
Abstract
Background
Genetic variation increases the risk of lung cancer, but the extent to which smoking amplifies this effect remains unknown. Therefore, we aimed to investigate the risk of lung cancer in people with different genetic risks and smoking habits.
Methods
This prospective cohort study included 345,794 European ancestry participants from the UK Biobank and followed up for 7.2 [6.5–7.8] years.
Results
Overall, 26.2% of the participants were former smokers, and 9.8% were current smokers. During follow-up, 1687 (0.49%) participants developed lung cancer. High genetic risk and smoking were independently associated with an increased risk of incident lung cancer. Compared with never-smokers, HR per standard deviation of the PRS increase was 1.16 (95% CI, 1.11–1.22), and HR of heavy smokers (≥40 pack-years) was 17.89 (95% CI, 15.31–20.91). There were no significant interactions between the PRS and the smoking status or pack-years. Population-attributable fraction analysis showed that smoking cessation might prevent 76.4% of new lung cancers.
Conclusions
Both high genetic risk and smoking were independently associated with higher lung cancer risk, but the increased risk of smoking was much more significant than heredity. The combination of traditional risk factors and additional PRS provides realistic application prospects for precise prevention.
Similar content being viewed by others


Background
Lung cancer is the most commonly diagnosed cancer and has the highest mortality worldwide among the general population and males, and it has the second leading mortality and the third incidence among females. In 2018, there were more than 2 million new cases and 1.7 million deaths from lung cancer [1]. Tobacco exposure is the leading cause of lung cancer, despite differences in the intensity of smoking and the type of cigarettes, and ~90% of lung cancers are attributed to smoking [2]. In addition, genetic factors also play essential roles in cancer development. Twin studies [3] and heritability estimation based on genome-wide association studies (GWASs) [4, 5] indicated that genetic factors contribute far less to incident lung cancer than environmental factors, including smoking. However, population-based prospective studies of smoking and genetic risk in lung cancer have not been fully validated.
Over the past decade, GWASs have identified multiple susceptibility loci associated with lung cancer risk, including TP63, TERT, CDKN2A/B and CHRNA3/5 [6,7,8,9]. However, while consistently and significantly associated with the lung cancer risk, each common variant’s impact is modest. Aggregating multiple single-nucleotide polymorphisms (SNPs) with tiny functions to generate a composite polygenic risk score (PRS) may explain the genetic risk of complex diseases [10]. In addition, multiple genes, including CHRNA3/5, were strongly associated with lung cancer, smoking behaviours [11], and nicotine addiction [12]. Although previous studies have reported a significant association with lung cancer based on case-control designs [13, 14], the relevance of combining these risk scores and smoking for individual subjects and whether smoking and genetic risk have a synergistic effect remains uncertain. Therefore, we hypothesised that smoking and genetic risk are independently associated with incident lung cancer.
This study’s primary purpose was to investigate whether there are differences in the association between smoking and new-onset lung cancer among individuals with low, intermediate or high genetic risk in a large population-based cohort. The second aim was to investigate the possible interaction between genetic risk and smoking for incident lung cancer.
Methods
Study design
The UK Biobank study started in 2006 and, until 2010, recruited >500,000 participants aged 40–69 years from the general population at 22 assessment centres throughout the UK [15]. Participants provided information on smoking and other potentially health-related aspects through extensive baseline questionnaires, verbal interviews and physical measurements. Moreover, blood samples were collected for genotyping.
Participants were excluded if they withdrew from the study (n = 1298), their genotype data does not meet the quality control conditions, related to another one more than second-degree, or were non-European ancestry (n = 44,072). Besides, participants with missing data on smoking or covariates were excluded (n = 75,546). Participants with a history of cancer at baseline were also excluded (n = 35,814).
Polygenic risk score
Polygenic risk scores were created following an additive model for previously published common genetic variants associated with lung cancer. To identify relevant risk loci, we began by searching the NHGRI-EBI GWAS Catalog of published GWAS [16]. Then, we reviewed both the original manuscript and supplementary materials to identify SNPs, risk alleles, and effect sizes. SNPs were selected for each locus according to the criteria of independent (r2 < 0.1), common (minor allele frequencies [MAF] > 0.01 in 1000 Genomes Project European population), UK Biobank available, large sample size in the development cohort, and smallest P value. The number of risk alleles (0, 1 or 2) for everyone was summed after multiplication with the effect size between the SNPs and each trait. A total of 33 SNPs from eight studies were used (eTable 1 in the Supplement) [8, 9, 17,18,19,20,21,22]. This polygenic risk score was then z-standardised based on values for all individuals and categorised into low (lowest quintile), intermediate (quintiles 2–4) and high (highest quintile) risk.
Smoking status and pack-years
Touchscreen questionnaires collected information on smoking status and pack-years at baseline. Detailed definitions of smoking status and the pack-years of smoking were provided in eTable 2 in the Supplement. All participants were categorised as never, former or current smoking according to their smoking status, and as no (0), light (0.1–19.9), intermediate (20–39.9), or heavy (≥40) smoking according to the pack-years of smoking.
Outcomes
Participants with incident lung cancer were identified as having a diagnosis in national cancer registries after baseline assessment. Diagnoses were recorded using the International Classification of Diseases-9 (ICD-9) and ICD-10 coding system (eTable 3 in Supplement). Death was ascertained via linkage to death registries. We calculated the follow-up time from the date of attendance to the date of first diagnosis, date of death, March 31, 2016 for Wales and England, and October 31, 2015 for Scotland, whichever occurred first.
Covariates
All models were adjusted for age, sex, education, socioeconomic status (household income and Townsend deprivation index [23]), body mass index (BMI), physical activity, diet, alcohol consumption, passive smoking, occupational exposure, the relatedness of individuals in the sample and first 20 principal components of ancestry. Body mass index (BMI) (kg/m2) was calculated for all UK Biobank participants based on their measured weight and height. Duration and intensity of physical activity were ascertained by touchscreen questionnaires based on the validated International Physical Activity Questionnaire [24]. A healthy diet was calculated based on the Dietary Approaches to Stop Hypertension (DASH) recommendation, associated with multiple cancer types [25, 26]. Alcohol consumption was calculated based on US Dietary Guidelines for Americans 2015–2020 [27]. Exposure to tobacco smoke from others at home or outside for more than an hour per week was considered passive smoking. Occupational exposure is based on self-reported exposure to asbestos, paints, thinners, glues, pesticides, diesel exhaust, or other chemical smog at work.
Statistical analyses
Baseline characteristics of participants were summarised across incident lung cancer status as a percentage for categorical variables, mean (standard deviation [SD]) for normally distributed variables, and median (interquartile range) for skewed variables. The association between genetic-risk categories, smoking categories, and the combination of genetic and smoking categories (nine categories with low genetic risk and never-smoking as a reference, 12 categories with low genetic risk and no smoking pack-years as a reference) and incident lung cancer were explored using multivariable Cox proportional hazard models. The assumption for proportional hazards was evaluated by tests based on Schoenfeld residuals [28]; violation of this assumption was not observed in our analyses. The area under the curve (AUC) of receiver operating characteristic (ROC) curves was used to assess each model’s predictive ability, including PRS, smoking, and the combination. The associations between PRS and incident lung cancer were evaluated on a continuous scale with restricted cubic spline curves based on multivariable Cox proportional hazards models. Moreover, interactions between polygenic risk scores and smoking status or pack-years were tested. The population-attributable fractions (PAFs), which estimate the proportion of events that would have been prevented if all individuals had been in the never-smoking category, were calculated [29]. The distribution of smoking status in the Health Survey for England (HSE) [30] and European Prospective Investigation into Cancer and Nutrition (EPIC) [31] with better representation to England and the European population were included in the analysis to deal with the incomplete representation of the UK Biobank [32].
Several sensitivity analyses were conducted to verify the robustness of the results. The risk of incident lung cancer was analysed using genetic-risk quintiles and pack-years of smoking in more subdivided groups. The association was also adjusted for self-reported and hospital diagnosed chronic obstructive pulmonary disease (COPD) and chronic pulmonary infections (definitions in eTable 3) at baseline, which may be important confounding factors [33, 34]. The sensitivity analysis excluded participants who had third-degree or higher relatedness to further reduce non-random distribution of risk genes, developed outcomes within the first two years of follow-up to avoid reverse causality, and had a mismatch between calculation and self-reported never-smoking. Moreover, stratified analyses were performed to estimate potential modification effects according to sex (female or male), age (<60 or ≥60 years). Analyses were undertaken using R v3.6.1 (R Center for Statistical Computing, Vienna, Austria). P value < 0.05 (two-sided) was considered significant.
Results
Participants characteristics
A total of 345,794 European individuals with a complete genotype and phenotype were included in the analysis of incident lung cancer, and their detailed information is shown in Fig. 1. Their mean (SD) age was 56.3 (8.0) years, and 186,330 (53.9%) were female. The PRS was normally distributed among all participants (eFigure 1 in Supplement). There were 90,727 (26.2%) former smokers and 33,994 (9.8%) current smokers, among which 40,889 (11.8%) individuals had intermediate smoking exposure (20–39.9 pack-years) and 19,027 (5.5%) individuals had heavy smoking exposure (≥40 pack-years). The participant characteristics are provided in Table 1.

BMI body mass index, TDI Townsend deprivation index.
Over 2,454,915 person-years of follow-up (median [interquartile range] length of follow-up, 7.2 [6.5–7.8] years), there were 1687 cases of incident lung cancer. Participants who developed incident lung cancer were slightly older, more likely to be male, had more smoking exposure, had less physical activity, and had an unhealthy diet. Meanwhile, they also had higher genetic risks.
Associations of genetic risk with incident lung cancer
With the increase in genetic risk, the incidence rate and hazard ratio (HR) of lung cancer gradually increased. After additional adjustment for smoking status or pack-years, the HRs of the high genetic-risk group were 1.73 (95% confidence interval [CI], 1.48–2.02) and 1.69 (95% CI, 1.44–1.97) compared with the low genetic-risk group, and the HRs per SD of PRS increase were 1.16 (95% CI, 1.11–1.22) and 1.16 (95% CI, 1.10–1.21). This result was almost the same as before the adjustment (Table 2). When genetic-risk quintiles were used instead of categories, the same results trend was observed (eTable 4 in Supplement). Figure 2a shows the cumulative risk of incident lung cancer in each genetic-risk group during follow-up.

Cumulative risk of incident lung cancer during follow-up according to genetic risk (a), smoking status (b) and smoking pack-years (c).
Associations of smoking with incident lung cancer
With the changing smoking status and increasing pack-years, the incidence and HR of lung cancer were also increased. After additional adjustment for PRS, the HRs of the current or heavy smoking group were 14.54 (95% CI, 12.47–16.94) and 17.80 (95% CI, 15.23–20.81), respectively, compared with the never-smoking group. This result was almost the same as before the adjustment (Table 3). When the number of smoking pack-years was given in more subdivided categories, the same trend of results was observed (eTable 5 in Supplement). Figure 2b and c shows the cumulative risk of incident lung cancer in each smoking status and pack-year group during follow-up.
Associations of smoking and genetic risk with incident lung cancer
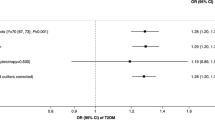
In each genetic-risk group, the incidence and HR of lung cancer increased with the smoking status deteriorating and pack-years increasing. Compared with the low genetic risk and never-smoking group, there was no significant difference of incident lung cancer risk in the high genetic risk but never-smoking group, while the HR of the low genetic risk but the current smoking group was 11.31 (95% CI, 7.84–16.33). A similar pattern was observed among genetic risk and smoking pack-year groups. The highest risks were observed among individuals with high genetic risk and current smoking (HR, 22.46 [95% CI, 15.99–31.53]) compared with low genetic risk and never-smoking. Individuals with high genetic risk and heavy smoking had a much higher risk of incident lung cancer (HR, 27.02 [95% CI, 19.28–37.88]) compared with those with low genetic risk and no smoking (Fig. 3). There was no significant interaction between the PRS and the smoking status or pack-years (both P for interaction > 0.05).

Risk of incident lung cancer according to genetic risk and smoking status (a) or genetic risk and smoking pack-years (b). The vertical line indicates the reference value of 1.
Further analyses stratified by genetic-risk category showed that the association between smoking and lung cancer appeared to increase with increasing genetic risk (Table 4). In the low, intermediate and high genetic-risk groups, the HRs of current smoking were 10.75 (95% CI, 7.28–15.88), 14.86 (95% CI, 12.22–18.07), and 16.85 (95% CI, 12.25–23.19), respectively, compared with never-smoking. Similarly, the HRs of heavy smoking were 16.22 (10.97–23.97), 17.06 (13.97–20.84) and 21.22 (15.34–29.35) compared with no smoking.
The same pattern of associations was observed in a series of sensitivity analyses with additional adjustment for COPD and chronic pulmonary infections, excluding participants who had third-degree or higher relatedness, excluding participants who developed outcomes within two years of baseline, and those who had a mismatch between calculation and self-reported never-smoking. (eTables 6 and 7 in the Supplement). Stratified analyses were performed by age and sex (eTables 8 and 9 in the Supplement), but the results were not markedly different among male and female or the <60 years and ≥60 years groups.
Population-attributable fractions
Since there was no significant interaction between PRS and smoking, the population-attributable fractions were calculated regardless of genetic risk. If all individuals had never smoked, 76.4% (95% CI, 73.4–79.2, based on smoking status) to 75.3% (95% CI, 72.0–78.2, based on smoking pack-years) new-onset lung cancer events might have been prevented during follow-up. If all current smokers quit smoking and the former smokers remained, the new-onset events might have been reduced by 26.4% (95% CI, 25.8–27.0). Further analyses stratified by genetic-risk category showed that 73.4% (95% CI, 64.5–80.4), 76.1% (95% CI, 72.2–79.6), and 79.1% (95% CI, 73.0–83.9) of incident lung cancer cases were attributed to smoking among the low, intermediate and high genetic-risk populations. When the smoking status proportional in HSE and EPIC were included, the PAFs of smoking were 83.2% (95% CI, 80.9–85.3) and 85.1% (95% CI, 83.1–87.0), respectively (eTable 10 in the Supplement).
Discussion
In this large population-based prospective cohort study of more than 345,000 European individuals, high genetic risk and smoking status were independently associated with an increased risk of incident lung cancer events. Among never-smokers, there was no significant difference in the incident risk between each genetic group. The high genetic risk was two-fold higher than that of low genetic risk for current smokers. A similar pattern was observed for genetic risk and smoking pack-year groups. Meanwhile, there was no significant interaction between the PRS and smoking status or pack-years for incident lung cancer, and smoking cessation or reduction can provide similar protection against lung cancer regardless of genetic risk. The PAF analysis hypothesised that ~76% of new-onset lung cancer events might have been prevented if all individuals had never smoked.
To our knowledge, this study is by far the most extensive and fully adjusted prospective study of lung cancer incidence treating smoking as a single modifiable factor and incorporating multiple genetic-risk factors. Many common variants with minor effects have been identified as associated with a high risk of lung cancer, and the PRS can indicate their combined impact. Previous studies used 19 SNPs to construct a PRS for non-small cell lung cancer and showed predictive effects in a prospective study of 95,408 individuals [9]. Compared with this previous study, the present study included a larger sample size and more SNPs to increase the power for risk estimation. Meanwhile, we used the upper and lower quintiles to categorise the high and low genetic-risk groups [35, 36], which may reduce the accuracy for the high genetic-risk group but warn a broader population that they need to carry out PRS-informed disease screening or life planning for life-threatening lung cancer. It also ensured that the comparison between the combined smoking and genetic-risk subgroups had sufficient statistical power.
Compared with another study based on the UK Biobank [37], the current PRS contains fewer highly independent SNPs in each locus to avoid overinflation of the GWAS summary results caused by many linkage disequilibrium SNPs. Therefore, this PRS may have better generalisations in other populations [38]. The current results showed similar HRs after adjusting for confounding factors (economic and social background, lifestyle factors, occupational exposure). Compared with case-control studies [39, 40], prospective studies may lose some statistical power, but estimates of the absolute risk support using the PRS to predict incident lung cancer [10, 41]. Regarding the role of PRS in never-smokers, our results suggest that their incident risk did not achieve statistical significance as the PRS group increased. Among never-smokers, the post hoc study powers for incident lung cancer in those with intermediate and high genetic risk were only 0.243–0.293. Therefore, we speculate that more outcome events may bring different results with the extension of follow-up time. To sum up, we believe that PRS could be a powerful tool for lung cancer risk assessment as it provides additional information independent of smoking and combining it with traditional risk factors could contribute to a better prediction of lung cancer.
We observed a strong association between smoking and incident lung cancer, independent of genetic risk, and the increased risk was much greater than the genetic risk. This means that smoking will significantly offset low genetic-risk benefits, consistent with a previous study [9]. However, we followed the same grouping method and found that the risk values were much more significant than those in a previous study (eTable 11 in the Supplement). Sample size, confounding factors, subtle differences in smoking habits, and outcome data sources may be the reasons for the differences. We observed similar associations between smoking and lung cancer with other relevant studies [42, 43]. Based on a study of the contemporary population, although smoking, a long-recognised risk factor has undergone tremendous changes in production, composition and use method [44], it still plays a decisive role in lung cancer occurrence. Therefore, smoking cessation is still the most significant and cost-effective way to prevent lung cancer.
Previous studies believed that smoking was responsible for 80%~90% of lung cancer [2, 43, 45], and a study showed that 63.6% of lung cancer are attributable to comprehensive modifiable factors, including smoking and air pollution [37]. We found that the entire population would avoid 76.4% of lung cancer cases by becoming never-smokers. The slight reduction in this proportion is probably because of the reduction in smoking prevalence (23.3% of individuals were current smokers in The European Prospective Investigation into Cancer and Nutrition cohort [43]), manifesting the achievement of tobacco use control. In addition, differences in sample, methodology, and confounders’ representativeness also contribute to the different PAFs between studies. Furthermore, we also estimated the attribution of smoking by a more natural form of PAFs called the generalised impact fraction [46]. Our results showed that if all current smokers stop smoking and former smokers remain, the expected reduction in lung cancer cases would be 26%, again highlighting the efficiency of smoking cessation.
GWASs have shown that a locus may be simultaneously associated with smoking preference and lung cancer [12, 47, 48]. The interaction between smoking and genetic risk for lung cancer is a topic worth discussing, as it may help explain some of the missing heritability in lung cancer susceptibility [49]. Variants at the 15q25 locus have been confirmed by several studies associated with increased tobacco addiction and lung cancer risk [47, 48], but a significant gene-environment interaction is controversial [50, 51]. Some studies suggested that there were significant gene-smoking interactions at 10q25 [52], 14q22, 15q22 [53] and 19q13 [54]. In this study, there was no significant PRS-smoking interaction for lung cancer. This may be because the combination of multiple loci may mask the potential interaction, and the model selection and the specific definition of smoking habits may also affect the results. Besides, the number of positive cases observed in this cohort was far less than in large-scale GWASs, so there may be insufficient statistical power. However, based on the analysis of adjusting for extensive potential confounding factors and using the two smoking measures, we still believe that PRS and smoking promote lung cancer independently.
Strengths and limitations
This study has several strengths. Many participants from the UK Biobank study provided complete exposure information, and the extensive phenotype information provided many covariates that could be adjusted in the model to eliminate potential confounders. A more detailed grouping of lifetime tobacco exposure showed a typical dose-response relationship. Furthermore, the study population was utterly independent of previous GWASs that identified the risk loci and their effect sizes, which avoided overfitting to some extent.
Several limitations also need to be considered. First, the analysis was conducted on overall lung cancer without constructing PRS and assessing their effects for more detailed lung cancer classifications, which may mask their heterogeneity. Second, additional variants or genetic patterns associated with lung cancer are likely to be identified in the future, which may refine estimates of genetic risk. Third, PRS based on GWASs of European ancestry may limit its application in a larger population due to the differences in risk alleles, allele frequency, and the effect sizes of risk alleles. Fourth, smoking behaviours were self-reported and may have recall and misclassification bias, and there may be differences in the distribution of individuals excluded due to lacking smoking information. Fifth, smoking was not randomly assigned. Although analyses were adjusted for several covariates and sensitivity analyses, the possibility of unmeasured confounding remained. Sixth, the current study included 936 (0.27%) participants with inconsistent information on never-smoking and 0 pack-years of smoking. This may be due to the difference between the self-reported state and participants’ calculated state with minimal smoking exposure. Although we excluded these people in the sensitivity analysis, there may still be potential inconsistencies. Finally, the potential “healthy volunteer” selection bias in the UK biobank may be accompanied by a lower proportion of the smoking population and underestimated PAF. A mild increase in PAF was found using representative England and European population structures.
Conclusion
In conclusion, high genetic risk and smoking were independently associated with higher lung cancer risk, and there were no interactions between these risk factors. Polygenic risk assessment can provide important information beyond a variety of environmental exposures. This study provided new insights to quantitatively evaluate the role of smoking and genetics in lung cancer.
Data availability
The dataset supporting the conclusions of this article is available in the UK Biobank upon request (https://www.ukbiobank.ac.uk/).
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Tyczynski JE, Bray F, Parkin DM. Lung cancer in Europe in 2000: epidemiology, prevention, and early detection. Lancet Oncol. 2003;4:45–55.
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl J Med. 2000;343:78–85.
Dai J, Shen W, Wen W, Chang J, Wang T, Chen H, et al. Estimation of heritability for nine common cancers using data from genome-wide association studies in Chinese population. Int J Cancer. 2017;140:329–36.
Sampson JN, Wheeler WA, Yeager M, Panagiotou O, Wang Z, Berndt SI, et al. Analysis of heritability and shared heritability based on genome-wide association studies for thirteen cancer types. J Natl Cancer Inst. 2015;107:djv279.
Bossé Y, Amos CI. A decade of GWAS results in lung cancer. Cancer Epidemiol Biomark Prev. 2018;27:363–79.
Fehringer G, Kraft P, Pharoah PD, Eeles RA, Chatterjee N, Schumacher FR, et al. Cross-cancer genome-wide analysis of lung, ovary, breast, prostate, and colorectal cancer reveals novel pleiotropic associations. Cancer Res. 2016;76:5103–14.
McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49:1126–32.
Dai J, Lv J, Zhu M, Wang Y, Qin N, Ma H, et al. Identification of risk loci and a polygenic risk score for lung cancer: a large-scale prospective cohort study in Chinese populations. Lancet Respir Med. 2019;7:881–91.
Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19:581–90.
Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42:436–40.
Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–42.
Zhang YD, Hurson AN, Zhang H, Choudhury PP, Easton DF, Milne RL, et al. Assessment of polygenic architecture and risk prediction based on common variants across fourteen cancers. Nat Commun. 2020;11:3353.
Kiyohara C, Horiuchi T, Takayama K, Nakanishi Y. IL1B rs1143634 polymorphism, cigarette smoking, alcohol use, and lung cancer risk in a Japanese population. J Thorac Oncol. 2010;5:299–304.
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–d1012.
Broderick P, Wang Y, Vijayakrishnan J, Matakidou A, Spitz MR, Eisen T, et al. Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res. 2009;69:6633–41.
Wang M, Liu H, Liu Z, Yi X, Bickeboller H, Hung RJ, et al. Genetic variant in DNA repair gene GTF2H4 is associated with lung cancer risk: a large-scale analysis of six published GWAS datasets in the TRICL consortium. Carcinogenesis. 2016;37:888–96.
Poirier JG, Brennan P, McKay JD, Spitz MR, Bickeböller H, Risch A, et al. Informed genome-wide association analysis with family history as a secondary phenotype identifies novel loci of lung cancer. Genet Epidemiol. 2015;39:197–206.
Dong J, Hu Z, Wu C, Guo H, Zhou B, Lv J, et al. Association analyses identify multiple new lung cancer susceptibility loci and their interactions with smoking in the Chinese population. Nat Genet. 2012;44:895–9.
Shiraishi K, Kunitoh H, Daigo Y, Takahashi A, Goto K, Sakamoto H, et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat Genet. 2012;44:900–3.
Wang Y, McKay JD, Rafnar T, Wang Z, Timofeeva MN, Broderick P, et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet. 2014;46:736–41.
Townsend P. Deprivation. J Soc Policy. 1987;16:125–46.
Craig CL, Marshall AL, Sjöström M, Bauman AE, Booth ML, Ainsworth BE, et al. International physical activity questionnaire: 12-country reliability and validity. Med Sci Sports Exerc. 2003;35:1381–95.
Ali Mohsenpour M, Fallah-Moshkani R, Ghiasvand R, Khosravi-Boroujeni H, Mehdi Ahmadi S, Brauer P, et al. Adherence to dietary approaches to stop hypertension (DASH)-style diet and the risk of cancer: a systematic review and meta-analysis of cohort studies. J Am Coll Nutr. 2019;38:513–25.
Appel LJ, Moore TJ, Obarzanek E, Vollmer WM, Svetkey LP, Sacks FM, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N. Engl J Med. 1997;336:1117–24.
U.S. Department of Health and Human Services and U.S. Department of Agriculture. 2015-2020 Dietary Guidelines for Americans. 8th Edition. 2015. https://health.gov/our-work/food-nutrition/previous-dietary-guidelines/2015.
Schoenfeld D. Partial residuals for the proportional hazards regression model. Biometrika. 1982;69:239–41.
Knudsen TB, Thomsen SF, Nolte H, Backer V. A population-based clinical study of allergic and non-allergic asthma. J Asthma. 2009;46:91–94.
NHS. Health Survey for England—2010, Trend tables 2011, https://digital.nhs.uk/data-and-information/publications/statistical/health-survey-for-england/health-survey-for-england-2010-trend-tables.
Riboli E, Kaaks R. The EPIC project: rationale and study design. European Prospective Investigation into Cancer and Nutrition. Int J Epidemiol. 1997;26:S6–14.
Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, et al. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am J Epidemiol. 2017;186:1026–34.
Tockman MS, Anthonisen NR, Wright EC, Donithan MG. Airways obstruction and the risk for lung cancer. Ann Intern Med. 1987;106:512–8.
Schottenfeld D, Beebe-Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. CA Cancer J Clin. 2006;56:69–83.
Lourida I, Hannon E, Littlejohns TJ, Langa KM, Hyppönen E, Kuzma E, et al. Association of lifestyle and genetic risk with incidence of dementia. J Am Med Assoc. 2019;322:430–7.
Said MA, Verweij N, van der Harst P. Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK biobank study. JAMA Cardiol. 2018;3:693–702.
Kachuri L, Graff RE, Smith-Byrne K, Meyers TJ, Rashkin SR, Ziv E, et al. Pan-cancer analysis demonstrates that integrating polygenic risk scores with modifiable risk factors improves risk prediction. Nat Commun. 2020;11:6084.
Choi SW, Mak TS, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15:2759–72.
Qian DC, Han Y, Byun J, Shin HR, Hung RJ, McLaughlin JR, et al. A novel pathway-based approach improves lung cancer risk prediction using germline genetic variations. Cancer Epidemiol Biomark Prev. 2016;25:1208–15.
Weissfeld JL, Lin Y, Lin HM, Kurland BF, Wilson DO, Fuhrman CR, et al. Lung cancer risk prediction using common SNPs located in GWAS-identified susceptibility regions. J Thorac Oncol. 2015;10:1538–45.
Hung RJ, Warkentin MT, Brhane Y, Chatterjee N, Christiani DC, Landi MT, et al. Assessing lung cancer absolute risk trajectory based on a polygenic risk model. Cancer Res. 2021. https://doi.org/10.1158/0008-5472.Can-20-1237.
Tindle HA, Stevenson Duncan M, Greevy RA, Vasan RS, Kundu S, Massion PP, et al. Lifetime smoking history and risk of lung cancer: results from the Framingham Heart Study. J Natl Cancer Inst. 2018;110:1201–7.
Agudo A, Bonet C, Travier N, González CA, Vineis P, Bueno-de-Mesquita HB, et al. Impact of cigarette smoking on cancer risk in the European prospective investigation into cancer and nutrition study. J Clin Oncol. 2012;30:4550–7.
U.S. Department of Health and Human Services. The health consequences of smoking: 50 years of progress. A report of the surgeon general. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2014. p. 151–54.
U.S. Department of Health and Human Services. The health consequences of smoking: a report of the surgeon general. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2004. p. 42–61.
Drescher K, Becher H. Estimating the generalized impact fraction from case-control data. Biometrics. 1997;53:1170–6.
Saccone NL, Culverhouse RC, Schwantes-An TH, Cannon DS, Chen X, Cichon S et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet. 2010;6: e1001053.
Lips EH, Gaborieau V, McKay JD, Chabrier A, Hung RJ, Boffetta P, et al. Association between a 15q25 gene variant, smoking quantity and tobacco-related cancers among 17 000 individuals. Int J Epidemiol. 2010;39:563–77.
Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21.
VanderWeele TJ, Asomaning K, Tchetgen Tchetgen EJ, Han Y, Spitz MR, Shete S, et al. Genetic variants on 15q25.1, smoking, and lung cancer: an assessment of mediation and interaction. Am J Epidemiol. 2012;175:1013–20.
David SP, Wang A, Kapphahn K, Hedlin H, Desai M, Henderson M, et al. Gene by environment investigation of incident lung cancer risk in African-Americans. EBioMedicine. 2016;4:153–61.
Li Y, Xiao X, Han Y, Gorlova O, Qian D, Leighl N, et al. Genome-wide interaction study of smoking behavior and non-small cell lung cancer risk in Caucasian population. Carcinogenesis. 2018;39:336–46.
Zhang R, Chu M, Zhao Y, Wu C, Guo H, Shi Y, et al. A genome-wide gene-environment interaction analysis for tobacco smoke and lung cancer susceptibility. Carcinogenesis. 2014;35:1528–35.
Zhou W, Liu G, Miller DP, Thurston SW, Xu LL, Wain JC, et al. Gene-environment interaction for the ERCC2 polymorphisms and cumulative cigarette smoking exposure in lung cancer. Cancer Res. 2002;62:1377–81.
Acknowledgements
We are grateful to UK Biobank participants. This research has been conducted using the UK Biobank resource (https://www.ukbiobank.ac.uk) under application number 43795.
Funding
This work was supported by the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2019), the National Natural Science Foundation of China (82103931 and 82003443), the Guangzhou Science and Technology Project (202002030255), and Young Elite Scientists Sponsorship Program by CAST (2019QNRC001). The funders had no role in the study design or implementation; data collection, management, analysis or interpretation; manuscript preparation, review or approval; or the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
Prof. Mao had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. PDZ and CM contributed to the study design and supervised the whole project. PDZ, ZHL, PLC, AZ and CM contributed to the data interpretation, data analysis, and manuscript writing. CM, PDZ, PLC, XRZ, YJZ and DL contributed to the data curation and funding acquisition. PDZ and PLC contributed equally to this work. All the authors reviewed or revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The UK Biobank received ethical approval from the research ethics committee (REC reference for UK Biobank 11/ NW/0382), and participants provided written informed consent. Any additional ethical approval was adjudged unnecessary for the present study.
Consent to publish
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, P., Chen, PL., Li, ZH. et al. Association of smoking and polygenic risk with the incidence of lung cancer: a prospective cohort study. Br J Cancer 126, 1637–1646 (2022). https://doi.org/10.1038/s41416-022-01736-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-01736-3
This article is cited by
-
Bioelectrical impedance analysis—derived phase angle (PhA) in lung cancer patients: a systematic review
BMC Cancer (2024)
-
Tobacco-induced hyperglycemia promotes lung cancer progression via cancer cell-macrophage interaction through paracrine IGF2/IR/NPM1-driven PD-L1 expression
Nature Communications (2024)
-
Health effects associated with smoking: a Burden of Proof study
Nature Medicine (2022)
