
Abstract
Over the past 15 years, there has been great interest in the potential to repurpose the diabetes drug, metformin, as a cancer treatment. However, despite considerable efforts being made to investigate its efficacy in a number of large randomised clinical trials in different tumour types, results have been disappointing to date. This perspective article summarises how interest initially developed in the oncological potential of metformin and the diverse clinical programme of work to date including our contribution to establishing the intra-tumoral pharmacodynamic effects of metformin in the clinic. We also discuss the lessons that can be learnt from this experience and whether a further clinical investigation of metformin in cancer is warranted.
Similar content being viewed by others

Introduction
A drug is described as being ‘repurposed’ when it exhibits clinical benefit for the treatment of cancer patients, despite being initially developed for an unrelated indication. As drug development costs rise for new entities, there is a growing attraction in looking to off-patent medicines that have established safety and pharmacokinetic profiles potentially reducing the time to entry into the oncology clinic. Indeed, a recent estimate suggested that the median cost to develop a single cancer drug is $648 million [1], although other analyses have suggested far higher sums [2]. This outlay directly results in the great expense of drug therapy for patients and health systems, often more than $100,000 per year for new cancer drugs, and contributes to inequality of access to cancer treatment. For rare cancers, where low commercial returns may be prohibitive for drug development, repurposing may be of particular value.
Metformin is the most widely prescribed medicine for type 2 diabetes worldwide and on the World Health Organisation’s list of essential medicines. A series of epidemiological studies which suggested that metformin may reduce cancer incidence in diabetic populations sparked great interest in its potential as a cancer treatment. However, there remains some debate as to its pharmacodynamic effects in tumours and recent randomised trials have not clearly demonstrated clinical benefit for any cancer indication to date. In this perspective article, we present a summary of the history of preclinical and clinical studies that informed the repurposing of metformin, discuss how this programme of work could have been better focused and coordinated and lastly describe oncological indications where there remains a strong rationale for investigation.
Metformin’s mechanism of action
As has typically been the case drug repurposing programmes, the interest in metformin was serendipitous. Two decades ago, researchers trying to understand the metabolic effects of metformin in diabetic patients discovered that it inhibits Complex 1 of the mitochondrial respiratory transport chain, the consequence of which was the activation of the cellular regulator of energy homoeostasis, AMP-activated protein kinase (AMPK) [3, 4]. AMPK is known to be a tumour suppressor that regulates a number of downstream anabolic pathways critical to tumour cell proliferation. On this basis, a pilot epidemiological case–control study was carried out and the analysis suggested that patients with Type 2 Diabetes Mellitus on metformin were less likely to develop cancer compared to patients on other diabetes drugs [5]. This finding led to a host of preclinical studies, which suggested that under laboratory conditions and with doses substantially greater than peak plasma level in patients, metformin possessed a number of anti-cancer properties including synergy with cytotoxic chemotherapy and radiotherapy.
Despite great effort and multiple published preclinical studies, the actual mechanism of action of metformin in tumour cells remains a topic of debate. As described above it is clear, at least when cells are treated with high doses, that metformin inhibits Complex-1 activity and cellular respiration. This was demonstrated in a series of elegant experiments in which metformin-resistant Saccharomyces cerevisiae NADH dehydrogenase NDI1 was overexpressed. In the same study the administration of metformin to mice inhibited the growth of control xenografts but not those expressing NDI1 [6]. In models, the consequences of inhibiting Complex 1 and hence the tricarboxylic acid cycle has been shown to check the funnelling of carbon from glucose to the anabolic building blocks needed for cell proliferation [7]. However, in patients, it remains unclear as to the degree to which the disruption of carbon metabolism or induction of energy stress and subsequent AMPK activation might be most critical to any antiproliferative effects. The further preclinical investigation has suggested a host of pleiotropic effects of metformin in cancer cells but it seems likely that many if not all of these are downstream consequences of metformin inhibiting complex 1 and subsequent AMPK activation, rather than the drug engaging multiple targets. For example, metformin has been shown to inhibit AKT/mTOR signalling and suppress fatty acid synthesis in an AMPK-dependent manner [8,9,10]. Metformin-induced AMPK activation has also been shown to reduce cancer cell proliferation through several other mechanisms, including activation of cMYC, HIF-1α and DICER1 [11].
However, an alternative hypothesis focussed on metformin’s effects on systemic ‘host’ metabolism has been proposed. Within the liver, activation of AMPK has been shown to reduce gluconeogenic gene expression in hepatocytes [12], increase insulin receptor activity and enhance translocation of glucose transporters [13]. It has also been proposed that biguanides inhibit glucagon signalling in the liver in an AMPK-independent manner, possibly by increasing AMP levels secondary to inhibition of Complex 1. AMP then by binding to adenylate cyclase downregulates cAMP-PKA activity suppressing gluconeogenesis [14]. Activation of the insulin receptor promotes downstream PI3K-AKT-mtor signalling and growth in tumour models [15] and increased insulin levels are associated with higher cancer incidence and mortality [16]. Hence, by reducing circulating insulin and glucose levels it is postulated that metformin may reduce insulin-mediated tumorigenesis and cancer progression, perhaps most relevant to patients with metabolic syndrome or type 2 diabetes although metformin has been shown to reduce insulin levels in cancer patients without these conditions [17].
Pharmacodynamic clinical studies
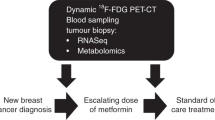
In the clinic, metformin’s anti-cancer effects were initially evaluated in several small pharmacodynamic clinical trials. These typically used ‘window of opportunity’ designs often prior to surgery and compared assays using the diagnostic and surgical tumour sample with a course of metformin in between to assess the drug’s effects on cancer biology. The endpoints and findings of these studies are described in Table 1 but in summary almost all of these early studies designated the well-validated marker of proliferation, Ki67, as the primary endpoint. Other immunohistochemical markers were often assayed, in particular for immunohistochemical markers of apoptosis, AMPK and mTOR pathway activation, however detailed characterisation of metformin’s effects on tumour biology was not evaluated. To address this, we undertook a radiogenomic ‘window-of-opportunity’ study in 40 non-diabetic patients with primary breast cancer, linking FDG–PET tumour uptake to tumour transcriptomic and metabolomic profiling. Here, we observed the upregulation of several transcriptomic pathways linked to mitochondrial metabolism and change in the levels of a number of mitochondrial metabolites suggesting that metformin disrupted mitochondrial metabolism at clinical dosing. A reactive increase in mitochondrial oxidative phosphorylation gene transcription linked to metformin resistance and two distinct metabolic responses in breast cancer were observed. Furthermore, we showed that metformin increases 18-FDG flux in primary breast cancer concomitant to the increased expression of multiple genes regulating glycolysis, glucose transport and glutamine metabolism. This was consistent with breast tumours upregulating well-described mitochondrial metabolic resistance pathways in response to metformin adding weight to the potential of previously proposed strategies to target these pathways, in combination with metformin [17, 18].
Efficacy studies
Results have now been presented from a number of randomised trials in different settings which in most cases have not demonstrated clinical benefit for metformin as a cancer treatment. These studies have assessed the combination of metformin with chemotherapy, endocrine and other targeted therapy in a number of different tumour types as summarised in Table 2. Most notably, the MA.32 study was a Phase III randomised trial that recruited over 3600 patients with high-risk operable breast cancer randomised to 850 mg metformin or placebo for 5 years. The investigators concluded that the addition of adjuvant metformin did not lead to an improvement in disease-free survival for either oestrogen receptor-positive or negative breast cancer [19]. An exploratory analysis did suggest that there might be some benefit in patients with HER2-positive disease and who genotyped for the C allele of the rs11212617 single-nucleotide polymorphism although the authors concluded that this would need to be confirmed with further prospective study [19, 20]. Tumour hypoxia is strongly linked to radiotherapy resistance and metformin has been shown to improve tumour oxygenation and radiotherapy response in xenograft models [21]. Hence, the combination of metformin and chemoradiotherapy has been investigated in patients with non-small lung cancer in two randomised trials but again with no evidence of benefit [22, 23]. A handful of studies have had encouraging results but with insufficient sample sizes to be firmly conclusive [24,25,26,27,28].
Can lessons be learnt from the metformin experience?
When the MA.32 study commenced recruitment in 2010 there was very limited information regarding the pharmacodynamic effects of metformin in breast cancer at therapeutic doses from the clinic. This impacted on the ability to develop rationale clinical trial designs that took into account markers of selection, resistance, treatment combination and dosing. As has been frequently been demonstrated in drug development without appropriate patient selection clinical benefit may be masked.
Initially, there was little effort to establish the tolerability and possible advantage of higher dose levels of metformin in the context of treatment for cancer where a greater risk/side effect profile might be acceptable. Efforts have now been made to evaluate different dose levels for metformin and its biguanide cousin phenformin in various therapeutic combinations [29,30,31]. However, to our knowledge, a well-designed dose escalation study of metformin with detailed tumour pharmacodynamic assessment is still awaited.
An example of a well-structured programme of work that could be taken as template to repurpose an anti-mitochondrial agent for cancer therapy is the ongoing evaluation of the anti-parasitic drug atorvaquone as a radio-sensitiser. A decade ago, a group of investigators carried out a high throughput screen for drugs that reduced oxygen consumption and hence, potentially tumour hypoxia. Atovaquone is an anti-malarial agent and ubiquinone analogue that inhibits mitochondrial complex III and was identified as a ‘top hit’ in this screen. In vivo, atovaquone reduced tumour hypoxia and sensitised xenograft models to radiotherapy [32]. To determine whether atovaquone could reduce tumour hypoxia in patients, a pharmacodynamic clinical study compared 15 atovaquone treated versus 15 untreated non-small cell lung cancer (NSCLC) patients, recruited sequentially. Here, [18F]-fluoromisonidazole (FMISO) PET-CT demonstrated a significant reduction in hypoxia in the atovaquone group and this was corroborated using a transcriptomic hypoxia gene expression signature [33]. An ongoing dose escalation study, the ‘ARCADIAN trial’ is designed to ascertain the recommended phase 2 dose of atovaquone in combination with concurrent chemoradiotherapy in locally advanced NSCLC.
The contrast here with the approach to metformin is clear. The work was led by a team of collaborators who worked together throughout each stage of the drug development project to ensure that each step was informed by the prior. At an early point during clinical evaluation detailed pharmacodynamic assessment of the drug was carried out at several dose levels. We believe this stepwise approach to pharmacodynamic characterisation prior to Phase II/III efficacy trials optimises the chances of success in a drug repurposing programme.
Future directions: is further clinical investigation of metformin in cancer warranted?
Given the results of the randomised efficacy trials enthusiasm to develop further clinical studies of metformin as a treatment for established cancers is waning. However, preclinical and clinical pharmacodynamic data obtained since the design of early clinical efficacy studies has now informed new avenues of investigation.
Markers are now being established that may define response to Complex-1 inhibitors such as mutations in the SWI-SNF complex [34]. Mitochondrial mutations in genes encoding for Complex 1 have also been proposed as markers of sensitivity for biguanide therapy [35] although mitochondrial heteroplasmy and the dynamic negative and positive enrichment of mitochondrial mutations may prevent their application as biomarkers. The transcription factor STAT3 is frequently activated in a variety of malignancies and emerging data points toward STAT3-mediated upregulation of OXPHOS as a mechanism of survival in drug-resistant tumours and a potential marker for drugs targeting mitochondrial metabolism [36,37,38]. The biobanking of translational samples from the trials already carried out to date may facilitate exploratory research to evaluate some of these emerging markers of susceptibility to anti-mitochondrial therapy with the opportunity for future trials with appropriate stratification. We suggest ‘window’ studies over short time frames for selected tumours may allow stratification of patients by evaluating dynamic response and highlight additional drug combination opportunities. If these had been performed a priori for metformin it may have aided trial design and outcome.
A number of animal and human studies have shown that metformin can alter the metabolism of gut microbiota [39, 40]. Transfer of faeces from obese mice treated with metformin into untreated mice inhibited tumour growth independently of changes in body mass, blood glucose or serum insulin. The study authors proposed that metformin treatment led to a proportionate increase in short-chain fatty acid-producing microbes and faecal transfer then led to reprogramming of tumour metabolism specifically changes in lipid homoeostasis [41]. To date, these approaches have been unexplored in the clinic.
Metformin, by inhibiting oxidative respiration and hence oxygen consumption has been shown to reduce hypoxia in tumour models [6] and more recently in a clinical study of patients with advanced cervical cancer using fluoroazomycin arabinoside (FAZA) PET-CT [42]. Via a number of mechanisms, hypoxia has been shown to suppress the anti-tumour immune response and this may be a significant mechanism of resistance to immune checkpoint immunotherapy [43]. Preclinical data have suggested that by remodelling the hypoxic tumour microenvironment metformin could potentiate the effect of anti PD-1 immunotherapy [44]. Metformin may enhance tumour immunosurveillance in ways other than reducing hypoxia in the tumour microenvironment. AMPK activation in immune cells leads to phosphorylation of PD-L1, subsequent PD-L1 glycosylation and its accumulation in the endoplasmic reticulum and degradation [45]. In syngeneic in vivo cancer models metformin enhanced the anti-tumour effect of anti-CTLA-4 therapy [45]. In another model metformin-induced AMPK activation was shown to inhibit PD-1 gene expression in CD8 + T lymphocytes and in metformin-treated lung cancer patients there was an increase in the frequency of memory stem and central memory T cells [46]. Metformin-induced AMPK activation may downregulate CD39 and CD79 gene expression thereby reducing myeloid-derived suppressor cell-driven immunosuppression [47]. Tumour-associated macrophages have been shown to be immunosuppressive through production of specific immunomodulatory cytokines promoting tumour growth. The preclinical investigation has shown that metformin can alter macrophage polarisation from an M2 to M1-like phenotype inhibiting tumour growth and angiogenesis and that this may be driven by activation of AMPK/ NF-κB signalling [48, 49].
Metformin and its role in cancer prevention is an area that has been underexplored in prospective studies. Indeed, the epidemiological data provide a strong rationale for testing this hypothesis in selected groups of patients, for example, obese or insulin-resistant individuals and now early clinical trial data is emerging in support. One clinical study investigated metformin’s potential utility in preventing tamoxifen-induced endometrial hyperplasia showing reduced endometrial thickness on transvaginal ultrasound for metformin-treated patients compared to the placebo control group [50]. Metformin has been shown to suppress intestinal polyp growth in a murine model of familial adenomatous polyposis coli [51] and a subsequent randomised clinical trial showed that metformin reduced the prevalence and number of metachronous adenomas or polyps after polypectomy following 12 months of treatment with metformin [52].
However, prevention studies designed to identify differences in cancer incidence are notoriously difficult to execute given the numbers of patients needed to properly power such a trial and the length of time it takes to complete adequate follow-up. However, opportunity lies in investigating the potential of metformin as cancer preventative for patients with cancer predisposition syndromes which will allow for smaller studies and shorter follow-up. For example, Li-Fraumeni syndrome (LFS) is a rare inherited cancer predisposition syndrome with a lifetime risk of cancer close to 100% by age 60 years in women and 73% in men. LFS is caused by germline pathogenic variants in the TP53 tumour suppressor gene [53] and in studies of mice carrying a knock-in missense mutation of TP53, metformin increases their cancer-free survival [54, 55]. This has been attributed to metformin’s direct anti-mitochondrial effect, supported by clinical evidence of attenuated mitochondrial respiration in peripheral blood mononuclear cells (PBMCs) from metformin-treated mTP53 carriers. On this basis, randomised clinical trials are now moving forward to evaluate whether metformin can reduce cancer incidence in this high-risk population.
In summary, outcomes from late-phase efficacy studies testing metformin as a repurposed cancer therapeutic have been disappointing. In a rush to establish its potential utility, such trials were designed prior to due diligence with regard to patient selection, mechanism of action and appropriate combination. New avenues of investigation in selected populations including the assessment of combination with immunotherapy, and potential as a cancer preventative agent still warrant well-designed clinical investigation.
Data availability
Not applicable.
References
Prasad V, Mailankody S. Research and development spending to bring a single cancer drug to market and revenues after approval. JAMA Intern Med. 2017;177:1569–75.
DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33.
Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348:607–14.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Investig. 2001;108:1167–74.
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5.
Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242.
Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73:4429–38.
Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–52.
Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17:351–60.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26.
Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun. 2012;3:865.
Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes. 2000;49:896–903.
Gunton JE, Delhanty PJ, Takahashi S, Baxter RC. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J Clin Endocrinol Metab. 2003;88:1323–32.
Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–60.
Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 2018;560:499–503.
Hopkins BD, Goncalves MD, Cantley LC. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol. 2020;16:276–83.
Lord SR, Cheng WC, Liu D, Gaude E, Haider S, Metcalf T, et al. Integrated pharmacodynamic analysis identifies two metabolic adaption pathways to metformin in breast cancer. Cell Metab. 2018;28:679–688.e674.
Lord SR, Collins JM, Cheng WC, Haider S, Wigfield S, Gaude E, et al. Transcriptomic analysis of human primary breast cancer identifies fatty acid oxidation as a target for metformin. Br J Cancer. 2020;122:258–65.
Goodwin PJ, Chen BE, Gelmon KA, Whelan TJ, Ennis M, Lemieux J, et al. Effect of metformin vs placebo on invasive disease-free survival in patients with breast cancer: the MA.32 randomized clinical trial. J Am Med Assoc. 2022;327:1963–73.
Goodwin PJ, Dowling RJO, Ennis M, Chen BE, Parulekar WR, Shepherd LE, et al. Effect of metformin versus placebo on metabolic factors in the MA.32 randomized breast cancer trial. NPJ Breast Cancer. 2021;7:74.
Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res. 2013;19:6741–50.
Tsakiridis T, Pond GR, Wright J, Ellis PM, Ahmed N, Abdulkarim B, et al. Metformin in combination with chemoradiotherapy in locally advanced non-small cell lung cancer: the OCOG-ALMERA randomized clinical trial. JAMA Oncol. 2021;7:1333–41.
Skinner H, Hu C, Tsakiridis T, Santana-Davila R, Lu B, Erasmus JJ, et al. Addition of metformin to concurrent chemoradiation in patients with locally advanced non-small cell lung cancer: the NRG-LU001 phase 2 randomized clinical trial. JAMA Oncol. 2021;7:1324–32.
Alghandour R, Ebrahim MA, Elshal AM, Ghobrial F, Elzaafarany M, MA EL. Repurposing metformin as anticancer drug: Randomized controlled trial in advanced prostate cancer (MANSMED). Urol Oncol. 2021;39:831.e810.
Arrieta O, Barron F, Padilla MS, Aviles-Salas A, Ramirez-Tirado LA, Arguelles Jimenez MJ, et al. Effect of metformin plus tyrosine kinase inhibitors compared with tyrosine kinase inhibitors alone in patients with epidermal growth factor receptor-mutated lung adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5:e192553.
El-Haggar SM, El-Shitany NA, Mostafa MF, El-Bassiouny NA. Metformin may protect nondiabetic breast cancer women from metastasis. Clin Exp Metastasis. 2016;33:339–57.
Marrone KA, Zhou X, Forde PM, Purtell M, Brahmer JR, Hann CL, et al. A randomized phase II study of metformin plus paclitaxel/carboplatin/bevacizumab in patients with chemotherapy-naive advanced or metastatic nonsquamous non-small cell lung cancer. Oncologist. 2018;23:859–65.
Yee D, Isaacs C, Wolf DM, Yau C, Haluska P, Giridhar KV, et al. Ganitumab and metformin plus standard neoadjuvant therapy in stage 2/3 breast cancer. NPJ Breast Cancer. 2021;7:131.
Gulati S, Desai J, Palackdharry SM, Morris JC, Zhu Z, Jandarov R, et al. Phase 1 dose-finding study of metformin in combination with concurrent cisplatin and radiotherapy in patients with locally advanced head and neck squamous cell cancer. Cancer. 2020;126:354–62.
Trucco M, Barredo JC, Goldberg J, Leclerc GM, Hale GA, Gill J, et al. A phase I window, dose escalating and safety trial of metformin in combination with induction chemotherapy in relapsed refractory acute lymphoblastic leukemia: metformin with induction chemotherapy of vincristine, dexamethasone, PEG-asparaginase, and doxorubicin. Pediatr Blood Cancer. 2018;65:e27224.
Khawaja MR, Nick AM, Madhusudanannair V, Fu S, Hong D, McQuinn LM, et al. Phase I dose escalation study of temsirolimus in combination with metformin in patients with advanced/refractory cancers. Cancer Chemother Pharm. 2016;77:973–7.
Ashton TM, Fokas E, Kunz-Schughart LA, Folkes LK, Anbalagan S, Huether M, et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat Commun. 2016;7:12308.
Skwarski M, McGowan DR, Belcher E, Di Chiara F, Stavroulias D, McCole M, et al. Mitochondrial inhibitor atovaquone increases tumor oxygenation and inhibits hypoxic gene expression in patients with non-small cell lung cancer. Clin Cancer Res. 2021;27:2459–69.
Lissanu Deribe Y, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med. 2018;24:1047–57.
Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–12.
Lin CC, Yeh HH, Huang WL, Yan JJ, Lai WW, Su WP, et al. Metformin enhances cisplatin cytotoxicity by suppressing signal transducer and activator of transcription-3 activity independently of the liver kinase B1-AMP-activated protein kinase pathway. Am J Respir Cell Mol Biol. 2013;49:241–50.
Deng XS, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367–76.
Tosic I, Frank DA. STAT3 as a mediator of oncogenic cellular metabolism: Pathogenic and therapeutic implications. Neoplasia. 2021;23:1167–78.
Lee H, Ko G. Effect of metformin on metabolic improvement and gut microbiota. Appl Environ Microbiol. 2014;80:5935–43.
Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24:1919–29.
Broadfield LA, Saigal A, Szamosi JC, Hammill JA, Bezverbnaya K, Wang D, et al. Metformin-induced reductions in tumor growth involves modulation of the gut microbiome. Mol Metab. 2022;61:101498.
Han K, Fyles A, Shek T, Croke J, Dhani N, D’Souza D, et al. A phase II randomized trial of chemoradiation with or without metformin in locally advanced cervical cancer. Clin Cancer Res. 2022;28:5263–71.
Fu Z, Mowday AM, Smaill JB, Hermans IF, Patterson AV. Tumour hypoxia-mediated immunosuppression: mechanisms and therapeutic approaches to improve cancer immunotherapy. Cells. 2021;10:1006.
Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. 2017;5:9–16.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–20.e607.
Zhang Z, Li F, Tian Y, Cao L, Gao Q, Zhang C, et al. Metformin enhances the antitumor activity of CD8(+) T lymphocytes via the AMPK-miR-107-Eomes-PD-1 pathway. J Immunol. 2020;204:2575–88.
Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018;78:1779–91.
Chiang CF, Chao TT, Su YF, Hsu CC, Chien CY, Chiu KC, et al. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-kappaB signaling. Oncotarget. 2017;8:20706–18.
Wang JC, Sun X, Ma Q, Fu GF, Cong LL, Zhang H, et al. Metformin’s antitumour and anti-angiogenic activities are mediated by skewing macrophage polarization. J Cell Mol Med. 2018;22:3825–36.
Davis SR, Robinson PJ, Jane F, White S, Brown KA, Piessens S, et al. The benefits of adding metformin to tamoxifen to protect the endometrium-A randomized placebo-controlled trial. Clin Endocrinol. 2018;89:605–12.
Tomimoto A, Endo H, Sugiyama M, Fujisawa T, Hosono K, Takahashi H, et al. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008;99:2136–41.
Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016;17:475–83.
Malkin D. Li-fraumeni syndrome. Genes Cancer. 2011;2:475–84.
Wang PY, Li J, Walcott FL, Kang JG, Starost MF, Talagala SL, et al. Inhibiting mitochondrial respiration prevents cancer in a mouse model of Li-Fraumeni syndrome. J Clin Investig. 2017;127:132–6.
Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N Engl J Med. 2013;368:1027–32.
Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, et al. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat. 2011;128:783–94.
Hadad SM, Coates P, Jordan LB, Dowling RJ, Chang MC, Done SJ, et al. Evidence for biological effects of metformin in operable breast cancer: biomarker analysis in a pre-operative window of opportunity randomized trial. Breast Cancer Res Treat. 2015;150:149–55.
Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. 2012;30:2593–2600.
Cazzaniga M, DeCensi A, Pruneri G, Puntoni M, Bottiglieri L, Varricchio C, et al. The effect of metformin on apoptosis in a breast cancer presurgical trial. Br J Cancer. 2013;109:2792–7.
Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. 2012;135:821–30.
Dowling RJ, Niraula S, Chang MC, Done SJ, Ennis M, McCready DR, et al. Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res. 2015;17:32.
Kalinsky K, Crew KD, Refice S, Xiao T, Wang A, Feldman SM, et al. Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer Investig. 2014;32:150–7.
Laskov I, Drudi L, Beauchamp MC, Yasmeen A, Ferenczy A, Pollak M, et al. Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecol Oncol. 2014;134:607–14.
Mitsuhashi A, Kiyokawa T, Sato Y, Shozu M. Effects of metformin on endometrial cancer cell growth in vivo: a preoperative prospective trial. Cancer. 2014;120:2986–95.
Schuler KM, Rambally BS, DiFurio MJ, Sampey BP, Gehrig PA, Makowski L, et al. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015;4:161–73.
Sivalingam VN, Kitson S, McVey R, Roberts C, Pemberton P, Gilmour K, et al. Measuring the biological effect of presurgical metformin treatment in endometrial cancer. Br J Cancer. 2016;114:281–9.
Kitson SJ, Maskell Z, Sivalingam VN, Allen JL, Ali S, Burns S, et al. PRE-surgical metformin in uterine malignancy (PREMIUM): a multi-center, randomized double-blind, placebo-controlled phase III trial. Clin Cancer Res. 2019;25:2424–32.
Petchsila K, Prueksaritanond N, Insin P, Yanaranop M, Chotikawichean N. Effect of metformin for decreasing proliferative marker in women with endometrial cancer: a randomized double-blind placebo-controlled trial. Asian Pac J Cancer Prev. 2020;21:733–41.
Joshua AM, Zannella VE, Downes MR, Bowes B, Hersey K, Koritzinsky M, et al. A pilot 'window of opportunity' neoadjuvant study of metformin in localised prostate cancer. Prostate Cancer Prostatic Dis. 2014;17:252–8.
Nguyen MM, Martinez JA, Hsu CH, Sokoloff M, Krouse RS, Gibson BA, et al. Bioactivity and prostate tissue distribution of metformin in a preprostatectomy prostate cancer cohort. Eur J Cancer Prev. 2018;27:557–62.
Brown JR, Chan DK, Shank JJ, Griffith KA, Fan H, Szulawski R, et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight. 2020;5:e133247.
Curry J, Johnson J, Tassone P, Vidal MD, Menezes DW, Sprandio J, et al. Metformin effects on head and neck squamous carcinoma microenvironment: window of opportunity trial. Laryngoscope. 2017;127:1808–15.
Martin-Castillo B, Pernas S, Dorca J, Alvarez I, Martinez S, Perez-Garcia JM, et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: the METTEN study. Oncotarget. 2018;9:35687–704.
Nanni O, Amadori D, De Censi A, Rocca A, Freschi A, Bologna A, et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res Treat. 2019;174:433–42.
Zhao Y, Gong C, Wang Z, Zhang J, Wang L, Zhang S, et al. A randomized phase II study of aromatase inhibitors plus metformin in pre-treated postmenopausal patients with hormone receptor positive metastatic breast cancer. Oncotarget. 2017;8:84224–36.
Pimentel I, Lohmann AE, Ennis M, Dowling RJO, Cescon D, Elser C, et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast. 2019;48:17–23.
Pujalte Martin M, Borchiellini D, Thamphya B, Guillot A, Paoli JB, Besson D, et al. TAXOMET: a French prospective multicentric randomized phase II study of docetaxel plus metformin versus docetaxel plus placebo in metastatic castration-resistant prostate cancer. Clin Genitourin Cancer. 2021;19:501–9.
Zheng Y, Zhu J, Zhang H, Liu Y, Sun H. Metformin plus first-line chemotherapy versus chemotherapy alone in the treatment of epithelial ovarian cancer: a prospective open-label pilot trial. Cancer Chemother Pharm. 2019;84:1349–57.
Bae-Jump VL, Sill M, Gehrig PA, Moxley K, Hagemann AR, Waggoner SE, et al. A randomized phase II/III study of paclitaxel/carboplatin/metformin versus paclitaxel/carboplatin/placebo as initial therapy for measurable stage III or IVA, stage IVB, or recurrent endometrial cancer: an NRG Oncology/GOG study. Gynecol Oncol. 2020;159:7–7.
Li L, Jiang L, Wang Y, Zhao Y, Zhang XJ, Wu G, et al. Combination of metformin and gefitinib as first-line therapy for nondiabetic advanced NSCLC patients with EGFR mutations: a randomized, double-blind phase II trial. Clin Cancer Res. 2019;25:6967–75.
Lee Y, Joo J, Lee YJ, Lee EK, Park S, Kim TS, et al. Randomized phase II study of platinum-based chemotherapy plus controlled diet with or without metformin in patients with advanced non-small cell lung cancer. Lung Cancer. 2021;151:8–15.
Kordes S, Pollak MN, Zwinderman AH, Mathot RA, Weterman MJ, Beeker A, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–47.
Reni M, Dugnani E, Cereda S, Belli C, Balzano G, Nicoletti R, et al. (Ir)relevance of metformin treatment in patients with metastatic pancreatic cancer: an open-label, randomized phase II trial. Clin Cancer Res. 2016;22:1076–85.
Funding
SL is funded by Cancer Research UK, National Institute for Health and Care Research and Against Breast Cancer.
Author information
Authors and Affiliations
Contributions
SL wrote and edited the manuscript. ALH edited the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lord, S.R., Harris, A.L. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic?. Br J Cancer 128, 958–966 (2023). https://doi.org/10.1038/s41416-023-02204-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02204-2
This article is cited by
-
Metformin use correlated with lower risk of cardiometabolic diseases and related mortality among US cancer survivors: evidence from a nationally representative cohort study
BMC Medicine (2024)
-
The combined treatment with ketogenic diet and metformin slows tumor growth in two mouse models of triple negative breast cancer
Translational Medicine Communications (2024)
-
Metronomic cyclophosphamide and metformin inhibited tumor growth and repopulated tumor-infiltrating lymphocytes in an experimental carcinoma model
BMC Research Notes (2024)
-
Cancer Precision-Prevention trial of Metformin in adults with Li Fraumeni syndrome (MILI) undergoing yearly MRI surveillance: a randomised controlled trial protocol
Trials (2024)
-
Implications of obesity and insulin resistance for the treatment of oestrogen receptor-positive breast cancer
British Journal of Cancer (2024)
