
Abstract
The possibility to analyse the tumour genetic material shed in the blood is undoubtedly one of the main achievements of translational research in the latest years. In the modern clinical management of advanced non-small cell lung cancer, molecular characterisation plays an essential role. In parallel, immunotherapy is widely employed, but reliable predictive markers are not available yet. Liquid biopsy has the potential to face the two issues and to increase its role in advanced NSCLC in the next future. The aim of this review is to summarise the main clinical applications of liquid biopsy in advanced non-small cell lung cancer, underlining both its potential and limitations from a clinically driven perspective.
Similar content being viewed by others


Introduction
The role of precision medicine in advanced non-small cell lung cancer (NSCLC) has been continuously increasing in the latest years. The path to achieve complete molecular information is certainly long and challenging, as it is affected by the evolving and ever-changing nature of the neoplastic phenomenon itself. Cells mutability, metabolic changes and micro-environmental features defining different metastatic sites are able to influence the spawn and the growth of divergent tumoural sub-clones [1, 2], while cancer therapy itself affects clonal selection and host-tumour interaction [3].
Ideally, molecular information should derive from different anatomic sites, both at baseline and recurrence. In daily practice, however, tumour accessibility and patients’ clinical conditions often dictate the timing, quantity and final quality of tumour sampling [4, 5]. Molecular characterisation obtained from tissue biopsy is intrinsically partial and often not feasible due to low quantity or poor quality tumour DNA.
Tumour cells are able to shed macromolecules into the bloodstream, both from primary and metastatic sites. Assays capable of sampling, isolating and testing analytes from a biological fluid are referred to as a liquid biopsy. Liquid biopsy is minimally invasive and easily repeatable. Various biologic analytes could be isolated from peripheral blood, i.e. circulating tumour DNA (ctDNA), circulating tumour cells (CTCs), circulating exosomes, platelet RNA and ctRNA. ctDNA is certainly the most studied one [6, 7]. Cell-free DNA (cfDNA) in blood refers to degraded DNA fragments, derived mostly from normal white blood cells and stromal cells; in cancer patients, a fraction of cfDNA is represented by ctDNA, released from tumour cells through apoptotic and necrotic cell death or via active processes [8, 9]. ctDNA is amenable to be tested for the presence of somatic mutations, gene copy number variations (CNVs) and gene rearrangements and has a good concordance rate with tissue analysis [10,11,12,13].
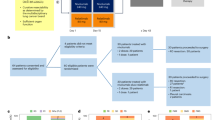
Historically, the first clinical application of liquid biopsy in advanced NSCLC was the detection of sensitising EGFR mutations [14,15,16,17,18,19] (Fig. 1). These pioneer studies were followed by the advent and widespread application of high-throughput sequencing methods, such as next-generation sequencing (NGS), which broadened our capacity for genomic profiling. NGS widened the spectrum of detection of somatic mutations from ctDNA, leading to different opportunities. First, whenever a druggable alteration was found, matched therapy might be offered [20,21,22]. Second, the presence of certain mutations or co-mutations could provide prognostic and predictive information [23, 24]. Last, the detection of tumour-specific genetic alterations at baseline and the measurement of their variation during treatment could be useful to monitor the course of the disease [25,26,27]. Large (>1.6 Mb) NGS assays allow also quantification of the tumour mutational burden (TMB), which is the total number of somatic mutations per coding area of a tumour genome. TMB is considered as a surrogate for tumour neo-antigen load and therefore a potential predictive marker for immune checkpoint inhibitors (ICIs) [28,29,30,31].

Development of liquid biopsy, from the discovery of cell-free DNA in plasma (cfDNA) to the capacity of detecting and analyzing tumour-associated genetic alterations.
Considering the feasibility of liquid biopsy as a diagnostic tool and the huge amount of data obtainable, it is crucial to integrate this technique into daily clinical workflow.
The aim of this review is to analyse currently available data and the main ongoing clinical trials, in order to provide a view on potential future applications in the clinical practice of advanced NSCLC.
The achievements of yesterday: from the discovery of cfDNA to the ability to characterise tumours in plasma
The main historical steps at the basis of the development of liquid biopsy as a tool in clinical practice are summarised in Fig. 1.
The presence of nucleic acids in the circulation was first reported by Mandel and Metais in 1948 [32]. Since then, many studies have reported a relative high concentrations of cell-free nucleic acid in the blood of cancer patients [33, 34]. Different mechanisms have been proposed concerning the source of ctDNA: cell lysis, breakdown of circulating tumour cells, destruction of tumour micro-metastases and active release by tumour cells [35]. ctDNA is frequently highly diluted in plasma and its total concentration is also influenced by the clearance and degradation by the nuclease activity.
After the discovery of ctDNA and its likely correlation with metastatic potential, research efforts mainly focused on the characterisation of tumour-associated genetic alterations in plasma. In this context, highly sensitive analytical techniques, as well as stringent and standardised pre-analytical procedures, are needed, in order to face the short half-life of cfDNA and the detection of low-frequency somatic mutations against a high background level of wild-type cfDNA fragments. The recommended workflow for plasma management considers the timing for its collection particularly crucial, as the contamination with DNA from leukocytes occurs if blood remains unprocessed for a long time (up to 4 h) [36, 37]. In order to overcome the need for rapid processing of plasma samples, commercial cell stabiliser tubes have been recently developed to prevent white blood cells degradation and to inhibit nuclease activity up to several days after blood draw [38]. Moreover, in order to safeguard the cfDNA stability and prevent its degradation in ex vivo plasma, immediate cooling at 4 °C and then storage in frozen conditions are strictly required to minimise nuclease activity [36].
Over time, various analytical methods have been developed and applied for the molecular characterisation of ctDNA. They can be divided into narrow and broad approaches.
The former methods involve assays that are able to detect genetic alterations in selected regions of cfDNA and are PCR-based techniques. A prominent PCR-based platform is the cobas EGFR mutation test (Roche diagnostics): a real-time PCR (rt-PCR) test able to provide quantitative information about the presence of specific EGFR gene alterations [39,40,41]. This assay is currently approved by U.S. Food and Drug Administration (FDA) for the detection of common sensitising EGFR alterations and of acquired resistance mutations T790M in ctDNA. Although not FDA approved, highly sensitive assays, such as the droplet digital PCR (ddPCR), can also detect additional genetic alterations in ctDNA with higher sensitivity and specificity [26, 42].
ddPCR is an approach based on water-oil emulsion droplet technology: sample is partitioned so that each droplet has from one to five molecules of DNA, and PCR amplification of the template molecules occurs in each individual droplet [43]. Such methods have several pros; the most important ones are sensitivity, affordability and short turn-around time. Notably, their analytical specificity reaches 98% [39,40,41]. Anyway, even though the detection of EGFR mutations in ctDNA is possible even at low mutated allele abundance (generally 0.5–1%), the sensitivity might be relatively low (70–80%), translating into a rate of false negatives of at least one out of five cases [44, 45]. This phenomenon is believed to be linked to the amounts of ctDNA shed into the plasma, which could be lower when tumour burden or shedding capacity is limited [6, 46].
The main limit of the narrow approach is related to its possibility to interrogate a very limited number of loci and are intrinsically unable to provide comprehensive molecular characterisation.
In parallel, data on PCR-based techniques for plasma detection of other important druggable gene alterations, such as ALK rearrangements, are very limited, they show very low sensitivity rates for fusion genes and prospective validation studies are not available yet [47,48,49].
On the other hand, high-throughput NGS-based multi-gene tests can be performed in order to interrogate in a single workflow several genomic alterations, including single nucleotide variant (SNV), small insertions and deletions (indels), gene rearrangements, CNVs, and define their variant allelic frequency (VAF). The amount of information a single NGS analysis can provide is strictly linked to the size of the gene panel analysed. Moreover, the sequencing coverage could significantly affect costs, turn-around times and also the sensitivity of NGS assays [50]. Historically NGS-based methods have been considered less sensitive than PCR-based ones; however, recent studies using ultra-deep NGS have described a similar sensitivity for the detection rate of certain driver mutations [51]. High analytical specificity is the main feature also of NGS-based genotyping assays, with a positive-predictive value of over 99%. In this context, the phenomenon of clonal hematopoiesis is noteworthy: some mutations detectable in the ctDNA are attributable to the DNA of white blood cells, rather than to tumour genetic material [52]. This issue might affect the interpretation of the test, especially when the VAF of the genetic variant called by the test is low; therefore, some assays have now incorporated paired sequencing of ctDNA and white blood cell DNA to overcome this hurdle [51].
Overall, NGS-based multi-target approaches have the advantage of allowing the simultaneous analysis of multiple genetic alterations, but they are more expensive and technically demanding and they probably require centralisation of testing for clinical use.
Liquid biopsy as the challenge of today
To date, undoubtedly, tissue biopsy represents the gold standard for lung cancer diagnosis and cannot be replaced by liquid biopsy. However, scarcity of tumour tissue is common, with over 40% of patients needing more than one procedure in order to achieve a definitive diagnosis of lung cancer [53]. In tissue, the failure rate of adequate molecular characterisation, essential for the proper treatment of lung cancer, revolves around 30% of cases [5] (Fig. 2). Over time, the number of molecular information needed is increasing. ESMO guidelines currently recommend testing at least EGFR mutations, BRAF mutations, ALK fusions, ROS-1 fusions, MET exon 14 skipping mutations, RET rearrangements and PD-L1 expression levels in non-squamous advanced NSCLC [54]. This panel could be further implemented considering KRAS mutations, HER2 mutations, MET amplification and NTRK rearrangements [54].

A simplified summary of potential advantages and limits of liquid biopsy, when compared to standard approach (tissue biopsy).
The first application of liquid biopsy in clinical practice concerns EGFR-mutated advanced NSCLC (Fig. 1). Testing EGFR mutation in plasma at baseline when tissue analysis is not feasible has been accepted in clinical practice to select patients for first-line treatment with EGFR-TKIs [54].
The second clinical application of liquid biopsy in EGFR mutation-positive NSCLC is the evaluation of the most common acquired resistance mechanism to first and second-generation EGFR-TKIs (erlotinib, gefitinib and afatinib): T790M mutation [55, 56] (Fig. 3).

Blue colour is used for static applications of liquid biopsy in clinical practice, orange colour is used for dynamic applications while in white squares validation tests are included.
In AURA3 trial, demonstrating the effectiveness of osimertinib in patients with acquired EGFR T790M mutation, tissue re-biopsy was mandatory to assess T790M mutational status, but a pre-defined subgroup analysis in patients with T790M positive in plasma confirmed the clear superiority of osimertinib over chemotherapy in this subgroup of patients [15]. A pivotal retrospective analysis in patients treated with osimertinib in phase I AURA study demonstrated that the sensitivity of detecting EGFR mutation in plasma by using BEAMing technology, a PCR-based method, was 82% for exon 19 deletion and 86% for L858R mutation and confirmed the predictive role of T790M mutation detected in plasma [57]. The sensitivity for EGFR T790M mutation detection in plasma can vary according to the technology, but might reach 93% with ddPCR [58, 59].
In addition to technical issues, EGFR T790M detection rate might be associated with different biological reasons, being tumour heterogeneity and low tumour burden. Liquid biopsy in this context could also provide additional information: if T790M ratio to activating mutation in tumour may correlate with the response to osimertinib [57, 60], the allele frequency of EGFR-activating mutations and the ratio T790M/sensitising mutation in plasma have been associated with response to osimertinib [61].
Recently, a relatively large real-world retrospective analysis has confirmed the correlation between T790M status and tumour burden. Probably, for this reason, patients with T790M positive in plasma had a worse disease control rate (DCR) when compared to negative ones [62].
The presence of a high-prevalence resistance mechanism to first- and second-generation EGFR-TKI led to the optimisation of liquid biopsy in order to detect EGFR T790M in cfDNA. International guidelines include liquid biopsy as the initial test for detection of T790M mutation in patients with evidence of TKI resistance. However, owing to the limited sensitivity of liquid biopsy [63], when a negative result is obtained, tissue biopsy is necessary, whenever feasible [54].
NGS assays are also approved as companion diagnostics to identify EGFR mutations and ALK rearrangements that predict benefits from EGFR-TKIs and ALK-TKIs, respectively. However, the most appealing application of NGS in liquid biopsy is the study of additional druggable alterations (Fig. 3). The first study on 93 consecutive patients with advanced NSCLC with insufficient or inadequate tumour samples for standard molecular characterisation showed how NGS ctDNA genotyping with Guardant360 CDx was able to detect potentially actionable genomic alterations in 53 cases [64]. Twelve patients received matched therapies, deriving significant clinical benefits [64]. Another large experience reported the use of Guardant360 CDx to genotype over 8000 advanced NSCLC patients on ctDNA and 879 were considered as “undergenotyped” after tissue-based analyses, i.e. not evaluable for all guideline-recommended genetic alterations [65]. We also recently described a real-life experience with the prospective evaluation of liquid biopsy genetic screening using Guardant360 CDx in over 200 advanced NSCLC patients, already tested in tissue with standard molecular techniques, and 42 cases were tested positive for previously undetected druggable alterations. The administration of matched targeted agents was associated with improved outcomes [22].
All these experiences underline how the application of targeted-gene NGS panels to analyse ctDNA could actually improve clinical practice, allowing the detection of actionable genomic alterations and the consequent administration of a tailored systemic anti-cancer treatment. However, one of the most relevant issues to address is the assessment of gene fusions in liquid biopsy. Their detection by the NGS-based cfDNA analysis is challenging and the sensitivity of detecting in cfDNA is currently debated [66, 67]. Moreover, recent reports describe an incidence of detection of gene fusions in plasma samples lower than expected in NSCLC [68, 69]. To this date, the largest, prospective, real-world study evaluating the clinical relevance of liquid biopsy in ALK and ROS-1 rearranged advanced NSCLC patients, showed a sensitivity rate of 67% for the detection of such fusions by amplicon-based NGS [70]. Gene fusions arise from the inter-chromosomal or intra-chromosomal conjunction of different introns. For this reason, they are difficult to detect by DNA-based NGS, especially when the introns are large, or contain repetitive sequences, and genes present several fusion partners [71, 72]. In this context, analysis of the circulating cell-free (tumour) RNA (cfRNA/ctRNA) is not influenced by the previously described limitations and could complement ctDNA for the investigation of fusion gene abnormalities. In fact, analysis of cfRNA in circulation or embedded in vesicles or tumour-educated platelets (TEPs) has shown widely applicability in the detection of several cancer-associated aberrations [73, 74]. The sensitivity of cfRNA-based assays could reach over 77% [67]; however, its clinical implementation is to be improved and needs standardisation of liquid biopsy workflow and optimisation of pre-analytical conditions.
Overall, the availability of liquid biopsy and NGS techniques for ctDNA analysis are already affecting our clinical approach to advanced NSCLC management. In particular, the great amount of information obtained by using NGS in clinical practice and the integration of off-label treatments in therapeutic pathways are one of the main challenges for modern oncology and should be managed by a specifically organised multidisciplinary team, termed molecular tumour board [75]. In the next future, the optimisation of standardised techniques could also let us face other questions about the possibility of treating patients according to liquid biopsy analysis in the absence of tissue-based genetic characterisation and even in the absence of a sure pathological diagnosis. Anyway, genetic information obtained from liquid biopsy analysis does not provide a complete molecular characterisation. In this context, the tissue biopsy is the optimal and not replaceable source for analysing the expression of PD-L1 for routine use in clinical practice and permit to investigate the role of tumour immune microenvironment. Consequently, in our opinion, the integration of tissue and plasma characterisation represents the preferred approach for advanced NSCLC management in the next future.
Liquid biopsy in the clinical practice of tomorrow
Based on available evidence, liquid biopsy applications that we feel almost ready for prime-time are the detection of acquired resistance mechanisms to EGFR- and ALK-TKI (Fig. 3). On the other hand, growing evidence is available about the potentiality of blood-based TMB (bTMB), even though its clinical applications have not been clearly defined yet.
While the detection of T790M resistance mutation in plasma has led to the spread of liquid biopsy in clinical practice, the phase III FLAURA trial assessed the superiority of osimertinib over gefitinib or erlotinib in first-line setting [76, 77] and established osimertinib as standard treatment for first-line therapy in EGFR-mutated advanced NSCLC, thus opening new perspectives for liquid biopsy applications.
Acquired resistance mechanisms to osimertinib are more heterogeneous, than those to first- and second-generation EGFR-TKIs.
Most information stemmed from studies in patients treated with osimertinib in second- or third-line settings. The revision of these studies shows the increased role of liquid biopsy in this setting. Acquired resistance mechanisms to osimertinib can be classified as on- or off-target. Multiple EGFR acquired mutations have been identified, but the most frequent is EGFR C797S mutation, occurring at osimertinib binding site [78,79,80]. Off-target alterations are more frequent than in acquired resistance to first- and second-generation TKIs. The most frequent one is MET amplification, concerning about 20% of cases [78, 81, 82]. In patients progressing to osimertinib, the persistence of T790M mutation has also been associated with improved outcomes [83, 84]. Among patients enrolled in AURA3 trial, 83 had paired plasma samples for comparing NGS analyses by using Guardant360 at baseline and at the time of progression to osimertinib: 15% of patients acquired an EGFR mutation, mainly C797S, while MET amplification was detected in 19% of patients [85]. Smaller studies were based on tissue samples and they confirmed the persistence of EGFR founder mutation at the time of progression to osimertinib and main resistance mechanisms found through liquid biopsy, but they underlined the role of histological transformation [83, 86]. In two studies analyzing tissue re-biopsies, three out of 32 and six out 41 patients developed histological transformations [83, 86].
Acquired resistance mechanisms to first-line osimertinib are less investigated and they might be different from those to second-line osimertinib. To this extent, among patients enrolled in FLAURA trial, 91 were evaluated by liquid biopsy (NGS Guardant360 73 gene panel or Omni 500 gene panel) and the most common described mechanism was MET amplification (15%), followed by EGFR C797S mutation (7%); acquired HER2 amplification, PIK3CA and RAS mutations were also described [87]. The first tissue-based series describing first-line osimertinib resistance mechanisms included 27 patients and highlighted 15% of histological transformation [88]. The study of acquired resistance mechanism to first-line osimertinib is one of the key issues in the way to further improve the outcome of EGFR-mutated patients.
No targeted treatments are currently approved for osimertinib acquired resistance, although several clinical trials are ongoing, especially in the setting of MET-amplified patients. Different strategies are being tested in the context: amivantamab and lazertinib combination (Chrysalis-2, NCT04077463) [89], osimertinib plus savolitinib association (TATTON, NCT02143466) [90, 91] and tepotinib plus osimertinib combination (INSIGHT 2, NCT03940703). These ongoing trials require mandatory tissue re-biopsy, but include also systematic plasma collection, which could validate liquid biopsy for MET-positive acquired resistance detection. Interestingly, INSIGHT 2 also include a cohort of patients treated with positive liquid biopsy and negative/not evaluable tissue biopsy [92]. In this context, small experiences evaluating liquid biopsy-based strategy to assess MET amplification showed a high level of concordance with tissue (91.67%), with a sensitivity rate of over 85% [93].
Other acquired resistance mechanisms detectable through liquid biopsy have potential clinical application in the next future and wide NGS analysis in plasma at the time of progression is able to identify additional druggable alterations, such as ALK and RET rearrangements and BRAF V600E mutations, and strategies with a combination of targeted agents have been reported [94, 95].
Another potentially druggable acquired resistance mechanism concerns resistance to second-line osimertinib through the development of EGFR C797S is in trans with the T790M mutation cells. A combination of first- and third-generation EGFR-TKIs might be effective, while fourth generation EGFR inhibitors are under development. Since cis mutations are found to be resistant to EGFR inhibition [96], the potential of plasma NGS to detect if C797S tertiary mutations are in cis or trans might of potential clinical usefulness [96, 97].
Small-cell lung cancer (SCLC) transformation has been described as another resistance mechanism, accounting for 3 to 10% of all the EGFR-TKI-resistant cases [98]. However, this might be underestimated, due to the absence of re-biopsy at the time of progression. The most common mutations associated with SCLC transformation included TP53, RB1 and PIK3CA. Small experiences, applying complementary testing of tumour tissue and liquid biopsy, showed how ctDNA analysis could suggest neuroendocrine evolution, when typical SCLC-associated genetic alterations become detectable [99, 100]. Squamous cell carcinoma transformation has also been described in different case reports after EGFR-TKI treatment [101]. Such phenomenon seemed to be linked to the emergence of PTEN alterations and with considerable genomic complexity, including co-occurring gene mutations, amplification and chromosomal rearrangements [88]. In this context, tissue re-biopsy appears to be irreplaceable for detecting histologic transformation [102].
Overall, acquired resistance mechanisms to osimertinib are heterogeneous and require screening with methods able to detect multiple genetic alterations. NGS appear to be the best approach in this setting, but requires a relatively high amount of DNA, not always easy to obtain by tissue re-biopsy. Nevertheless, the relatively high fraction of histological transformation underlines the importance of paired tissue re-biopsy to get as much information as possible in prospective trials on acquired resistance to osimertinib.
One of the challenges for the future of our patients is to validate in clinical practice NGS panels including the most frequent genetic alterations related to acquired resistance to osimertinib, considering their potential therapeutic impact. This panel could then be routinely used and associated with tumour re-biopsy when feasible.
Prospective systematic analyses of resistance mechanisms to osimertinib are ongoing: MELROSE (NCT03865511) and ELIOS study (NCT03239340) are assessing genetic tumour profile in tissue and plasma at the time of disease progression to first-line osimertinib. On the other side, ORCHARD trial (NCT03944772) is an open-label, biomarker-directed phase II platform study, including targeted treatment options for patients progressing on first-line osimertinib according to molecular characterisation performed in tissue re-biopsy.
One of the most important settings displaying the huge potentiality of liquid biopsy is ALK-rearranged disease. Several ALK-TKIs have been approved for advanced NSCLCs harboring ALK translocations. Crizotinib, a first-generation ALK-TKI, was the first approved ALK inhibitor, while second-generation TKIs, ceritinib, alectinib, brigatinib and ensartinib, were initially evaluated for patients progressing to crizotinib and subsequently moved to first-line following phase III trials [103]. Lorlatinib, a third-generation ALK-TKI, demonstrated to be active in patients progressing to first- and second-generation ALK-TKI and later to be superior over crizotinib in first-line [104, 105].
While the ALK-rearranged treatment scenario is enriching, several points still need to be improved to optimise ALK-rearranged treatment options: no selection criteria are currently available to choose among the different ALK-TKIs available in the first-line setting. Different variants of ALK rearrangements, potentially affecting the response to ALK-inhibitors, are not routinely tested in clinical practice and, above all, molecular analysis of acquired resistance mechanisms are not routinely evaluated and are not currently leading treatment choice at the time of progression.
Liquid biopsy has the potential to provide tools to fill these gaps and its role might be even more relevant than in EGFR-mutated disease, due to the heterogeneity of resistance mechanisms.
NGS technique permits the detection of ALK status at baseline and might be useful when tissue biopsy is not adequate for immunohistochemistry (IHC) and fluorescent in situ hybridisation (FISH) analysis, but also to detect different kinds of ALK rearrangements, including those not detectable by using FISH method. This approach has been investigated by the BFAST trial, a phase I/II study screening 2,219 patients with FoundationOne Liquid CDx assay and 87 out of 119 ALK-positive patients received alectinib and the radiological response rate was 87.4% [106]. This was the formal confirmation of the predictive role of ALK rearrangements detected in plasma through NGS, while a concordance of 91% between tissue and plasma evaluation was shown in patients enrolled in eXalt2 phase I/II trial investigating safety and efficacy of ensartinib [107].
Anyway, the most important applications of liquid biopsy in ALK-positive advanced NSCLC are likely to concern the study of acquired resistance to ALK inhibition.
Acquired resistance mechanisms to ALK-TKIs are both on-target and off-target, but on-target mechanisms are highly heterogeneous, thus including different ALK mutations and ALK amplifications. First analyses were retrospective and performed in tissue small series at progression to crizotinib using targeted approaches; they showed on-target alterations in less than one-third of cases and described different ALK acquired mutations. Off-targets alterations included EGFR mutation/amplification, KIT overexpression, KRAS mutation and MET amplification [108,109,110]. Later, a pivotal study performed wide NGS on re-biopsies from 51 patients progressing on crizotinib and second-generation TKIs. Interestingly, the study enhanced different patterns of acquired resistance mechanisms following first- or second-generation TKI. Notably, a higher fraction of patients progressing to second-generation TKIs developed G1202R resistance mutation, associated with sensitivity to lorlatinib [111].
More recent studies on acquired resistance mechanisms to ALK-TKIs were performed mainly by using liquid biopsy and wider use of NGS analysis increased the level of information obtained. In particular, these studies showed heterogeneity of ALK mutations detected, increased number of mutations acquired after second- and third-generation TKIs, potential underestimation of resistance mutations in tissue and potential predictive role of ALK mutations in patients treated with lorlatinib after at least one second-generation TKIs [112,113,114]. Among off-target mechanisms, MET amplification has been demonstrated to be more likely when patients are treated with new-generation TKIs in first-line setting, potentially opening new treatment perspectives for the next future [110].
Finally, the aim of evaluating resistance mechanisms to ALK-TKIs is to apply a tailored approach to further-line therapies. In this context, a systematic approach is under evaluation in the NCI-NRG ALK study (NCT03737994), a phase II study including tissue and plasma genotyping by NGS after progression on a second-generation TKI (ceritinib, alectinib, ensartinib and brigatinib). Treatment options at the time of progression are designed according to molecular characterisation: type of acquired single or compound resistance mutations, MET amplification, absence of resistance mutations.
In parallel with the discovery of activity and efficacy of new targeted agents against other driver genetic alterations [115,116,117,118,119,120,121,122,123], translational research focuses on the study of acquired resistance mechanisms. In this context, liquid biopsy seems the preferred approach associated with tissue re-biopsy, whenever feasible. In particular, the first reports are available concerning acquired resistance alterations developed after treatment with KRAS [124], MET [125] and RET inhibitors [126, 127]. Both on-target and off-target alterations were described, thus opening further therapeutic perspectives to be developed in the next future.
Another application of liquid biopsy investigated in advanced NSCLC and potentially not so far from clinical practice is bTMB evaluation. Several phase III trials demonstrated the role of ICIs, alone or in combination strategies, in the management of the vast majority of advanced NSCLC [54, 128], but response and duration of clinical benefit following ICIs are highly heterogeneous and even potential detrimental effects have been depicted [129]. Treatment selection is needed in order to improve the treatment approach and minimise the cost of cancer therapy and useless toxicity. TMB is defined as the total number of non-synonymous somatic mutations per Mb in the coding region of the cancer genome and it is associated with the neoplastic production of neoantigens to be recognised by T immune cells [130]. For this reason, TMB has the potential to mirror the capacity of tumour cells to elicit an immune response and it has been correlated to response to ICIs [131]. TMB is optimally determined by using NGS to sequence all the protein-coding regions of the genes in the genome, a technique called whole-exome sequencing (WES). However, WES is highly expensive, requires a high amount of good quality DNA and high specific expertise for results analysis. Estimates of TMB can be given by sequencing a more limited number of genes and selected gene panels have been validated for this purpose [131]. The predictive role of tissue TMB (tTMB) was assessed in several retrospective studies, by using it both as a continuous variable and as a categorical one, by defining different cut-off points [132,133,134,135]. Efficacy by tTMB was also prospectively evaluated among patients treated with nivolumab plus ipilimumab in Checkmate 568 phase II trial as a secondary endpoint [136]. Promising results of this trial were not confirmed in larger trials. In the Checkmate 227 phase III trial, tTMB was tested by using the same method and cut-off value but its predictive value was not confirmed when considering overall survival as outcome endpoint; [137, 138] on the other hand, no predictive role has been observed among patients treated with chemo-immunotherapy [139].
Smaller evidence is provided concerning the potential clinical value of bTMB. Pivotal retrospective analysis indicated the predictive role of bTMB by using three different cut-off values and evaluating two independent groups of patients treated with second-line atezolizumab versus docetaxel [31]. Prospective data on the potential predictive role of bTMB are available in patients treated with ICIs in first-line setting atezolizumab or combination treatments including durvalumab plus tremelimumab or chemotherapy [29, 140,141,142,143].
Overall, even though data about the potential predictive role of tTMB are still considered controversial and far from clinical applicability; technical limits to perform tTMB in small biopsies suggest that bTMB could be more promising and deserves further validation in clinical practice. Standardisation of technologies, cut-off definition and significance of cases with no genetic alterations detected in plasma need to be pursued, in order not to lose the opportunity to exploit biological knowledge and the potential role of TMB in this setting [131, 144].
The potential relevance of TMB is also mirrored by the performance of harmonisation studies comparing several assays in order to face over-mentioned technical issues [131, 145,146,147].
In addition to bTMB, NGS analysis in plasma permits individuate genetic alterations associated with resistance to ICIs, such as LKB1/KRAS, LKB1/KEAP1 or LKB1/KEAP1/SMARCA4 co-mutations [23, 148, 149].
The day after tomorrow: what we are looking for
Despite the increasing amount of evidence available, what we currently know is likely only the tip of the iceberg of future applications of liquid biopsy in advanced NSCLC.
What we are looking for is to have as much information as possible at diagnosis, in order to optimise treatment options and get predictive information, but also to monitor biological changes of the disease during treatment.
To this extent, technical improvements are under evaluation, to permit adequate static and dynamic analysis of multiple genetic alterations such as CNVs and genomic rearrangements [150].
In the setting of oncogene-addicted disease, the presence of co-mutations found in plasma at baseline and the persistence of EGFR mutations in plasma after a short course of treatment could address the choice of combination treatment, being potentially associated with shorter benefit from targeted agents [62, 151, 152]. For example, the presence of TP53 mutations, or PIK3CA mutations, RB1 alterations, FAT tumour suppressor homolog 1 (FAT1), or ATP-binding cassette sub-family B member 1 (ABCB1) mutations in EGFR-mutated patients, potential negative predictive and prognostic biomarkers, could lead to choose to administer EGFR-TKIs combined with chemotherapy (and/or antiangiogenic agents) [151,152,153]. In this context, future applications of liquid biopsy are likely to concern the study of primary resistance to treatment in order to optimise treatment personalisation, also taking into account upcoming treatment options.
On the other side, the systematic evaluation of genetic alterations in plasma at baseline and at the time of progression has the potential to provide information concerning predictive markers and acquired resistance mechanisms in a shorter period compared to what has happened before by using retrospective evaluation in tissue. This approach should be encouraged early in the development of new targeted agents and pursued through prospective studies in patients treated according to clinical practice.
In the field of immunotherapy also the liquid microenvironment deserves further study as a potential predictive marker. As a matter of fact, pre-treatment T-cell receptor repertoire metrics could predict response to pembrolizumab and immune-related toxicity [154].
By the way, the role of liquid biopsy could not be limited to baseline evaluation. Recent experiences demonstrated a potential role for quantitative changes in tumour-associated mutations during treatment as able to mirror changes induced by ICIs and therefore anticipate outcomes to ICIs [26, 155]. Our first experience has been obtained by tracking KRAS mutations by ddPCR, but confirmation by using NGS is ongoing and, interestingly, early evaluation in plasma could be able to anticipate potential detrimental effects of ICIs [26].
On the other hand, the interest in liquid biopsy in lung cancer surely goes towards early-stage disease. For surgically resected patients, a reliable risk assessment evaluation is one of the major needs, also taking into account the new upcoming treatment options in the adjuvant settings. Minimal residual disease (MRD) refers to the persistence of residual cancer cells after treatment, in the absence of evidence detectable through conventional investigation, such as medical imaging [156]. The detection of tumour genetic material in plasma is a promising tool to assess MRD and information stemming from different sources of genetic material could optimise the model. Several trials are currently evaluating MRD assessment in NSCLC patients treated with curative intent, including the phase III MERMAID-1 (NCT04385368) [157] and MERMAID-2 trial (NCT04642469) [158].
Besides, liquid biopsy has potential applications even in the field of lung cancer screening. If low-dose computerised tomography (LDCT) has demonstrated a role in lung cancer screening for heavy-smoker patients [159, 160], the detection of false-positive cases is one of the major concerns [161]. To overcome this aspect, a recent study proposed a cfDNA-based machine-learning method, termed lung cancer likelihood in plasma (Lung-CLiP) to discriminate early-stage lung cancer patients from risk-matched controls [162]. Liquid biopsy approaches tested for application in the field of lung cancer screening include the analysis of cfDNA methylation to distinguish between non-tumour and tumour cfDNA [163], the study of plasma microRNA [164] or the analysis of plasma metabolites through mass spectrometry [165].
Future directions for liquid biopsy in lung cancer are also related to the study of other analytes in plasma beyond ctDNA, which could open new perspectives in the next future. CTCs are not routinely used in clinical practice and in main translational studies planned for large clinical trials, due to the low number of CTCs usually detected in NSCLC. In addition, the detection and analysis of CTC, although biologically extremely fascinating, has major technical challenges and require high specific expertise and more expensive approaches, when compared to ctDNA, and standardised pre-analytical procedures are not available yet [166, 167]. CTCs might have negative prognostic value in early-stage disease; moreover, they could be potentially useful for the study of PD-L1 expression. Their heterogeneous phenotype and genotype yield CTCs analysis advantageous to investigate genetic heterogeneity through a dynamic approach. Finally, the development of CTC-cultures and CTC-derived xenograft could be of help in the research field and even in personalisation of the treatment approaches [166, 167]. As mentioned above, other analytes with potentially interesting applications are TEPs, being a potential source of abundant high-quality RNA. Extracellular vescicles are also a source of more abundant genetic information when compared with ctDNA and they are characterised by high stability, even though DNA extraction procedures are technically more complex. Finally, microRNA are small non-coding RNA, frequently deregulated in tumours and they can be detected and analysed in plasma, even though several technical issues have to be faced before validation for clinical practice [168,169,170].
In conclusion, liquid biopsy has huge potentialities in advanced NSCLC treatment and systematic prospective evaluation of its role, in parallel to validation of techniques specifically designed for clinical needs, are one of the main challenges to improve advanced NSCLC management in the next future.
Data availability
Not applicable.
References
McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–28.
López S, Lim EL, Horswell S, Haase K, Huebner A, Dietzen M, et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat Genet. 2020;52:283–93.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13.
Chouaid C, Dujon C, Do P, Monnet I, Madroszyk A, Le Caer H, et al. Feasibility and clinical impact of re-biopsy in advanced non small-cell lung cancer: a prospective multicenter study in a real-world setting (GFPC study 12-01). Lung Cancer. 2014;86:170–3.
VanderLaan PA, Yamaguchi N, Folch E, Boucher DH, Kent MS, Gangadharan SP, et al. Success and failure rates of tumor genotyping techniques in routine pathological samples with non-small-cell lung cancer. Lung Cancer. 2014;84:39–44.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20:71–88.
Nikolaev S, Lemmens L, Koessler T, Blouin J-L, Nouspikel T. Circulating tumoral DNA: Preanalytical validation and quality control in a diagnostic laboratory. Anal Biochem. 2018;542:34–39.
Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347–76.
Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA. Clin Chim Acta. 2001;313:139–42.
Sung JS, Chong HY, Kwon N-J, Kim HM, Lee JW, Kim B, et al. Detection of somatic variants and EGFR mutations in cell-free DNA from non-small cell lung cancer patients by ultra-deep sequencing using the ion ampliseq cancer hotspot panel and droplet digital polymerase chain reaction. Oncotarget. 2017;8:106901–12.
Thompson JC, Yee SS, Troxel AB, Savitch SL, Fan R, Balli D, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin Cancer Res. 2016;22:5772–82.
Schwartzberg LS, Horinouchi H, Chan D, Chernilo S, Tsai ML, Isla D, et al. Liquid biopsy mutation panel for non-small cell lung cancer: analytical validation and clinical concordance. npj Precis Oncol. 2020;4:15.
Basher F, Saravia D, Fanfan D, Cotta JA, Lopes G. Concordance of next-generation sequencing between tissue and liquid biopsies in non-small cell lung cancer. J Clin Oncol. 2020;38:e21547–e21547.
Kwapisz D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med. 2017;5:46–46.
Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med. 2017;376:629–40.
Papadimitrakopoulou VA, Mok TS, Han J-Y, Ahn M-J, Delmonte A, Ramalingam SS, et al. Osimertinib versus platinum–pemetrexed for patients with EGFR T790M advanced NSCLC and progression on a prior EGFR-tyrosine kinase inhibitor: AURA3 overall survival analysis. Ann Oncol. 2020;31:1536–44.
Lee JY, Qing X, Xiumin W, Yali B, Chi S, Bak SH, et al. Longitudinal monitoring of EGFR mutations in plasma predicts outcomes of NSCLC patients treated with EGFR TKIs: Korean Lung Cancer Consortium (KLCC-12-02). Oncotarget. 2016;7:6984–93.
Zhou C, Imamura F, Cheng Y, Okamoto I, Cho BC, Lin MC, et al. Early clearance of plasma EGFR mutations as a predictor of response to osimertinib and comparator EGFR-TKIs in the FLAURA trial. J Clin Oncol. 2019;37:9020.
Thress KS, Markovets A, Barrett JC, Chmielecki J, Goldberg SB, Shepherd FA, et al. Complete clearance of plasma EGFR mutations as a predictor of outcome on osimertinib in the AURA trial. J Clin Oncol. 2017;35:9018.
Tannock IF, Hickman JA. Molecular screening to select therapy for advanced cancer? Ann Oncol. 2019;30:661–3.
Trédan O, Wang Q, Pissaloux D, Cassier P, de la Fouchardière A, Fayette J, et al. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: analysis from the ProfiLER trial. Ann Oncol. 2019;30:757–65.
Bonanno L, Pavan A, Ferro A, Calvetti L, Frega S, Pasello G, et al. Clinical impact of plasma and tissue next‐generation sequencing in advanced non‐small cell lung cancer: a real‐world experience. Oncologist. 2020. https://doi.org/10.1634/theoncologist.2020-0148.
Pavan A, Boscolo Bragadin A, Calvetti L, Ferro A, Zulato E, Attili I, et al. Role of next generation sequencing-based liquid biopsy in advanced non-small cell lung cancer patients treated with immune checkpoint inhibitors: impact of STK11, KRAS and TP53 mutations and co-mutations on outcome. Transl Lung Cancer Res. 2021;10:202–20.
Rizvi N, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn M, et al. OA04.07 mutations associated with sensitivity or resistance to immunotherapy in mNSCLC: analysis from the MYSTIC trial. J Thorac Oncol. 2019;14:S217.
Cabel L, Riva F, Servois V, Livartowski A, Daniel C, Rampanou A, et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: a proof-of-concept study. Ann Oncol. 2017;28:1996–2001.
Zulato E, Attili I, Pavan A, Nardo G, Del Bianco P, Boscolo Bragadin A, et al. Early assessment of KRAS mutation in cfDNA correlates with risk of progression and death in advanced non-small-cell lung cancer. Br J Cancer. 2020;123:81–91.
Bratman SV, Yang SYC, Iafolla MAJ, Liu Z, Hansen AR, Bedard PL, et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat Cancer. 2020;1:873–81.
Galvano A, Gristina V, Malapelle U, Pisapia P, Pepe F, Barraco N, et al. The prognostic impact of tumor mutational burden (TMB) in the first-line management of advanced non-oncogene addicted non-small-cell lung cancer (NSCLC): a systematic review and meta-analysis of randomized controlled trials. ESMO Open. 2021;6:100124.
Socinski M, Velcheti V, Mekhail T, Chae YK, Leal TA, Dowell JE, et al. Final efficacy results from B-F1RST, a prospective phase II trial evaluating blood-based tumour mutational burden (bTMB) as a predictive biomarker for atezolizumab (atezo) in 1L non-small cell lung cancer (NSCLC). Ann Oncol. 2019;30:v919–v920.
Si H, Kuziora M, Quinn KJ, Helman E, Ye J, Liu F, et al. A blood-based assay for assessment of tumor Mutational burden in first-line metastatic NSCLC treatment: results from the MYSTIC study. Clin Cancer Res. 2021;27:1631–40.
Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018. https://doi.org/10.1038/s41591-018-0134-3.
Mandel P, Metais P. Les acides nucléiques du plasma sanguin chez l’homme [Nuclear Acids In Human Blood Plasma]. C R Seances Soc Biol Fil. 1948;142:241–3.
Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–37.
Leon S, Shapiro B, Sklaroff D, Yaros M. Free DNA in the serum of cancer patients and the effect of therapy - PubMed. Cancer Res. 1977;37:646–50.
Stroun M, Anker P, Lyautey J, Lederrey C, Maurice PA. Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol. 1987;23:707–12.
Meddeb R, Pisareva E, Thierry AR. Guidelines for the preanalytical conditions for analyzing circulating cell-free DNA. Clin Chem. 2019;65:623–33.
Normanno N, Denis MG, Thress KS, Ratcliffe M, Reck M. Guide to detecting epidermal growth factor receptor (EGFR) mutations in ctDNA of patients with advanced non-small-cell lung cancer. Oncotarget. 2017;8:12501–16.
Warton K, Yuwono NL, Cowley MJ, McCabe MJ, So A, Ford CE. Evaluation of streck BCT and PAXgene stabilised blood collection tubes for cell-free circulating DNA studies in plasma. Mol Diagn Ther. 2017;21:563–70.
Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O’Connell A, Feeney N, et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2016;2:1014.
Wu Y-L, Lee V, Liam C-K, Lu S, Park K, Srimuninnimit V, et al. Clinical utility of a blood-based EGFR mutation test in patients receiving first-line erlotinib therapy in the ENSURE, FASTACT-2, and ASPIRATION studies. Lung Cancer. 2018;126:1–8.
Jenkins S, Yang JC-H, Ramalingam SS, Yu K, Patel S, Weston S, et al. Plasma ctDNA analysis for detection of the EGFR T790M mutation in patients with advanced non–small cell lung cancer. J Thorac Oncol. 2017;12:1061–70.
Buder A, Setinek U, Hochmair MJ, Schwab S, Kirchbacher K, Keck A, et al. EGFR mutations in cell-free plasma DNA from patients with advanced lung adenocarcinoma: improved detection by droplet digital PCR. Target Oncol. 2019;14:197–203.
Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10:1003–5.
Thress KS, Brant R, Carr TH, Dearden S, Jenkins S, Brown H, et al. EGFR mutation detection in ctDNA from NSCLC patient plasma: a cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 2015;90:509–15.
Gray JE, Okamoto I, Sriuranpong V, Vansteenkiste J, Imamura F, Lee JS, et al. Tissue and plasma EGFR mutation analysis in the FLAURA trial: osimertinib versus comparator EGFR tyrosine kinase inhibitor as first-line treatment in patients with EGFR-mutated advanced non–small cell lung cancer. Clin Cancer Res. 2019;25:6644–52.
Passiglia F, Rizzo S, Rolfo C, Galvano A, Bronte E, Incorvaia L, et al. Metastatic site location influences the diagnostic accuracy of ctDNA EGFR- mutation testing in NSCLC patients: a pooled analysis. Curr Cancer Drug Targets. 2018;18:697–705.
Nilsson RJA, Karachaliou N, Berenguer J, Gimenez-Capitan A, Schellen P, Teixido C, et al. Rearranged EML4-ALK fusion transcripts sequester in circulating blood platelets and enable blood-based crizotinib response monitoring in non-small-cell lung cancer. Oncotarget. 2016;7:1066–75.
Park C-K, Kim J-E, Kim M-S, Kho B-G, Park H-Y, Kim T-O, et al. Feasibility of liquid biopsy using plasma and platelets for detection of anaplastic lymphoma kinase rearrangements in non-small cell lung cancer. J Cancer Res Clin Oncol. 2019;145:2071–82.
Heeke S, Benzaquen J, Vallee A, Allegra M, Mazieres J, Fayada J, et al. Detection of ALK fusion transcripts in plasma of non-small cell lung cancer patients using a novel RT-PCR based assay. Ann Transl Med. 2021;9:922.
Petrackova A, Vasinek M, Sedlarikova L, Dyskova T, Schneiderova P, Novosad T, et al. Standardization of sequencing coverage depth in NGS: recommendation for detection of clonal and subclonal mutations in cancer diagnostics. Front Oncol. 2019. https://doi.org/10.3389/fonc.2019.00851.
Li BT, Janku F, Jung B, Hou C, Madwani K, Alden R, et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: results from the actionable genome consortium. Ann Oncol. 2019;30:597–603.
Chan HT, Nagayama S, Chin YM, Otaki M, Hayashi R, Kiyotani K, et al. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol Oncol. 2020;14:1719–30.
Zhang Y, Shi L, Simoff MJ, Wagner OJ, Lavin J. Biopsy frequency and complications among lung cancer patients in the United States. Lung Cancer Manag. 2020. https://doi.org/10.2217/lmt-2020-0022.
Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv192–iv237.
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26–75ra26.
Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–5.
Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol. 2016;34:3375–82.
Buder A, Hochmair MJ, Schwab S, Bundalo T, Schenk P, Errhalt P, et al. Cell-free plasma DNA-guided treatment with osimertinib in patients with advanced EGFR-mutated NSCLC. J Thorac Oncol. 2018;13:821–30.
Papadimitrakopoulou VA, Han JY, Ahn MJ, Ramalingam SS, Delmonte A, Hsia TC, et al. Epidermal growth factor receptor mutation analysis in tissue and plasma from the AURA3 trial: osimertinib versus platinum-pemetrexed for T790M mutation-positive advanced non–small cell lung cancer. Cancer. 2020;126:373–80.
Ariyasu R, Nishikawa S, Uchibori K, Oh-hara T, Yoshizawa T, Dotsu Y, et al. High ratio of T790M to EGFR activating mutations correlate with the osimertinib response in non-small-cell lung cancer. Lung Cancer. 2018;117:1–6.
Del Re M, Bordi P, Rofi E, Restante G, Valleggi S, Minari R, et al. The amount of activating EGFR mutations in circulating cell-free DNA is a marker to monitor osimertinib response. Br J Cancer. 2018;119:1252–8.
Ding PN, Roberts TL, Chua W, Becker TM, Caixeiro N, de Souza P, et al. Plasma pre-treatment T790M relative allelic frequency in patients with advanced EGFR-mutated non-small cell lung cancer predicts treatment response to subsequent-line osimertinib. Transl Lung Cancer Res. 2021;10:1623–34.
Rolfo C, Mack PC, Scagliotti GV, Baas P, Barlesi F, Bivona TG, et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): a statement paper from the IASLC. J Thorac Oncol. 2018;13:1248–68.
Zugazagoitia J, Ramos I, Trigo JM, Palka M, Gómez-Rueda A, Jantus-Lewintre E, et al. Clinical utility of plasma-based digital next-generation sequencing in patients with advance-stage lung adenocarcinomas with insufficient tumor samples for tissue genotyping. Ann Oncol. 2019;30:290–6.
Mack PC, Banks KC, Espenschied CR, Burich RA, Zill OA, Lee CE, et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non–small cell lung cancer: analysis of over 8000 cases. Cancer. 2020;126:3219–28.
Supplee JG, Milan MSD, Lim LP, Potts KT, Sholl LM, Oxnard GR, et al. Sensitivity of next-generation sequencing assays detecting oncogenic fusions in plasma cell-free DNA. Lung Cancer. 2019;134:96–99.
Hasegawa N, Kohsaka S, Kurokawa K, Shinno Y, Takeda Nakamura I, Ueno T, et al. Highly sensitive fusion detection using plasma cell‐free RNA in non‐small‐cell lung cancers. Cancer Sci. 2021;112:4393–403.
Aggarwal C, Thompson JC, Black TA, Katz SI, Fan R, Yee SS, et al. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non-small cell lung cancer. JAMA Oncol. 2019;5:173–80.
Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24:3539–49.
Mezquita L, Swalduz A, Jovelet C, Ortiz-Cuaran S, Howarth K, Planchard D, et al. Clinical relevance of an amplicon-based liquid biopsy for detecting ALK and ROS1 fusion and resistance mutations in patients with non–small-cell lung cancer. JCO Precis Oncol. 2020;4:272–82. https://doi.org/10.1200/PO.19.00281.
Wong D, Yip S, Sorensen PH. Methods for identifying patients with tropomyosin receptor kinase (TRK) fusion cancer. Pathol Oncol Res. 2020;26:1385–99.
Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46.
Best MG, Wesseling P, Wurdinger T. Tumor-educated platelets as a noninvasive biomarker source for cancer detection and progression monitoring. Cancer Res. 2018;78:3407–12.
Sorber L, Zwaenepoel K, Jacobs J, De Winne K, Goethals S, Reclusa P, et al. Circulating cell-free DNA and RNA analysis as liquid biopsy: optimal centrifugation protocol. Cancers (Basel). 2019. https://doi.org/10.3390/CANCERS11040458.
Danesi R, Fogli S, Indraccolo S, Del Re M, Dei Tos AP, Leoncini L, et al. Druggable targets meet oncogenic drivers: opportunities and limitations of target-based classification of tumors and the role of Molecular Tumor Boards. ESMO Open. 2021. https://doi.org/10.1016/j.esmoop.2020.100040.
Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378:113–25.
Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR -mutated advanced NSCLC. N Engl J Med. 2020;382:41–50.
Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121:725–37.
Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non–small cell lung cancer patients. Clin Cancer Res. 2018;24:3097–107.
Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–2.
Le X, Puri S, Negrao MV, Nilsson MB, Robichaux J, Boyle T, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR -mutant NSCLC. Clin Cancer Res. 2018;24:6195–203.
Schoenfeld AJ, Yu HA. The evolving landscape of resistance to osimertinib. J Thorac Oncol. 2020;15:18–21.
Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of resistance mechanisms and clinical implications in patients With EGFR T790M–positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018;4:1527.
Zhao S, Li X, Zhao C, Jiang T, Jia Y, Shi J, et al. Loss of T790M mutation is associated with early progression to osimertinib in Chinese patients with advanced NSCLC who are harboring EGFR T790M. Lung Cancer. 2019;128:33–39.
Papadimitrakopoulou VA, Wu Y-L, Han J-Y, Ahn M-J, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29:viii741.
Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR -mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018;8:1529–39.
Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29:viii740.
Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. 2020;26:2654–63.
Bauml J, Cho BC, Park K, Lee KH, Cho EK, Kim D-W, et al. Amivantamab in combination with lazertinib for the treatment of osimertinib-relapsed, chemotherapy-naïve EGFR mutant (EGFRm) non-small cell lung cancer (NSCLC) and potential biomarkers for response. J Clin Oncol. 2021;39:9006.
Oxnard GR, Yang JCH, Yu H, Kim SW, Saka H, Horn L, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31:507–16.
Oxnard GR, Cantarini M, Frewer P, Hawkins G, Peters J, Howarth P, et al. SAVANNAH: A Phase II trial of osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-driven (MET+), locally advanced or metastatic non-small cell lung cancer (NSCLC), following disease progression on osimertinib. J Clin Oncol. 2019;37:TPS9119.
Yang JC-H, Ellers-Lenz B, Straub J, Johne A, Wu Y-L. INSIGHT 2: Tepotinib plus osimertinib in patients with EGFR-mutant NSCLC having acquired resistance to EGFR TKIs due to MET-amplification: a phase II trial in progress study. Ann Oncol. 2019;30:ix181.
Mondelo-Macía P, Rodríguez-López C, Valiña L, Aguín S, León-Mateos L, García-González J, et al. Detection of MET alterations using cell free DNA and circulating tumor cells from cancer patients. Cells. 2020;9:522.
Rehman M, Kim C, Reuss JE, Kiedrowski LA, Garg RJ, Liu SV. Divergent RET- and BRAF-mediated resistance to osimertinib in EGFR -mutant NSCLC: a case report. JCO Precis Oncol. 2021;5:939–42. https://doi.org/10.1200/PO.21.00083.
Hou H, Sun D, Zhang C, Liu D, Zhang X. <scp> ALK </scp> rearrangements as mechanisms of acquired resistance to osimertinib in <scp> EGFR </scp> mutant non‐small cell lung cancer. Thorac Cancer. 2021;12:962–9.
Wang Z, Yang JJ, Huang J, Ye JY, Zhang XC, Tu HY, et al. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J Thorac Oncol. 2017;12:1723–7.
Arulananda S, Do H, Musafer A, Mitchell P, Dobrovic A, John T. Combination osimertinib and gefitinib in C797S and T790M EGFR-mutated non–small cell lung cancer. J Thorac Oncol. 2017;12:1728–32.
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–7.
Vendrell JA, Quantin X, Serre I, Solassol J. Combination of tissue and liquid biopsy molecular profiling to detect transformation to small cell lung carcinoma during osimertinib treatment. Ther Adv Med Oncol. 2020;12:175883592097419.
Pizzutilo EG, Pedrani M, Amatu A, Ruggieri L, Lauricella C, Veronese SM, et al. Liquid biopsy for small cell lung cancer either de novo or transformed: systematic review of different applications and meta-analysis. Cancers (Basel). 2021;13:2265.
Jukna A, Montanari G, Mengoli MC, Cavazza A, Covi M, Barbieri F, et al. Squamous cell carcinoma “Transformation” concurrent with secondary T790M mutation in resistant EGFR -mutated adenocarcinomas. J Thorac Oncol. 2016;11:e49–e51.
Uruga H, Fujii T, Nakamura N, Moriguchi S, Kishi K, Takaya H. Squamous cell transformation as a mechanism of acquired resistance to tyrosine kinase inhibitor in EGFR‐mutated lung adenocarcinoma: a report of two cases. Resp Case Rep. 2020. https://doi.org/10.1002/rcr2.521.
Elliott J, Bai Z, Hsieh S-C, Kelly SE, Chen L, Skidmore B, et al. ALK inhibitors for non-small cell lung cancer: a systematic review and network meta-analysis. PLoS ONE. 2020;15:e0229179.
Shaw AT, Bauer TM, de Marinis F, Felip E, Goto Y, Liu G, et al. First-line lorlatinib or crizotinib in advanced ALK -positive lung cancer. N Engl J Med. 2020;383:2018–29.
Chuang C-H, Chen H-L, Chang H-M, Tsai Y-C, Wu K-L, Chen I-H, et al. Systematic review and network meta-analysis of anaplastic lymphoma kinase (ALK) inhibitors for treatment-naïve ALK-positive lung cancer. Cancers (Basel). 2021;13:1966.
Gadgeel SM, Mok TSK, Peters S, Alexander JAA, Leighl NB, Sriuranpong V, et al. Phase II/III blood first assay screening trial (BFAST) in patients (pts) with treatment-naïve NSCLC: initial results from the ALK+ cohort. Ann Oncol. 2019;30:v918.
Horn L, Whisenant JG, Wakelee H, Reckamp KL, Qiao H, Leal TA, et al. Monitoring therapeutic response and resistance: analysis of circulating tumor DNA in patients with ALK+ lung cancer. J Thorac Oncol. 2019;14:1901–11.
Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Cancer: Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012. https://doi.org/10.1126/scitranslmed.3003316.
Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82.
Dagogo-Jack I, Yoda S, Lennerz JK, Langenbucher A, Lin JJ, Rooney MM, et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin Cancer Res. 2020;26:2535–45.
Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK -rearranged lung cancer. Cancer Discov. 2016;6:1118–33.
Shaw AT, Solomon BJ, Besse B, Bauer TM, Lin CC, Soo RA, et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non–small-cell lung cancer. J Clin Oncol. 2019;37:1370–9.
Dagogo-Jack I, Rooney M, Lin JJ, Nagy RJ, Yeap BY, Hubbeling H, et al. Treatment with next-generation ALK inhibitors fuels plasma ALK mutation diversity. Clin Cancer Res. 2019;25:6662–70.
Pailler E, Faugeroux V, Oulhen M, Mezquita L, Laporte M, Honore A, et al. Acquired resistance mutations to ALK inhibitors identified by single circulating tumor cell sequencing in ALK-rearranged non–small-cell lung cancer. Clin Cancer Res. 2019;25:6671–82.
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371–81.
Jänne P, Rybkin I, Spira A, Riely G, Papadopoulos K, Sabari J, et al. KRYSTAL-1: Activity and safety of adagrasib (MRTX849) in advanced/ metastatic non–small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Presented at the 32nd EORTC-NCI-AACR Symposium, vir- tual meeting, October 24–25, 2020: LBA3. abstract. Eur J Cancer. 2020;138:S1–S2.
Drilon A, Clark J, Weiss J, Ou S, Camidge DR, Solomon B, et al. OA12.02 updated antitumor activity of crizotinib in patients with MET exon 14-altered advanced non-small cell lung cancer. J Thorac Oncol. 2018;13:S348.
Landi L, Chiari R, Tiseo M, D\textquoterightIncà F, Dazzi C, Chella A, et al. Crizotinib in MET-deregulated or ROS1-rearranged pretreated non-small cell lung cancer (METROS): a phase II, prospective, multicenter, two-arms trial. Clin Cancer Res. 2019;25:7312–9.
Felip E, Sakai H, Patel J, Horn L, Veillon R, Griesinger F, et al. OA12.01 phase II data for the MET inhibitor tepotinib in patients with advanced NSCLC and MET exon 14-skipping mutations. J Thorac Oncol. 2018;13:S347.
Paik PK, Veillon R, Cortot AB, Felip E, Sakai H, Mazieres J, et al. Phase II study of tepotinib in NSCLC patients with METex14 mutations. J Clin Oncol. 2019;37:9005.
Wolf J, Seto T, Han J-Y, Reguart N, Garon EB, Groen HJM, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J Clin Oncol. 2019;37:9004.
Subbiah V, Hu MI-N, Gainor JF, Mansfield AS, Alonso G, Taylor MH, et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. J Clin Oncol. 2020;38:109.
Gainor JF, Curigliano G, Kim D-W, Lee DH, Besse B, Baik CS, et al. Registrational dataset from the phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET fusion+ non-small cell lung cancer (NSCLC). J Clin Oncol. 2020;38:9515.
Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med. 2021;384:2382–93.
Recondo G, Bahcall M, Spurr LF, Che J, Ricciuti B, Leonardi GC, et al. Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14 mutant NSCLC. Clin Cancer Res. 2020. https://doi.org/10.1158/1078-0432.CCR-19-3608.
Solomon BJ, Tan L, Lin JJ, Wong SQ, Hollizeck S, Ebata K, et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J Thorac Oncol. 2020;15:541–9.
Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann Oncol. 2020;31:1725–33.
Attili I, Passaro A, Pavan A, Conte PF, De Marinis F, Bonanno L. Combination immunotherapy strategies in advanced non-small cell lung cancer (NSCLC): does biological rationale meet clinical needs? Crit Rev Oncol Hematol. 2017;119:30–39.
Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. Hyperprogressive disease in patients with advanced non–small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncol. 2018;4:1543.
Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56.
Sholl LM, Hirsch FR, Hwang D, Botling J, Lopez-Rios F, Bubendorf L, et al. The promises and challenges of tumor mutation burden as an immunotherapy biomarker: a perspective from the International Association for the Study of Lung Cancer Pathology Committee. J Thorac Oncol. 2020;15:1409–24.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. N Engl J Med. 2017;376:2415–26.
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6.
Herbst RS, Lopes G, Kowalski DM, Nishio M, Wu Y-L, de Castro Junior G, et al. Association between tissue TMB (tTMB) and clinical outcomes with pembrolizumab monotherapy (pembro) in PD-L1-positive advanced NSCLC in the KEYNOTE-010 and -042 trials. Ann Oncol. 2019;30:v916–v917.
Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol. 2019;37:992–1000.
Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–104.
Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381:2020–31.
Garassino M, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, Speranza G, et al. OA04.06 evaluation of TMB in KEYNOTE-189: pembrolizumab plus chemotherapy vs placebo plus chemotherapy for nonsquamous NSCLC. J Thorac Oncol. 2019;14:S216–S217.
Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-line treatment of PD-L1–selected patients with NSCLC. N Engl J Med. 2020;383:1328–39.
Peters S, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn M-J, et al. Abstract CT074: Tumor mutational burden (TMB) as a biomarker of survival in metastatic non-small cell lung cancer (mNSCLC): blood and tissue TMB analysis from MYSTIC, a Phase III study of first-line durvalumab ± tremelimumab vs chemotherapy. Cancer Res. 2019;79:CT074–CT074.
Aggarwal C, Thompson J, Chien A, Quinn K, Lefterova M, Nagy R, et al. MA25.04 blood-based tumor mutation burden as a predictive biomarker for outcomes after pembrolizumab based first line therapy in metastatic NSCLC. J Thorac Oncol. 2019;14:S352.
Rizvi N, Chul Cho B, Reinmuth N, Lee K, Ahn M, Luft A, et al. Durvalumab with or without tremelimumab vs platinum-based chemotherapy as first-line treatment for metastatic non-small cell lung cancer: MYSTIC | OncologyPRO. Ann Oncol. 2018;29:x39–x43.
Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: is more always better? Clin Cancer Res. 2021;27:1236–41.
Vokes N, Liu D, Ricciuti B, Jimenez-Aguilar E, Rizvi H, Dietlein F, et al. Harmonization of tumor mutational burden quantification and association with response to immune checkpoint blockade in non-small-cell lung cancer. JCO Precis Oncol. 2019;3:1–12.
Merino DM, McShane LM, Fabrizio D, Funari V, Chen S-J, White JR, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020;8:e000147.
Stenzinger A, Endris V, Budczies J, Merkelbach-Bruse S, Kazdal D, Dietmaier W, et al. Harmonization and standardization of panel-based tumor mutational burden measurement: real-world results and recommendations of the quality in pathology study. J Thorac Oncol. 2020;15:1177–89.
Marinelli D, Mazzotta M, Scalera S, Terrenato I, Sperati F, D’Ambrosio L, et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann Oncol. 2020;31:1746–54.
Zhou H, Shen J, Liu J, Fang W, Zhang L. Efficacy of immune checkpoint inhibitors in SMARCA4-mutant NSCLC. J Thorac Oncol. 2020;15:e133–e136.
Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non–small-cell lung cancer. N Engl J Med. 2017;376:2109–21.
Duan J, Xu JC, Wang Z, Bai H, Cheng Y, An T, et al. Refined stratification based on baseline concomitant mutations and longitudinal circulating tumor DNA monitoring in advanced EGFR-mutant lung adenocarcinoma under Gefitinib treatment. J Thorac Oncol. 2020;15:1857–70.
Molina-Vila MA, Bertran-Alamillo J, Gascó A, Mayo-de-las-Casas C, Sánchez-Ronco M, Pujantell-Pastor L, et al. Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2014;20:4647–59.
Chen M, Xu Y, Zhao J, Zhong W, Zhang L, Bi Y, et al. Concurrent driver gene mutations as negative predictive factors in epidermal growth factor receptor-positive non-small cell lung cancer. EBioMedicine. 2019;42:304–10.
Abed A, Calapre L, Bowyer S, Millward M, Gray E. LBA68 Clinical value of pre-treatment T-cell receptors (TCR) repertoire in non-small cell lung cancer (NSCLC) patients treated with single agent immunotherapy. Ann Oncol. 2021;32:S1344.
Kruger S, Heinemann V, Ross C, Diehl F, Nagel D, Ormanns S, et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann Oncol. 2018;29:2348–55.
Cescon DW, Bratman SV, Chan SM, Siu LL. Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer 2020 13. 2020;1:276–90.
Peters S, Spigel D, Ahn M, Tsuboi M, Chaft J, Harpole D, et al. P03.03 MERMAID-1: A phase III study of adjuvant durvalumab plus chemotherapy in resected NSCLC patients with MRD+ post-surgery. J Thorac Oncol. 2021;16:S258–S259.
Spigel DR, Peters S, Ahn M-J, Tsuboi M, Chaft J, Harpole D, et al. 93TiP MERMAID-2: Phase III study of durvalumab in patients with resected, stage II-III NSCLC who become MRD+ after curative-intent therapy. J Thorac Oncol. 2021;16:S745–S746.
The National Lung Screening Trial Research Team. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395–409.
de Koning HJ, van der Aalst CM, de Jong PA, Scholten ET, Nackaerts K, Heuvelmans MA, et al. Reduced lung-cancer mortality with volume CT screening in a randomized trial. N Engl J Med. 2020;382:503–13.
Jemal A, Fedewa SA. Lung cancer screening with low-dose computed tomography in the United States—2010 to 2015. JAMA Oncol. 2017;3:1278–81.
Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–51.
Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Liu MC, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31:745–59.
Sozzi G, Boeri M, Rossi M, Verri C, Suatoni P, Bravi F, et al. Clinical utility of a plasma-based miRNA signature classifier within computed tomography lung cancer screening: a correlative MILD trial study. J Clin Oncol. 2014;32:768–73.
Zhang L, Zheng J, Ahmed R, Huang G, Reid J, Mandal R, et al. A high-performing plasma metabolite panel for early-stage lung cancer detection. Cancers (Basel). 2020. https://doi.org/10.3390/CANCERS12030622.
Hofman V, Heeke S, Marquette C-H, Ilié M, Hofman P. Circulating tumor cell detection in lung cancer: but to what end? Cancers (Basel). 2019. https://doi.org/10.3390/CANCERS11020262.
Guibert N, Pradines A, Favre G, Mazieres J. Current and future applications of liquid biopsy in nonsmall cell lung cancer from early to advanced stages. Eur Respir Rev. 2020. https://doi.org/10.1183/16000617.0052-2019.
Liu L, Lin F, Ma X, Chen Z, Yu J. Tumor-educated platelet as liquid biopsy in lung cancer patients. Crit Rev Oncol Hematol. 2020. https://doi.org/10.1016/J.CRITREVONC.2020.102863.
Li M-Y, Liu L-Z, Dong M. Progress on pivotal role and application of exosome in lung cancer carcinogenesis, diagnosis, therapy and prognosis. Mol Cancer. 2021;20:1–22.
Pedraz-Valdunciel C, Rosell R. Defining the landscape of circRNAs in non-small cell lung cancer and their potential as liquid biopsy biomarkers: a complete review including current methods. Extracell Vesicles Circ Nucleic Acids. 2021;2:179–201.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
LB: conception and design of the work, reviewed literature, drafted and revised the manuscript. ADM: reviewed literature, drafted and revised the manuscript. AP: reviewed literature, drafted and revised the manuscript. EZ: reviewed literature, drafted and revised the manuscript. LC: reviewed literature, drafted and revised the manuscript. GP: critical review. VG: critical review, editing and supervision. PFC: critical review, editing and supervision. SI: conception and design of the work, editing, critical review and supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
No ethics approval was required.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bonanno, L., Dal Maso, A., Pavan, A. et al. Liquid biopsy and non-small cell lung cancer: are we looking at the tip of the iceberg?. Br J Cancer 127, 383–393 (2022). https://doi.org/10.1038/s41416-022-01777-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-01777-8
This article is cited by
-
Longitudinal tracking of circulating rare events in the liquid biopsy of stage III–IV non-small cell lung cancer patients
Discover Oncology (2024)
-
Resistance to osimertinib in advanced EGFR-mutated NSCLC: a prospective study of molecular genotyping on tissue and liquid biopsies
British Journal of Cancer (2024)
-
Predicting response and toxicity to immune checkpoint inhibitors in lung cancer using antibodies to frameshift neoantigens
Journal of Translational Medicine (2023)
-
Economic Evaluations of Imaging Biomarker-Driven Companion Diagnostics for Cancer: A Systematic Review
Applied Health Economics and Health Policy (2023)
