
Abstract
Background
Biomarker-guided therapy in an experimental setting has been suggested to improve patient outcomes. However, trial-specific pre-screening tests are time and tissue consuming and complicate the personalised treatment of patients eligible for early-phase clinical trials. In this study the feasibility of whole-genome sequencing (WGS) as a one-test-for-all for guided inclusion in early-phase trials was investigated.
Methods
Phase I Molecular Tumor Board (MTB) at the Erasmus MC Cancer Institute reviewed patients with advanced cancer without standard-of-care treatment (SOC) options for a ‘fresh-frozen’ (FF) tumour biopsy for WGS based on clinical-pathological features. Clinical grade WGS was performed by Hartwig Medical Foundation. MTB matched the patient with a trial, if available.
Results
From September 2019–March 2021, 31 patients with highly diverse tumour types underwent a tumour biopsy for WGS. The median turnaround time (TAT) was 15 days [10–42 days]. At least one actionable event was found in 84% of the patients (26/31). One-third of the patients (11/31) received matched experimental treatment.
Conclusions
WGS on fresh FF biopsies is a feasible tool for the selection of personalised experimental therapy in patients with advanced cancer without SOC options. WGS is now possible in an acceptable TAT and thus could fulfil the role of a universal genomic pre-screening test.
Similar content being viewed by others


Introduction
Patients with advanced cancer without standard-of-care treatment (SOC) options can opt for experimental treatment in early-phase clinical trials (Phase I/II clinical trials). Early-phase clinical trials can be divided into three categories; all-comer design trials, enrichment design trials, i.e. targeted or biomarker-guided trials, and master protocol trials, i.e. basket and umbrella trials [1]. Traditionally, an all-comer design strategy has minimal inclusion criteria and low response rates [2, 3]. The onset of precision oncology increased the proportion of early-phase enrichment design clinical trials considerably. These trials with personalised inclusion might be beneficial for patient outcomes [4, 5]. To identify enough eligible candidates for enrichment design clinical trials many patients have to be pre-screened. The pre-screening diagnostics for these trials are usually limited to the trial-specific target or biomarker. So in this study-oriented approach patients might have to pre-screen in multiple trials to find a suitable match which is time and tissue consuming. Trial by trial pre-screening testing could be replaced by up-front molecular profiling and immunophenotyping to gather individual tumour-specific information on possible targets and biomarkers [6]. Currently, molecular diagnostics is generally performed by panel-based Next Generation Sequencing (NGS). However, the molecular profile is by definition limited to the number of genes and exons in the implemented panel and the size of the panels varies between institutes. In order to obtain an unbiased molecular profile of the tumour and to detect less common and novel informative genetic variants Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS) would be an attractive future-proof solution. In contrast to WES, WGS reports on both the coding as the non-coding regions and can therefore be used for the unbiased detection of large structural variants (SV), e.g. ALK and NTRK fusions [7], genomic rearrangements and mutational signatures. Immunophenotyping of the tumour through RNA sequencing or protein expression can supplement the genomic data to complete the individual tumour profile [8]. Intra-, and peritumoural CD8-positive, PD-L1 and CD68-positive cell counts are signs of tumour inflammation and are potential biomarkers for immune-modulating agents [9,10,11]. Exploratory up-front WGS in combination with immunophenotyping would enable patients and treating clinicians to discover the individual profile of the tumour and consequently explore suitable trial options.
Materials and methods
Study design
From September 2019, patients with advanced solid cancer without SOC or clinical trial options were offered a tumour biopsy for WGS analysis and subsequent clinical trial allocation as part of regular diagnostic care by the Molecular Tumor Board (MTB) of the Phase I unit, Erasmus MC Cancer Institute. Patients had to be in good clinical condition (ECOG performance score 0–1), have acceptable laboratory values, and be amenable to biopsy. At that point, patients provided written consent to this trial for data collection concerning patient characteristics, WGS, MTB and choice of trial in order to evaluate WGS as a tool for experimental treatment selection.
The primary endpoint of this study is the feasibility of tumour-agnostic WGS in fresh ‘fresh-frozen’ (FF) tumour biopsies for experimental (early-phase trials) treatment selection in patients with advanced solid cancers. WGS will be deemed feasible if the inclusion rate based on WGS is comparable to similar trials with up-front molecular diagnostics. The secondary endpoint is to determine the immunogenicity of these tumours by multiplex immunofluorescent (IF) staining.
For WGS analysis, the tumour biopsy was directly frozen. Based on the results the MTB matched the patient with a trial, if available. At the time of the FF biopsy, one or two extra biopsies were collected in formalin for IF staining, additional testing and if applicable for pharmaco-dynamic analysis (PD) for a potential trial. Immunophenotyping by multiplex IF staining was performed retrospectively to obtain possible biomarkers for clinical trials with immune checkpoint inhibitors (ICI), namely Forkhead Box P3 (FOXP3), a specific marker for regulatory T cells (Tregs), Cluster of Differentiation 8 (CD8), a marker for cytotoxic T cells, Cluster of Differentiation 68 (CD68), a marker for macrophages and Programmed Death-Ligand 1 (PD-L1) [12]. The results were analysed by descriptive statistics in SPSS (IBM SSPS Statistics 25).
Pathology
The extra parallel biopsy cores were sent to the Pathology department for a regular hematoxylin and eosin (H&E) stain after formalin fixation and paraffin embedding (FFPE) to confirm the presence of representative tumour tissue.
Whole-genome sequencing
The FF biopsies were analysed by the Hartwig Medical Foundation (HMF) as is previously described [13]. WGS performed on matching blood reference DNA was used to discriminate somatic mutations from the patients’ germline variants. Germline variants were only reported for a limited number of genes for which germline variants could have diagnostic or therapeutic consequences (e.g. BRCA1/2). To establish the single base mutational signatures (COSMIC v3) Mutational Patterns v3.0.1 was used, which was run using R version 4.0.3. The threshold for a dominant COSMIC signature was set at a relative contribution of more than 25% [14]. Tumour mutation burden (TMB) is reported and samples with a TMB of higher than 10 or a tumour mutational load of higher than 140 are considered TMB high [13].
Multiplex immunofluorescent staining and digital image analysis
To determine the immune subsets, quadruple staining with FOXP3, CD68, CD8 and PD-L1 was done by automated multiplex IF using the Ventana Benchmark Discovery (Ventana Medical Systems Inc.). The IF staining and imaging are described in Supplementary Data S1. Digital image analysis (DIA) was conducted using QuPath version 0.2.3. After applying QuPath’s cell detection algorithms to segment and train to identify the biomarkers, QuPath was able to count the number of positive cells. The methodology of training and validation of the QuPath algorithms can be found in Supplementary Data S1.
Data collection
Variables collected were age at the time of biopsy, gender, ECOG performance status, smoking history, medical history, date of diagnosis, TNM classification, last date of follow up, survival status (deceased or alive), previous and current treatments, laboratory results and genomic sequencing data. Outcome data were not collected as these data were part of ongoing Phase I and II trials. Royal Marsden Hospital prognostic score (RMH) was determined in retrospect on routinely performed laboratory tests with a 1-month window from biopsy [15]. Identified genomic alterations were classified according to the ESMO Scale of Clinical Actionability for molecular Targets (ESCAT). Furthermore, genomic targets were divided into actionable events (ESCAT I-III), potentially actionable events, i.e. theoretical potential sensitivity to targeted therapy (ESCAT IV) and institutional relevant events, i.e. (potentially) actionable events for which a drug or clinical trial is available at our institute or associated institutes. This data were obtained from medical records, anonymised and entered into the ALEA database. Written informed consent was obtained from all patients for the data collection. This study was approved by our local Medical Ethical Committee (METC 19-0446).
Statistical analyses
Descriptive statistics were used to explore patient and genomic characteristics. The difference in median cell count of CD8, FOXP3, PD-L1 and CD68 in TMB high and low groups was tested with a Mann–Whitney U test (non-parametric unpaired data). The relation between high CD68/PD-L1 double-positive, high CD8-positive and high CD8/PD-L1 double-positive cell count was compared using Fisher’s exact test. Similarly, the relationship between TMB high and high CD8 cell-positive count was tested. The correlation between the number of CD68/PD-L1 double-positive cells, CD8-positive cells and CD8/PD-L1 double-positive cells was tested with a Spearman’s correlation.
Results
Patient characteristics
From September 2019 to March 2021, 31 patients underwent a FF tumour biopsy for WGS. Clinical characteristics are described in Table 1. The median age was 59 years [range 32–79] and patients had a median RMH score of 1. None of the patients had known brain metastases at the time of the biopsy. The tumour types were highly diverse (Supplementary Data S2). All biopsies were taken from metastatic sites. Four patients had not received any previous anti-cancer treatment, as no standard-of-care treatment was available for these tumour types; anaplastic and poorly differentiated thyroid cancer, adenocarcinoma of the urinary bladder and parathyroid carcinoma. Of four patients, prior molecular data was available. One of these patients received prior targeted therapy with a tyrosine kinase inhibitor. This patient had a KIT exon 11 mutated gastro-intestinal tumour for which he received imatinib and consecutively sunitinib. Resistance analysis after treatment with sunitinib revealed a potential resistance mutation in KIT exon 17 for which the patient first received regorafenib and after progression ripretinib. At the time of progression to ripretinib, WGS was performed to analyse resistance mechanisms to prior targeted therapy.
Feasibility
No complications due to the biopsy procedure were seen. The histological review showed representative tumour tissue in all 31 simultaneous FFPE biopsy cores. The median turnaround time (TAT) from biopsy to WGS results was 15 calendar days [range 10–42 days]. Based on initial shallow sequencing (~10–15× coverage) four specimens were deemed not evaluable due to a very low abundance of genomic aberrations. Because the corresponding FFPE tissue of these specimens was assessed to contain a sufficient tumour cell percentage, full sequencing (~90–100×) was performed on all samples. Eventually, WGS did not yield evaluable results in two out of 31 samples (7%) due to low tumour purity (Supplementary Data S3). In one of these patients, WGS revealed a NRAS mutation (p.Q61K). However, mutation copy number and tumour corrected variant allele frequency (tVAF) could not be reliably assessed, so the sample remained classified as not evaluable.
Molecular findings
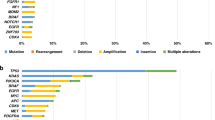
The majority of the samples (26/31) contained a molecular targetable alteration of ESCAT IV or lower (Fig. 1). Nine patients (29%) had a high TMB. One tumour was classified as homologous recombination deficiency (HRD) (defined as a HR deficiency score > 0.5) and one tumour as microsatellite unstable (MSI) (defined as a microsatellite stability score > 4.0). In addition, patient 31 had a microsatellite stability score of 3.15 in combination with a MSH2 variant that was classified as pathogenic. Additional DNA mismatch repair immunohistochemistry on the parallel FFPE biopsy material showed a loss of MSH2 and MSH6 expression. The borderline microsatellite stability score could be due to the low tumour cell percentage (18%) or could represent an MSI-low result. In five patients (16%) a driver fusion gene was detected (HNRNPA2B1-ETV1 fusion, FUS-DITT3 fusion, WHSC1L1-FGFR1 fusion, SS18-POU5F1 fusion, ADAM9-BRAF fusion). In three patients (10%), insertion of viral DNA was detected; human papillomavirus type 16 in patients with anal and penile cancer and alpha papillomavirus 7 in a patient with a neuro-endocrine carcinoma of the cervix. Supplementary Data S4 summarises special findings that warranted additional testing or discussion. In the four patients who had prior molecular testing done, we found additional alterations in three patients (RB1 mutation; BRAF fusion; KIT mutation).

Some tumours harboured multiple actionable alterations. Actionability of microsatellite instability and high tumour mutational burden/load are expressed in OncoKb level of evidence, since these are no biomarkers for targeted therapy. The CDKN2A mutations entail homozygous deletions. In Table 2 the exact mutations and therapy consequences are specified. AMP amplification, ESCAT ESMO Scale of Clinical Actionability for molecular Targets, HRD homologous recombinant deficient, HZD homozygous deletion, MSI microsatellite instability.
In 16 patients (52%), the tumour revealed a dominant mutational signature (Supplementary Data S5). Some dominant single base substitutions (SBS) signatures corresponded clinically with the proposed aetiology, e.g. ‘apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like’ (APOBEC) signature (SBS2/SBS13) and viral insertion, platinum chemotherapy signature (SBS31) and previous treatment with platinum chemotherapy [16, 17]. Other signatures have a still unknown aetiology or the proposed aetiology could not clearly be found in the patient’s history.
Immunophenotyping by multiplex immunofluorescent staining
Multiplex IF staining was completed in 30 out of the 31 tumour samples on the parallel FFPE biopsies. For one sample there was insufficient tissue material for IF staining. H&E and IF morphology were compared on a case-by-case basis to confirm the presence of representative tumour material in IF analysis. In one sample, the H&E staining did not correspond to the IF staining, and these data were excluded. So, in total, 29 samples were used for the statistical analyses. The details per sample can be found in Supplementary Data S6. Figure 2c illustrates an example of the immune staining.

a Boxplot of CD8-positive, PD-L1-positive and FOXP3-positive cells per mm2 tumour tissue according to TMB high or low. No significant differences were found in median cell count between TMB high and low. Ns not significant, TMB tumour mutational burden. b Boxplot of CD68/PD-L1 double-positive and -negative tumours plotted against CD8/PD-L1 double-positive cell count. The CD8/PD-L1 double-positive cell count was significantly higher in the CD68/PD-L1 double-positive sample than in the negative sample (p = 0.000).***p < 0.001. c Illustrative example of the multiplex immunofluorescent staining of one patient and corresponding cell counts.
For comparing the groups, we arbitrarily determined the cut-off for high cell count at the 50th percentile of the respective populations of high CD8/PD-L1 double-positive cell count (≥ 3 cells/mm2), high CD8-positive cell count (≥ 95 cells/mm2) and high CD68-positive cell count (≥133 cells/mm2). The median for CD68/PD-L1 double-positive cells was 0 cells/mm2, therefore samples were classified as CD68/PD-L1 double-positive or CD68/PD-L1 double-negative samples. Eleven tumour samples were CD68/PD-L1 double-positive.
There was no association between TMB and CD8-positive cell count (p = 1.000) (Fig. 2a). A significant association could be found between high and low CD8-positive cell count and high and low CD68-positive cell count (p = 0.027). Nine tumour samples contained both a high CD68/PD-L1 double-positive cell count and a high CD8/PD-L1 double-positive cell count. There was an association between CD68/PD-L1 double-positive and negative samples and high and low CD8/PD-L1 double-positive cell count (p = 0.008) and the median cell count of CD8/PD-L1 double-positive cells was significantly higher in the CD68/PD-L1 double-positive tumours than in the negative tumours (p = 0.000) (Fig. 2b). In addition, a strong positive correlation was found between the number of CD68/PD-L1 double-positive and CD8/PD-L1 double-positive cells (r = 0.80, p < 0.001). No association could be found between high CD68/PD-L1 double-positive cell count and high CD8-positive cell count (p = 0.128).
Allocation to treatment
For the majority of the patients with an (potentially) actionable event (18/26, 69%), a matched experimental treatment was available at our institute (Supplementary Data S2). However, for eight patients with an (potentially) actionable event no clinical trial with a matching compound was available in our or adjacent institutes. So, these patients could not receive any matched therapy. In total, eleven patients (11/31, 35%) received the matched experimental treatment. The patients were predominantly included in a Dutch nationwide initiative, the drug rediscovery protocol (DRUP, NCT02925234), in which patients with actionable aberrations can receive matched off-label drugs, either targeted therapy or checkpoint inhibition in a tumour-agnostic setting [18]. Based on high TMB or MSI, five patients received a registered ICI (pembrolizumab or nivolumab) or an experimental ICI. In the Netherlands, there is currently no indication for ICI as SOC based on high TMB or MSI. Four patients with a high TMB did not receive any therapy; three patients due to rapid clinical deterioration and one patient due to an absolute contra-indication for ICI treatment. Six patients started treatment based on a targetable mutation: palbociclib—CDKN2A homozygous deletion (n = 2) and CDK4 amplification (n = 1), experimental ATR inhibitor—ARID1A mutation (n = 1), trametinib—NRAS mutation (n = 1), gemcitabine—ATR mutation (n = 1). Patient 20 harboured a CDKN2A homozygous deletion but did not receive a CDK4/6 inhibitor due to the presence of additional driver mutations (TP53 mutation and MAP2K7 mutation). Patient 19, anaplastic thyroid carcinoma with an NRAS mutation, had spontaneous shrinkage of the tumour. Due to this atypical clinical course for anaplastic thyroid carcinoma, no therapy was started. Patient 25 had rapid clinical progression and was deemed not eligible for targeted treatment for an ADAM9 exon 19 - BRAF exon 8 fusion.
Discussion
WGS on fresh FF tumour biopsies is a feasible technique for the selection of personalised experimental therapy in patients with advanced cancer without SOC options. In our study, one-third of the patients received matched experimental treatment based on the WGS outcome. WGS performs well in comparison to similar trials with up-front molecular diagnostics (Table 2). In 84% of the patients at least one (potentially) actionable alteration or biomarker was found. As WGS is now possible with an acceptable TAT (15 days) and the analysis pipeline is open-source, WGS is a strong candidate to fulfil the role of universal and future-proof up-front genomic pre-screening test for trial inclusion [13]. Additionally, a full genome perspective in combination with open-source bioinformatics and reporting pipeline provides an advantage for transparent data sharing and combining of various datasets.
Due to the patient selection, the studied population consisted of a high variety of rare tumour types. No patients with canonical tumour types, e.g. colon carcinoma or non-small-cell lung cancer, were included because for these patients a molecular profile was usually already available as part of routine diagnostics. Our study shows that the dropout rate of molecular pre-screening was relatively low in comparison to other trials with similar patient populations, as we combined stringent patient selection based on performance scores and laboratory values with an acceptable TAT [19,20,–21]. Moreover, early-phase trial inclusion was facilitated using our approach of simultaneously acquiring a biopsy for FFPE material as it could effectively replace the study-required biopsies for pharmaco-dynamic analysis. This approach also shortens the screening period, another factor delaying treatment start.
Most tumour types that were included were rare and their genomic landscape has been essentially unexplored, so the genomic profiles of these tumours revealed useful therapeutic and etiological information, e.g. novel ADAM9-BRAF fusion, genomic algorithms, HRD without a DNA mismatch repair gene mutation and viral insertions. On the other hand, a significant proportion of the identified mutations and biomarkers could have been found by dedicated NGS panels, including TMB and MSI. It is difficult to quantify the exact number of extra alterations found by WGS as it is highly dependent on the molecular diagnostics used by pathology laboratories. Still many findings were rare and the clinical relevance of these aberrations was difficult to estimate. To help interpret the WGS data, the reports provided multi-layered information including all technical details and a clinical annotation of the findings by integration of knowledge bases (e.g. CIViC, JAX-CKB, OncoKB). An MTB with dedicated experts in medical oncology, molecular biology and pathology discussed and integrated the WGS results with clinicopathological features and available current evidence to ensure adequate interpretation and therapeutic decision making.
The rate of treatment allocation based on the WGS results in early-phase clinical trials was relatively high (35%) (Table 2). Still, more than half of the patients with an (potentially) actionable event did not receive a matched drug, mostly due to the unavailability of a matching trial. In addition, most patients had more than one driver variant and therefore combination therapies would be favourable [1, 22, 23]. This demonstrates an unmet need for accessible targeted treatment for patients with certain actionable targets. Another challenge for the development of personalised therapy is the ambiguous actionability of several molecular biomarkers. Even promising biomarkers, like TMB, are currently being revisited and we recognise that some biomarkers used to include patients in biomarker-guided trials might eventually be discarded as being actionable. On the other hand, the patients in this study would have otherwise not received any other treatment (either experimental or SOC) and they were well-informed about the experimental nature of these trials. In addition, identifying and including patients in these trials is important to study the mechanisms of these potentially actionable events [24, 25]. The genomic data generated by WGS could help understand the interpatient variability of responses to matched drugs. The response outcomes of the patients treated in this study were part of ongoing trials, therefore we were not at liberty to report on these outcomes. In this study, we therefore focused on the feasibility of WGS and not on the clinical validation of the biomarkers.
In this study, five patients were included in clinical trials with ICI treatment based on TMB or MSI. However, patients with low TMB but with inflammatory tumours can also benefit from ICI treatment [26]. TME features, e.g. CD8 + cell density and CD68/PD-L1 count, are associated with response to ICI treatment and might become a biomarker for trial inclusion [9,10,11]. Combining TME characteristics and genomic profiles could further improve the prediction models for an outcome of ICI [8, 27]. Based on the immune phenotype, we identified several patients with high CD68/PD-L1 double-positive cell count who might benefit from ICI therapy [11]. As CD68/PD-L1 double-positive cell count was correlated to CD8/PD-L1 double-positive cell count, CD8/PD-L1 double-positive cells might be involved in the sensitivity to ICI treatment. The immune phenotyping was done retrospectively and the data was not available when patients were being discussed in the MTB. Because the immune phenotype has the potential to become an important biomarker, we believe that optimisation of its diagnostics is pivotal. In this study, we show that multiplex immunofluorescence is an efficient manner to get an overview of the TME. A general challenge of immune phenotyping is that there are no standardised thresholds associated with treatment outcomes available yet.
Despite the obvious advantages of WGS, its implementation comes with some challenges, mainly due to the requirement of FF biopsy material but also because the biopsies need to contain at least 20% tumour cells. In our study we did not use micro-dissected tumour enriched regions for DNA isolation, which could improve the tumour cell percentage and increase the success rate. However, prior micro-dissection would hamper the standardisation of the methodology and is more challenging for FF material. Interpretation of tumour-specific DNA alterations and their variant allele frequency and bi-aIlelic status (to identify loss-of-heterozygosity) makes use of tumour purity assessment using the sequencing data [13]. Since this metric relies on the presence of (sufficient) DNA aberrations, this could occasionally result in the misclassification of cancer types with stable genomes, as they are falsely flagged to harbour insufficient tumour purity. It is important to be aware of this aspect, to prevent unneeded re-biopsies for repeat-analysis. A third consideration is the cost-effectiveness of WGS as a single pre-screening tool, which is currently being investigated [28]. WGS inherently requires substantially more sequencing capacity compared to panel-based NGS and thus it is more expensive. The real issue is whether the repeated and heterogeneous assays that are currently used to detect all relevant aberrations are cost-effective as a package. This comparison is hard to make as it depends heavily on the tumour type and the willingness to look for (very) rare alterations. In addition, updating DNA-based panels with new hotspot mutations or genes based on novel insights inherently causes a delay in clinical implementation and is accompanied by additional costs. While with WGS analysis all genomic information is readily available for clinical practice regardless of novel developments.
An addition to WGS is whole-transcriptome sequencing (WTS), which can provide us with more information about the overexpression of biomarkers (e.g. fusion genes), epigenetic changes, e.g. silencing of genes, and immunophenotypical gene expression profiles. But to get the complete tumour profile, both tests would be required, because important genomic information regarding mutational signature (e.g. COSMIC) is (largely) missed using WTS, as it is typically based on non-coding regions. This information is important for WGS-based tumour typing, such as in the case of a patient with carcinoma of unknown primary origin. Also, WTS misses structural variations that can provide information on possible inactivation mechanisms of tumour suppressor genes. Currently, we are exploring the clinical feasibility of combining WGS and WTS in this patient population.
We realise that currently WGS has some limitations regarding practicality and costs in comparison to smaller gene panels. However, with the continuing surge of molecular targets and targeted drugs we believe that in the near future extensive genomic testing, especially for less common tumour types, will play an important role in personalised treatment selection. WGS also has major scientific value as it reveals unbiased etiological information, which may answer both questions for patients as larger pathogenic questions, and even challenge the clinicians and researchers to ask new questions.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
References
Bui NQ, Kummar S. Evolution of early phase clinical trials in oncology. J Mol Med. 2018;96:31–8.
Italiano A, Massard C, Bahleda R, Vataire AL, Deutsch E, Magné N, et al. Treatment outcome and survival in participants of phase I oncology trials carried out from 2003 to 2006 at Institut Gustave Roussy. Ann Oncol. 2008;19:787–92.
Kimmelman J. Is participation in cancer phase I trials really therapeutic? J Clin Oncol. 2017;35:135–8.
Fontes Jardim DL, Schwaederle M, Wei C, Lee JJ, Hong DS, Eggermont AM, et al. Impact of a biomarker-based strategy on oncology drug development: a meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst. 2015;107:1–11.
Schwaederle M, Zhao M, Lee JJ, Eggermont AM, Schilsky RL, Mendelsohn J, et al. Impact of precision medicine in diverse cancers: a meta-analysis of phase II clinical trials. J Clin Oncol. 2015;33:3817–25.
Nelson AC, Yohe SL. Cancer whole-genome sequencing: the quest for comprehensive genomic profiling in routine oncology care. J Mol Diagn. 2021;23:784–7.
Yi K, Ju YS. Patterns and mechanisms of structural variations in human cancer. Exp Mol Med. 2018;50:98.
Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–62.
Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–e51.
Prelaj A, Tay R, Ferrara R, Chaput N, Besse B, Califano R. Predictive biomarkers of response for immune checkpoint inhibitors in non–small-cell lung cancer. Eur J Cancer. 2019;106:144–59.
Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res. 2020;26:970–7.
Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21:1346–58.
Roepman P, de Bruijn E, van Lieshout S, Schoenmaker L, Boelens MC, Dubbink HJ, et al. Clinical validation of whole genome sequencing for cancer diagnostics. J Mol Diagn. 2021;23:816–33.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Garrido-Laguna I, Janku F, Vaklavas C, Falchook GS, Fu S, Hong DS, et al. Validation of the royal marsden hospital prognostic score in patients treated in the phase I clinical trials program at the MD Anderson Cancer Center. Cancer. 2012;118:1422–8.
Sanger Institute. COSMIC v92, released 27-AUG-20. 2020. https://cancer.sanger.ac.uk/cosmic.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
van der Velden DL, Hoes LR, van der Wijngaart H, van Berge Henegouwen JM, van Werkhoven E, Roepman P, et al. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature. 2019;574:127–31.
Tuxen IV, Rohrberg KS, Oestrup O, Ahlborn LB, Schmidt AY, Spanggaard I, et al. Copenhagen prospective personalized oncology (CoPPO)—clinical utility of using molecular profiling to select patients to phase I trials. Clin Cancer Res. 2019;25:1239–47.
Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25:744–50.
Tsimberidou A-M, Hong DS, Wheler JJ, Falchook GS, Janku F, Naing A, et al. Long-term overall survival and prognostic score predicting survival: the IMPACT study in precision medicine. J Hematol Oncol. 2019;12:145.
Soldatos TG, Kaduthanam S, Jackson DB. Precision Oncology-The Quest for Evidence. J Person Med. 2019;9:1–17.
Park JJH, Hsu G, Siden EG, Thorlund K, Mills EJ. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J Clin. 2020;70:125–37.
Tannock IF, Hickman JA. Molecular screening to select therapy for advanced cancer? Ann Oncol. 2019;30:661–3.
Dittrich C. Basket trials: from tumour gnostic to tumour agnostic drug development. Cancer Treat Rev. 2020;90:102082.
Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer. 2018;6:157.
Lei JT, Zhang B. Proteogenomics drives therapeutic hypothesis generation for precision oncology. Br J Cancer. 2021;125:1–3.
Simons M, Van De Ven M, Coupé V, Joore M, Ijzerman M, Koffijberg E, et al. Early technology assessment of using whole genome sequencing in personalized oncology. Expert Rev Pharmacoecon Outcomes Res. 2021;21:343–51.
Bertucci F, Gonçalves A, Guille A, Adelaïde J, Garnier S, Carbuccia N, et al. Prospective high-throughput genome profiling of advanced cancers: results of the PERMED-01 clinical trial. Genome Med. 2021;13:87.
Riedl JM, Hasenleithner SO, Pregartner G, Scheipner L, Posch F, Groller K, et al. Profiling of circulating tumor DNA and tumor tissue for treatment selection in patients with advanced and refractory carcinoma: a prospective, two-stage phase II Individualized Cancer Treatment trial. Ther Adv Med Oncol. 2021;13:1758835920987658.
Réda M, Richard C, Bertaut A, Niogret J, Collot T, Fumet JD, et al. Implementation and use of whole exome sequencing for metastatic solid cancer. EBioMedicine. 2020;51:102624.
Keith TF, Robert JG, Alice PC, Shuli L, Lisa MM, David P, et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J Clin Oncol. 2020;38:3883–94.
Rodon J, Soria JC, Berger R, Miller WH, Rubin E, Kugel A, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25:751–8.
Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E, et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med. 2019;25:738–43.
Tredan O, Wang Q, Pissaloux D, Cassier P, de la Fouchardiere A, Fayette J, et al. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: analysis from the ProfiLER trial. Ann Oncol. 2019;30:757–65.
Rouven H, Anna-Lena G, Ralph F, Rainer C, Julius W, Patrick M, et al. Personalized clinical decision making through implementation of a molecular tumor board: a German Single-Center Experience. JCO Precision Oncol. 2018;2:1–16.
Massard C, Michiels S, Ferté C, Le Deley MC, Lacroix L, Hollebecque A, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7:586–95.
Cousin S, Grellety T, Toulmonde M, Auzanneau C, Khalifa E, Laizet Yh, et al. Clinical impact of extensive molecular profiling in advanced cancer patients. J Hematol Oncol. 2017;10:45.
Acknowledgements
We would like to thank M. Smid, Department of Medical Oncology, Erasmus MC, Rotterdam, NL for facilitating the mutational signature analyses. We would like to thank Hartwig Medical Foundation for facilitating and supporting whole-genome sequencing data generation, data analyses and data interpretation.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
All authors contributed to designing the work that led to the submission, acquiring data and interpreting the results. All authors contributed to drafting and revising the manuscript. All authors have approved the final version. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
A-MCD reports receiving research support from AstraZeneca, Bristol-Myers Squibb and Abbvie; receiving honoraria from Amgen, Bayer, Bristol-Myers Squibb, Novartis, Roche. A-MCD reports having honoraria from Eli Lily, AstraZeneca, Jansen, Chiesi, Pfizer and Takeda; receiving grants from Amgen; having participation on a data safety monitoring board or advisory board of Boehringer Ingelheim, Amgen, Bayer, Pharmamar and Sanofi; having leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid of Roche. JHvdT reports receiving grants from Bristol-Myers Squibb and AstraZeneca; consulting fees from Bristol-Myers Squibb, Eli Lily and Roche; honoraria from Johnson & Johnson and MSD; receiving medical equipment from Roche Diagnostics. MPL reports receiving grants from Janssen Cilag B.V., Sanofi and Merck; consulting fees from Pfizer, AstraZeneca, Astellas, Merck, Novartis, Julius Clinical and Stichting Treat Meds. The remaining authors declare no competing interests.
Ethics approval and consent to participate
Written informed consent was obtained from all patients for the data collection. This study was approved by our local Medical Ethical Committee (METC 19-0446), Erasmus Medical Center. The study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pruis, M.A., Groenendijk, F.H., Badloe, K.S. et al. Personalised selection of experimental treatment in patients with advanced solid cancer is feasible using whole-genome sequencing. Br J Cancer 127, 776–783 (2022). https://doi.org/10.1038/s41416-022-01841-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-01841-3
This article is cited by
-
Biomarker testing in cancer management- can one size fit all?
British Journal of Cancer (2022)
