
Downing, J. R. et al. The pediatric cancer genome project. Nat. Genet. 44, 619–622 (2012).
Tannock, I. F. & Hickman, J. A. Limits to personalized cancer medicine. N. Engl. J. Med. 375, 1289–1294 (2016).
Le Tourneau, C. et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 16, 1324–1334 (2015).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Westhoff, M. A., Marschall, N. & Debatin, K. M. Novel approaches to apoptosis-inducing therapies. Adv. Exp. Med. Biol. 930, 173–204 (2016).
Westhoff, M. A. et al. Sequential dosing in chemosensitization: targeting the PI3K/Akt/mTOR pathway in neuroblastoma. PLoS ONE 8, e83128 (2013).
Nonnenmacher, L. et al. RIST: A potent new combination therapy for glioblastoma. Int. J. Cancer 136, E173–E187 (2015).
Stroh, S., Debatin, K. M. & Westhoff, M. A. Darwinian principles in Cancer therapy. Eur. Oncol. Haematol. 10, 116–120 (2014).
Luria, S. E. & Delbruck, M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28, 491–511 (1943).
Nichol, D. et al. Steering evolution with sequential therapy to prevent the emergence of bacterial antibiotic resistance. PLOS Comput. Biol. 11, e1004493 (2015).
Franzetti G. A. et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 36, 3505–3514 (2017).
Hauer, K. et al. DKK2 mediates osteolysis, invasiveness, and metastatic spread in Ewing sarcoma. Cancer Res. 73, 967–977 (2013).
Giese, A. Glioma invasion--pattern of dissemination by mechanisms of invasion and surgical intervention, pattern of gene expression and its regulatory control by tumorsuppressor p53 and proto-oncogene ETS-1. Acta Neurochir. Suppl. 88, 153–162 (2003).
Hatzikirou, H., Basanta, D., Simon, M., Schaller, K. & Deutsch, A. Go or Grow: the key to the emergence of invasion in tumour progression? Math. Med. Biol. 29, 49–65 (2012).
Donson, A. M., Addo-Yobo, S. O., Handler, M. H., Gore, L. & Foreman, N. K. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatric Blood Cancer 48, 403–407 (2007).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Gore, L., DeGregori, J. & Porter, C. C. Targeting developmental pathways in children with cancer: what price success? Lancet Oncol. 14, e70–e78 (2013).
Mahmood, Z. & Shukla, Y. Death receptors: Targets for cancer therapy. Exp. Cell Res. 316, 887–899 (2010).
Barnhart, B. C. et al. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. The EMBO J. 23, 3175–3185 (2004).
Ishimura, N., Isomoto, H., Bronk, S. F. & Gores, G. J. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am. J. Physiol.– Gastrointest. Liver Physiol. 290, G129–G136 (2006).
Chen, L. et al. CD95 promotes tumour growth. Nature 465, 492–496 (2010).
Eggert, A. et al. Resistance to TRAIL-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Med. Pediatr. Oncol. 35, 603–607 (2000).
Grotzer, M. A. et al. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene 19, 4604–4610 (2000).
von Karstedt, S., Montinaro, A. & Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 17, 352–366 (2017).
Martin-Villalba, A., Llorens-Bobadilla, E. & Wollny, D. CD95 in cancer: tool or target? Trends Mol. Med. 19, 329–335 (2013).
Meadows A. T., Krejmas N. L., Belasco J. B. The medical cost of cure: sequelae in survivors of childhood cancer. In Proc. Status of the Curability of Childhood Cancers: the University of Texas System Cancer Center, M.D. Anderson Hospital and Tumor Institute 24th Annual Clinical Conference on Cancer (ed. van Eys J., Sullivan M. P) 263–276 (Raven Press: New York, USA, 1980).
Hartford, C., Volchenboum, S. L. & Cohn, S. L. 3+3 ≠ (Rolling) 6. J. Clin. Oncol. 26, 170–171 (2008).
Moreno L. et al. Early phase clinical trials of anticancer agents in children and adolescents—an ITCC perspective. Nat. Rev. Clin. Oncol. 14, 497-507 (2017).
Herrmann M. D. et al. Transitory dasatinib-resistant states in KITmut t(8;21) acute myeloid leukemia cells correlate with altered KIT expression. Exp. Hematol. 42, 90-100 (2013).
Gambacorti-Passerini, C. et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J. Natl Cancer Inst. 103, 553–561 (2011).
An, X. et al. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk. Res. 34, 1255–1268 (2010).
Kast, R. E., Karpel-Massler, G. & Halatsch, M. E. CUSP9* treatment protocol for recurrent glioblastoma: aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 5, 8052–8082 (2014).
Tomlinson, I., Sasieni, P. & Bodmer, W. How many mutations in a cancer? Am. J. Pathol. 160, 755–758 (2002).
Chaffer, C. L. & Weinberg, R. A. A perspective on cancer cell metastasis. Science 331, 1559–1564 (2011).
Padovani, L., Andre, N., Constine, L. S. & Muracciole, X. Neurocognitive function after radiotherapy for paediatric brain tumours. Nat. Rev. Neurol. 8, 578–588 (2012).
Brown, P. D. et al. Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro. Oncol. 15, 1429–1437 (2013).
Rapp, S. R. et al. Donepezil for irradiated brain tumor survivors: a phase iii randomized placebo-controlled clinical trial. J. Clin. Oncol. 33, 1653–1659 (2015).
Verhagen, A. M., Coulson, E. J. & Vaux, D. L. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol. 2, REVIEWS3009 (2001).
Berthelet, J. & Dubrez, L. Regulation ofApoptosis by Inhibitors of Apoptosis (IAPs). Cells 2, 163–187 (2013).
Fulda, S., Wick, W., Weller, M. & Debatin, K. M. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 8, 808–815 (2002).
Acknowledgements
The authors thank S. E. Barry for critically reading earlier versions of the manuscript. We are grateful to Professor Erdlenbruch for sharing his interest in Abraham Jacobi. K-M.D. would like to express his gratitude to B. Welz and S. Glatzel for their many years of outstanding secretarial assistance. M-A.W. and K.M.D. would also like to acknowledge the support from the Förderkreis für tumour- und leukämiekranke Kinder Ulm e.V.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Westhoff, MA., Marschall, N., Grunert, M. et al. Cell death-based treatment of childhood cancer. Cell Death Dis 9, 116 (2018). https://doi.org/10.1038/s41419-017-0062-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-017-0062-z
This article is cited by
-
Combinatorial targeting of MTHFD2 and PAICS in purine synthesis as a novel therapeutic strategy
Cell Death & Disease (2019)
-
Compare and contrast: pediatric cancer versus adult malignancies
Cancer and Metastasis Reviews (2019)
-
Precision medicine in pediatric oncology
Molecular and Cellular Pediatrics (2018)
-
Cell death-based treatment of various diseases: a fifty-year journey
Cell Death & Disease (2018)
