
Abstract
Genome-wide association studies coupled with large-scale replication and fine-scale mapping studies have identified more than 150 genomic regions that are associated with breast cancer risk. Here, we review efforts to translate these findings into a greater understanding of disease mechanism. Our review comes in the context of a recently published fine-scale mapping analysis of these regions, which reported 352 independent signals and a total of 13,367 credible causal variants. The vast majority of credible causal variants map to noncoding DNA, implicating regulation of gene expression as the mechanism by which functional variants influence risk. Accordingly, we review methods for defining candidate-regulatory sequences, methods for identifying putative target genes and methods for linking candidate-regulatory sequences to putative target genes. We provide a summary of available data resources and identify gaps in these resources. We conclude that while much work has been done, there is still much to do. There are, however, grounds for optimism; combining statistical data from fine-scale mapping with functional data that are more representative of the normal “at risk” breast, generated using new technologies, should lead to a greater understanding of the mechanisms that influence an individual woman’s risk of breast cancer.
Similar content being viewed by others


Background
Genome-wide association studies (GWAS, Table 1) coupled with large-scale replication and fine-scale mapping studies (Table 1) have led to the identification of more than 150 genomic regions that are associated with breast cancer risk [1,2,3,4,5,6]. Efforts to translate these findings to improve the lives of women who are at risk of developing breast cancer are focused on two main areas: risk prediction and a greater understanding of disease mechanism. The subject of this review is the latter.
Translating GWAS findings into a greater understanding of the mechanisms that influence an individual’s risk of breast cancer, requires the identification of functional variants (as opposed to correlated variants, Table 1) and the targets of these functional variants (the genes or non-coding RNAs that mediate the associations observed in GWAS). The output from a GWAS is an association signal between a marker single-nucleotide polymorphism (SNP, Table 1) and outcome; in short, a GWAS provides evidence that a particular region of the genome is associated with breast cancer risk but little or no information regarding the mechanism behind this association. The vast majority of GWAS signals map to non-protein-coding regions and are thought to influence transcriptional regulation [7, 8]. With a few exceptions (e.g., splice donor and acceptor sites, Table 1), our ability to predict the likely impact of non-coding variation is extremely limited. To compound this difficulty, marker SNPs are specifically selected to capture common variation at a given region of the genome, and by definition, therefore, will be highly correlated with many other variants, any of which might have a functional association with breast cancer risk. Identifying “targets”, that is, the genes or non-coding RNAs that mediate the associations observed in GWAS, also provides challenges. Regulatory elements can influence the expression of multiple genes, they can “skip over” nearby genes and can act over distances of at least 1 Mb [9, 10]. Indeed, many breast cancer GWAS signals map to gene deserts with the nearest-known protein-coding genes mapping several hundred kilobases (kb) away.
The Breast Cancer Association Consortium (BCAC, http://bcac.ccge.medschl.cam.ac.uk/) was set up as a multidisciplinary consortium of investigators, who are interested in investigating the inherited risk of breast cancer. Led by scientists at the University of Cambridge, BCAC published the first breast cancer GWAS [11] and have led a series of locus-specific follow-up studies [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29]. Most recently, BCAC carried out a fine-scale mapping analysis of 152 breast cancer risk regions in 109,900 breast cancer cases and 88,937 controls of European ancestry [5]. Using multivariate logistic regression, they confirmed associations for 150 of the 152 regions (P < 1 × 10−4); to define independent signals within each region, they used stepwise multinomial logistic regression, deriving the association of each variant, conditional on the more significant ones, in order of statistical significance. This analysis reported 352 independent signals, 196 for which the evidence for association was strong (conditional P < 10−6) and 156 for which the evidence was more modest (10−6 < conditional P < 10−4), with a range of one (n = 70) to nine (n = 2) independent signals per region. Finally, they defined credible causal variants (CCVs, Table 1) within each signal as variants with conditional P values within two orders of magnitude of the index variant [30]. The number of CCVs per signal ranged from one (n = 66) to 375, resulting in a total of 13,367 CCVs for follow-up analysis. Overall, these data provide some insight into the scale of the task required if we are to identify the truly functional variants from amongst the plethora of correlated variants, link these variants to their target genes and understand how the temporal, cell-type-specific response stimulated expression of these genes that influence breast cancer risk. Over the past 10 years, however, tools for the annotation of CCVs and the selection of target genes have been developed and publicly funded resources, such as the Encyclopedia of DNA Elements [31] (ENCODE, https://www.encodeproject.org/ Table 2) and Roadmap Epigenomics project (http://www.roadmapepigenomics.org/; Table 2), have coordinated efforts to generate genome-wide datasets for a wide range of the assay and cell types and made these rapidly publicly available, in an effort to facilitate the interpretation of GWAS signals.
In this review, we focus first on methods for selecting a subset of CCVs for which there is evidence of functionality; we have summarised high-throughput methods for identifying candidate-regulatory sequences (CRS) and then testing them using a functional output. We next describe techniques for prioritising putative target genes and finally methods for linking CCVs to those target genes. Where possible, we have reviewed these tools specifically in the context of breast cancer GWAS; where these tools have not yet been applied to breast cancer GWAS, we have illustrated their potential in the context of other disease outcomes or non-disease phenotypes.
Defining candidate-regulatory sequences (CRS)
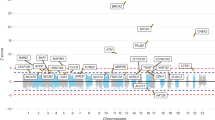
On the assumption that both the promoters (Table 1) and the more distal elements that regulate target genes (in particular enhancers, Table 1) will be active in one or more of the cell types that comprise breast tissue, a first step in the process of prioritising CCVs for follow-up studies is aligning the CCVs with markers of open chromatin (DNase-seq, ATAC-seq and FAIRE-seq, Table 1), active histone modifications (particularly H3K27ac, H3K4me1 and H3K4me3, Table 1) and transcription-factor- (TF, Table 1) binding sites generated in these cell types (Table 3). ENCODE, with the aim of building a comprehensive list of “regulatory elements that control cells and circumstances in which a gene is active” (https://www.encodeproject.org/), provides a rich source of these data for download and/or browsing through web portals such as the WashU Epigenome Browser (https://epigenomegateway.wustl.edu/; Table 2), the UCSC genome browser (https://genome.ucsc.edu/ Table 2) or Ensemble (https://www.ensembl.org/ Table 2). In addition to providing these datasets individually, ENCODE has generated a Chromatin State Segmentation by Hidden Markov Model (ChromHMM) function, which integrates ChIP-seq data for eight histone modifications and CTCF (Table 1) binding, to predict 15 chromatin states. Data generated in breast-relevant cell lines, tissue and primary cells are summarised in Fig. 1a and Supplementary Table 1. The majority of datasets and data types have been generated in MCF-7 cells (N = 267 out of a total 468 datasets), the most widely used cell-line model for oestrogen-receptor-positive breast cancer. In particular, there are ChIP-seq data for 117 TFs, including the three TFs that “define” the ER+ transcriptome (ESR1, FOXA1 and GATA3, Table 1) [32,33,34,35] generated, for example, in the presence and absence of oestradiol. By contrast, the Roadmap Epigenomics project [36] uses primary ex vivo tissues to generate normal epigenomes, these are arguably more relevant for analyses of breast cancer risk (see the eQTL section below), but the range of data types is, inevitably, more limited (Fig. 1b and Supplementary Table 1). While ENCODE and Roadmap Epigenomics are arguably the most comprehensive and widely used resources, other consortia-based resources using standardised sample preparation and assay protocols exist (summarised in ref. [37] and Table 2) and, now that data deposition is often a condition for publication, resources such as Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/; Table 2) provide access to many additional (non-standardised) datasets.

Datasets that are available through (a) ENCODE and (b) Roadmap Epigenomics are summarised as bar plots. Different data types are colour-coded as indicated in the keys. The cell or tissue types in which the data were generated are shown on the x axis with the number of datasets available in each of these cell or tissue types on the y axis.
These data are valuable resources that have already been widely used to define CRS and prioritise CCVs for follow-up studies (see locus-specific annotation studies below). Given that risk reflects early events that precede the somatic genome, the predominance of data generated in a breast cancer cell line (MCF-7) is potentially limiting progress in this area; in this context, Fachal et al. reported that 73% of active enhancer regions (as defined by ENCODE ChromHMM) that overlapped ER+ CCVs in MCF-7 cells were not active in normal human mammary epithelial cells [5]. Two additional rich resources have recently been reported; using genomic DNase-I footprinting integrated with TF-recognition sequences, condensed onto a common sequence axis for closely related TF family members, Vierstra et al. have generated a high-resolution genome-wide consensus TF-footprint index in 243 human cell and tissue types, including a breast cancer cell line (T-47D), a normal mammary epithelial cell line (MCF10a) and normal mammary fibroblasts [38]. Contemporaneously, Domcke et al. developed single-cell ATAC-seq (sci-ATAC-seq3), which they combined with single-cell gene expression data across a broad range of human foetal tissues, to create an atlas of linked cell-type-specific enhancers and genes that have the potential to inform our understanding of cell-fate specification and maintenance in normal tissue [39]. These data, which can be accessed through the ENCODE portal and the Descartes website, respectively (Table 2), have the potential to transform our ability to define CRS and evaluate CCVs.
Functional outputs for validating candidate-regulatory sequences
Markers of open chromatin, active histone modifications, TF binding and/or chromatin-interaction peaks (Table 3) have been used to map millions of sites with regulatory potential across the genome [31, 40]. The proportion of these predicted elements that truly function as, for example, enhancer elements, is not known, but estimates range from 12% to up to 90% [41,42,43,44]; clearly, functional validation is required. High-throughput methods for functional validation can assay expression of experimental constructs (massively parallel reporter gene assays (MPRA [45, 46]), self-transcribing active regulatory region sequencing (STARR-Seq [47])) or expression of nascent transcripts (eRNAs) from the predicted enhancer element in a “normal” genomic context (GRO-Seq [48], Table 3).
The classic method for assaying enhancer activity is the reporter gene assay [45]. Briefly, a candidate- regulatory sequence (CRS) is cloned into a reporter construct comprising a minimal promoter and a reporter gene with a quantifiable output such as green fluorescent protein (GFP), β-galactosidase (LacZ) or luciferase. In the context of breast cancer GWAS annotation, the reporter construct(s) are then transfected into a breast cancer cell line and reporter activity is assayed to determine whether the CRS enhances transcription and whether this activity is allele-specific. MPRA (46) and STARR-seq [47] were both developed to “high-throughput” reporter gene assays allowing several thousand putative CRS to be tested for enhancer activity simultaneously (Table 3). Both methods use RNA expression driven by the CRS either by pairing it to a transcribed barcode in the 3’ or 5’ UTR (Table 1) of the reporter gene (MPRA), or by using the CRS itself as a barcode (STARR-seq, CapStarr-seq [49]). Lenti-MPRA [50] (Table 3), a lentivirus-based version of MPRA, extends this technology to cell types that are “hard-to-transfect” and as lentiviruses integrate into the genome, produces “in-genome” readouts as opposed to episomal readouts (Table 1). To our knowledge, these high-throughput versions have not yet been used in the context of breast cancer GWAS, but the potential of lenti-MPRA to recapitulate an exquisitely regulated programme of temporal and cell-type-specific gene expression was demonstrated recently using neural induction from human pluripotent stem cells (hPSCs, Table 1) as a paradigm [43].
Non-coding transcription (eRNA, Table 1) is a defining feature of active enhancers [51]; these nascent RNAs can be assayed using high-throughput adaptations of a technique (nuclear run-on assays) that was originally developed to measure rates of transcription [52, 53]. GRO-seq [48], one of these high- throughput adaptations (Table 3), rather than incorporating radionucleotides (as used in the nuclear run- on assays) uses bromodeoxyuridine labelling of nascent RNA transcripts followed by immunoprecipitation using an antibody against bromodeoxyuridine. Subsequent methods (PRO-seq [54], mNET-seq [55], fastGRO-seq [56] and TTchem-seq [57], Table 3) have introduced modifications to this protocol that involve 4-thiouridine labelling, incorporating a biotin tag and/or hydrolysis rather than sonication to fragment the nascent RNAs (Table 3). In the context of breast cancer specifically, Franco et al. generated GRO-seq data in a series of 13 breast cell lines (11 cancer and two immortalised “normal” breast cell lines), and combined these with RNA-seq and ChIP-seq data to investigate whether subtype-specific gene expression programmes control breast cancer pathogenesis [58].
Reporter gene assays have been used to differentiate functional variants from correlated variants in several locus-specific studies (see locus-specific annotation studies below). Lenti-MPRA has several advantages that are likely to render these individual assays obsolete, specifically, by generating high- throughput data that capture the “in genome” activity of several thousand CCVs simultaneously in “hard-to-transfect” primary cells. Comparing GRO-seq with other enhancer marks (open chromatin and active histone modifications), Franco et al. demonstrated that GRO-seq identifies smaller numbers of high- specificity enhancers [58] and recent adaptations to the protocol reduce cell numbers, such that it should be possible to generate these data too, in primary cells [56]. However, without a formal comparison of these data types in the same cell types, and an understanding of ground truth (presumably in the form of extensive well-characterised positive and negative controls), it is not possible to say which methodology performs best in terms of providing a functional readout for bona fide regulatory elements.
Identifying putative target genes
The logical first step to identifying putative target genes is expression of quantitative trait locus (eQTL) analysis (Table 1), i.e., to test for association between genotype of a GWAS-risk SNP (or a correlated variant) and gene expression (generally steady-state levels of mRNA). It has been shown previously that levels of gene expression are genetically determined (reviewed in ref. [59]) and therefore steady-state levels of mRNA can be considered as an intermediate phenotype (Table 1) that potentially mediates a causal association between a regulatory variant and a complex disease such as breast cancer. As such, a statistically significant eQTL with a gene that maps locally to a GWAS signal (generally defined as within 1Mb) in breast tissue provides strong evidence of a causal association between this “target gene” and breast cancer risk. In their integrative eQTL-based analysis of 15 published breast cancer risk loci, Li et al. identified three cis-associations (2q35-IGFBP5, 5q11-SETD9 and 16q12-TOX3, Table 1) and three trans-associations, which they defined as associations with downstream genes for which there was evidence of regulation by a TF mapping locally to the GWAS locus [60] (6q25-ESR1, 9q31-KLF4 and 8q24-MYC).
Normal breast tissue and breast tumours have both been used as sources of gene expression data for eQTL analyses [2, 5, 60,61,62]; while normal tissue is arguably more relevant for analyses of breast cancer risk (reflecting early events that precede the somatic genome), there is greater availability of expression data from breast tumours. Publicly funded datasets that are available to researchers include the Genotype-Tissue Expression project (GTEx [63], https://www.gtexportal.org/home/), The Cancer Genome Atlas (TCGA, https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) METABRIC [64] and the Pan-Cancer Analysis of Whole Genomes (PCAWG [65], https://dcc.icgc.org/pcawg; Table 2). In addition to the germline variation (Table 1) that is being investigated by GWAS, gene expression in the somatic genome (Table 1) can be influenced by copy number (Table 1) and epigenetic changes such as promoter hypermethylation (Table 1); however, methods that adjust for these somatic events have been developed [59, 60], and breast tumours have been widely used in eQTL analyses to identify “target genes” of breast cancer GWAS-risk loci [2, 5, 60,61,62].
Colocalisation analysis provides an extension to individual SNP:eQTL lookup approaches, by using multiple variants and comparing the distribution of summary statistics from both eQTL and GWAS- association signals, colocalisation reduces false-positive associations and provides a greater degree of confidence that an association between a locus, gene expression and disease outcome is causal [66]. Using eQTL data generated in normal breast tissue from 396 individuals (GTEx v.8) and GWAS summary data, Beesley et al. [67] carried out a colocalisation analysis of the BCAC fine-scale mapping breast cancer risk regions [5]. They identified 17 genes at 14 loci at which the GTEx eQTL associations were statistically significant (defined as P < 10−6). For 11 of these genes, the eQTL SNPs colocalised with strong GWAS signals (P < 10−6, based on multinomial logistic-regression analysis) supporting a causal association. However, the extent to which these associations were replicated in TCGA data was limited and levels of orthogonal support varied [67].
Transcriptome-wide association studies (TWAS) further extend the concept of gene expression levels as an intermediate phenotype for both identifying target genes at known GWAS loci and the discovery of novel risk loci [68, 69]. Briefly, eQTL cohorts with gene expression and genotype data are used to develop models of expression variation on a per-gene (as opposed to per-SNP) basis. These models are subsequently used to predict levels of gene expression for individuals in a GWAS cohort, and test for the association between predicted levels of expression and, for example, breast cancer risk. TWAS methodology and tools for implementing this methodology have been published [68, 69] and were reviewed recently [70]. In the context of this current review specifically, breast cancer TWAS using breast tissue, whole blood, adipose tissue and immune cells as the gene expression cohorts has been reported [71,72,73,74,75], all report genes for which genetically regulated expression levels may be associated with breast cancer risk at both novel loci and known GWAS loci. While these analyses have exclusively used breast tissue and/or cell types that would be present in breast stroma (i.e., immune cells and adipocytes), Michaildou et al. [2] carried out a cell-type-specific enrichment analysis of genome-wide SNP heritability and found significant enrichment of active histone modifications in several non-breast- tissue types, including stomach, rectal and colonic mucosa.
Statistical methods that use gene expression and GWAS data to infer “causal tissues”, including colocalisation analysis [66], linkage-disequilibrium score regression applied to specifically expressed genes (LDSC-SEG [76]), driver-tissue estimation by selective expression (DESE [77]) and Composite likelihood-based Covariance regression Network model, (CoCoNet [78]) have been developed. For a comprehensive comparison of statistical approaches for integrating genome-wide datasets for the functional annotation of GWAS loci, the reader is referred to a recent review by Cano-Gamez and Trynka [79]. Briefly, these methods use gene expression data in multiple tissue types to determine whether disease heritability is directly associated with tissue-specific gene expression patterns (DESE [77]), enriched in regions surrounding genes that show high levels of tissue-specific expression (LDSC-SEG [76]) or co-expression in specific tissue types (CoCoNet [78]). Predicated on the assumption that driver genes will be “relatively” highly expressed in the most disease or trait-relevant tissue types, these methods integrate tissue-specific gene expression data with disease or trait-specific GWAS data to infer causal tissues and driver genes.
Breast tissue is heterogeneous; the parenchyma comprises a branched structure of ducts and lobules composed of specialised epithelial cells (an inner layer of luminal cells and an outer layer of myoepithelial cells) surrounded by stroma, connective tissue populated by fibroblasts, myofibroblasts, endothelial cells, adipocytes and immune cells [80]. Given that gene expression is cell-type-specific, not “tissue-specific”, several of the large data series have used microdissection to select out regions of the tumour that predominantly comprise cells of epithelial origin. This approach, however, assumes that the target gene(s) act in a cell-autonomous (Table 1) manner. Alternative in silico approaches to deconvolute cell-type-specific expression profiles have also been developed [81,82,83]. Whilst these have mainly been used to test for the association between clinical covariates and breast cancer prognosis [83, 84], Seo et al. used a deconvolution approach to examine gene expression in normal breast tissue [61]. Specifically, they modelled breast tissue as comprising four different cell types (adipocytes, epithelial, inflammatory and stromal), and identified eQTL associations at published breast cancer GWAS loci in two of these cell types—epithelial and stromal cells [61]. Notably, in their recent fine-mapping analysis of 150 breast cancer risk regions, Fachal et al. reported eQTL associations in normal breast tissue (NHS [85] or METABRIC [64]) at 72 of their fine-mapping regions, several of these stand out as associations with genes that are expressed in fibroblasts [86] or immune cells [87], including FBLN5 (fibroblasts), MEFV (monocytes and neutrophils) and APOC1 (macrophages) [5].
Exome (Table 1) and, more recently, whole-genome sequencing of a large series of matched cancer genes (Table 1) has been conducted for many different site-specific cancers, including breast cancer [65, 88,89,90] (Table 2). Several such genes map to published breast cancer risk loci, including 10q26-FGFR2, 6q25-ESR1 and 5q11-MAP3K1 and are a priori strong candidates for playing a functional role in the association between a GWAS locus and breast cancer risk [65, 88,89,90,91]. Accordingly, several large-scale annotation analyses have prioritised lists of putative target genes by comparing them with lists of somatically mutated cancer genes, both on an ad hoc basis [62] and more comprehensively [2, 5, 92, 93]. While finding agreement between somatically mutated cancer genes and putative target genes at GWAS-risk loci provides reassuring evidence that GWAS “work”, the strength of an unbiased GWAS approach is the potential for discovering novel cancer genes, and as such, it is arguable that the more interesting target genes are those that have not already been shown to be somatically mutated cancer genes.
Linking candidate-regulatory sequences with putative target genes
While the identification of a statistically significant eQTL between a GWAS SNP (or correlated variant) and a gene that maps locally to a GWAS signal provides strong evidence of a causal association, the absence of an eQTL does not preclude a gene from a functional association. Steady-state levels of mRNA will not capture expression during a particular developmental window, in response to an environmental stimulus or in a specific cell type that occurs at a relatively low frequency within the breast [59]. In addition, eQTL analyses alone cannot distinguish between functional variants and correlated variants.
In the first generation of GWAS, a “nearest gene”, a “nearest expressed gene” or even a “nearest plausible gene” approach was often used to infer the target gene(s) and define the locus. For example, the 10q26 breast cancer risk locus was referred to as the FGFR2 locus before Meyer et al. carried out functional studies that implicated regulation of FGFR2 expression through allele-specific binding of E2F1 and FOXA1 as the likely mechanism by which this locus influences risk [21, 94]. Linking potentially functional variants and/or the CRS to which they map, with the genes they regulate, requires consideration of the 3D genome [95]. Physical interactions between cis-acting regulatory elements and transcriptional start sites (TSS, Table 1) can occur over linear distances of ≥1 megabase (Mb), can skip over multiple intervening genes and are not exclusive; on average, each promoter interacts with 3.9 distal regulatory elements and each distal regulatory element interacts with 2.5 promoters [31]. The chromosome-conformation capture (3C, Table 1) family of methods is used to identify long-range interactions based on (3D) chromatin conformation in the cell. Briefly, spatially proximal segments of DNA are covalently linked using formaldehyde cross-linking of chromatin in intact nuclei, this is followed by restriction-enzyme fragmentation, ligation of linked DNA fragments and finally detection and quantification of ligation products. In the original 3C protocol, ligation products were identified one at a time using polymerase chain reaction (PCR) with locus-specific primers (a “one-by-one” approach); by contrast, Hi-C (Table 1) is the “all-by-all” method used to identify chromatin interactions genome-wide [95]. To generate the high-resolution data required for cataloguing interaction peaks at kilobase (or less) resolution, targeted chromatin-interaction methods focussed on GWAS linkage-disequilibrium (Table 1) blocks [62, 96] or annotated promoters [97] have been used (Table 3). We developed region-capture Hi-C (rCHi-C) specifically to identify target genes at three breast cancer-associated gene deserts [96]; we and others have expanded this approach to identify putative target genes at up to 139 independent breast cancer signals [62, 92]. Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET [98]) and HiChIP [10] are chromatin-interaction methods that combine 3C (ChIA-PET, Table 3) or Hi-C (HiChIP, Table 3) with an immunoprecipitation step targeting, for example, the histone modification H3K27ac. To our knowledge, there have been no ChIA-PET or HiChIP studies carried out in breast cancer or “normal” mammary epithelial cells. Chandra et al., however, demonstrated the potential of HiChIP to define functional eQTL associations; combining HiChIP for the histone modification H3K27ac in different types of primary immune cells with eQTL datasets from matched cell types, they identified a subset of “promoter interacting eQTLs” that were associated with cell-type-specific expression of target genes [10].
However, it is arguable that, based on the assays described above, the evidence that associations between CRS (harbouring one or more CCVs) and target gene expression are causal is at best circumstantial; direct evidence would require perturbation of the CRS, resulting in an alteration to levels of expression of the target gene. This type of direct evidence is achievable using CRISPR genome editing (Table 1). In a follow-up analysis of the 11q13 breast cancer risk locus, Betts et al. used CRISPR interference (CRISPRi) to introduce repressive histone modifications at an enhancer element (annotated by the most significant GWAS SNP at this locus) and demonstrated that this resulted in reduced levels of expression of two long noncoding RNAs (CUPID1 and 2) and the presumed target gene CCND1 [22]. We have recently shown that targeting a catalytically inactive Cas9 fused to an activating VPR domain (CRISPRa) to an enhancer element at the 2q35 breast cancer risk locus increases expression of IGFBP5 (mapping ~400 kb distal) but neither of the neighbouring genes IGFBP2 and RPL37A (~460 kb and ~600 kb, respectively) [99]. A genome-wide framework for mapping gene regulation using CRISPRi has been developed; in this approach, using a high multiplicity of infection, random combinations of CRS were perturbed in the erythroleukaemia cell line K562 and expression of target genes (defined as K562-expressed genes within 1Mb of the CRS) was assayed using single-cell RNA-seq [100]. To our knowledge, this type of genome-wide approach has not yet been used in the context of breast cancer GWAS loci.
Demonstrating an association between genotype of a GWAS-risk SNP (or correlated variant) and gene expression arguably still provides the most direct evidence that a gene plays a causal role in influencing disease risk. Statistical methods that consider multiple variants and compare the distribution of summary statistics (rather than individual eQTL:SNP lookups) provide more robust evidence and may contribute to our ability to infer causal tissues. However, these methods, which rely on steady-state levels of mRNA, will not capture expression during a particular developmental window, in response to an environmental stimulus or in a specific cell type. As the costs of single-cell RNA-seq continue to decrease, this may in part be addressed by increasing availability of large single-cell RNA-seq and genotype datasets for future eQTL-type analyses. In our view, chromatin-interaction methods and CRISPR perturbation can still add to, or detract from, the weight of evidence for a given variant influencing a particular “target gene”. There are advantages to CHi-C as a chromatin-interaction method; CHi-C makes no assumptions about the nature of the regulatory interaction and new kit-based methods (https://arimagenomics.com/, https://dovetailgenomics.com/) have the potential to improve resolution and reduce input in terms of numbers of cells. Ultimately identifying target genes and causal variants, robustly, is likely to require multiple data types; the most informative approaches will inevitably vary from locus to locus and depend on the mechanism that links variant, gene and disease risk.
Locus-specific functional annotation studies
Locus-specific functional annotation studies for at least 17 loci (defined for these purposes as chromosomal regions) have been reported by BCAC investigators and collaborators at 1p11.2 [25], 2q33 [27], 2q35 [12, 13, 99], 4q24 [14], 5p15.33 [28], 5p12 [24], 5q11.2 [15], 6q25 [16], 8q24 [17], 9q31.2 [18], 10q21.1 [19], 10q26 [21], 11q13 [23], 12p11 [26], 12q24 [92], 17q22 [20] and 19p13 [29]. These analyses, published predominantly prior to the recent global fine-mapping analysis, begin with locus-specific fine-scale mapping to define independent signals and CCVs. At the vast majority, this has resulted in too many signals and variants for individual functional assays without first prioritising a subset of CCVs by aligning them with regions of open chromatin, active histone modifications and/or TF-binding sites. Similarly, potential target genes (frequently defined as genes that map within 1 or 2Mb of the most significant SNP) tend to be selected on the basis of eQTL analyses and genome-wide chromatin- interaction data (ChIA-PET and/or Hi-C). On this basis alone, some studies have proposed possible target gene(s) and provided lists of variants that warrant further investigation [14, 17, 20, 25, 26]. Other studies have followed up a subset of variants and genes using functional assays and, in some instances, report more robust evidence for a causal variant (or variants), a target gene (or genes) and a mechanism by which the causal variant influences the expression of the target gene to impact breast cancer risk. Target genes include well-documented breast cancer genes (MAP3K1 at 5q11.2 [15], ESR1 at 6q25 [16], FGFR2 at 10q26 [21] and CCND1 at 11q13 [23]), TFs (KLF4 at 9q31.2 [18], NRBF2 at 10q21.2 [19] and TBX3 at 12q24 [92]), a putative tumour suppressor gene (IGFBP5 at 2q35 [12, 13, 99]), a methylcytosine dioxygenase (TET2 at 4q24 [14]) and a ribonucleoprotein polymerase that maintains telomere ends (TERT at 5p12 [24]). The majority of studies propose a mechanism in which allele-specific binding of a TF (or TFs) influences the expression of the target gene; most commonly, it is the allele-specific binding of one of the three factors that define the ER+ transcriptome (ESR1, FOXA1 and GATA3) [33,34,35] that is implicated. There is however an element of self-fulfilling prophecy to this: ESR1, FOXA1 and GATA3 ChIP-seq data in breast-relevant cell types are widely available and inevitably incorporated into the process for prioritising variants for follow-up studies.
While some of these locus-specific studies have provided insight into the mechanisms that influence risk at individual loci, it is clear, given the size of the task, that high-throughput approaches are required. In the global fine-scale mapping analysis recently published by the BCAC, Fachal et al. used two approaches to incorporate genome-wide functional data into their analyses [5]; they used a Bayesian approach (PAINTOR [101]) that combines genetic association, linkage disequilibrium and enriched genomic features to determine variants with high posterior probabilities of being causal (PPs) and then analysed both of these, and the CCVs from their fine-scale mapping by multinomial logistic regression, using their integrated-expression quantitative trait and in silico prediction of GWAS targets (INQUISIT). Inevitably, the range of assays and cell types used to generate the genomic features that are incorporated into PAINTOR, and those upon which INQUISIT predicts target genes, is limited by the available data: of the 811 genomic features incorporated into INQUISIT, 362 (44.6%) were generated in the oestrogen-receptor-positive breast cancer cell line MCF-7, and 191 (23.5%) were histone- modification ChIP-seq data. Overall, they reported 34 signals at 25 regions where there was either a single CCV or a variant for which the posterior probability was >80% (i.e., individual variants with a high a priori probability of being functional) and 191 high-confidence (level-1) target genes mapping to 88 regions. However, there remain multiple statistically indistinguishable CCVs at the majority of signals, multiple regions without high-confidence target genes and the high-confidence genes that have been predicted require validation and further (mechanistic) investigation.
Perspective
Over the last 15 years, GWAS has transformed our understanding of the genetic architecture of common diseases such as breast cancer. To date, however, the findings of breast cancer GWAS have not led to transformative insights into disease mechanism or new approaches to disease prevention and treatment. The recently published fine-scale mapping and functional annotation that was carried out by the BCAC constitutes a major step forward, but also highlights the challenges [5]; with 152 regions, 352 independent signals and 13,367 CCVs to characterise, there is a clear need for broad-scope systematic approaches integrating statistical and functional data. It is also clear, however, that based on the functional data that are currently available, the results of this type of systematic approach (exemplified by PAINTOR and INQUISIT), still fall a long way short of deciphering the mechanism by which each locus influences a woman’s risk of breast cancer. There are clearly some critical gaps in the range of genome-wide functional datasets that are available; there is an abundance of markers that correlate with enhancer marks (histone modification and TF ChiP-seq) but little or no data for the functional validation of these candidate-regulatory sequences (MPRA, eRNAs or CRISPR screens). In addition, the vast majority of data have been generated in a single oestrogen-receptor-positive breast cancer cell line—MCF-7. Regulation of gene expression can be highly specific in terms of timing (both with respect to development and/or a stimulus) and cell type; if it is arguable that normal tissue is more relevant for eQTL analyses of breast cancer risk, it must also be arguable that normal primary cells are more relevant for functional assays. Future efforts to generate breast-relevant functional data may be better focussed on normal primary cells rather than breast cancer cell lines. In addition, as the range of single-cell technologies increases, and the cost of these methods decreases, the opportunities for generating more sophisticated functional data that more accurately reflect the cellular heterogeneity within breast tissue are also opening up [102]. In conclusion, while much work has been done, there is still much to do. There are, however, grounds for optimism; combining statistical data from fine-scale mapping with functional data that are more representative of the normal “at risk” breast, generated using new technologies, should lead to a greater understanding of the mechanisms that influence an individual woman’s risk of breast cancer.
References
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353–61. 61e1-2.
Michailidou K, Lindstrom S, Dennis J, Beesley J, Hui S, Kar S, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92–4.
Milne RL, Kuchenbaecker KB, Michailidou K, Beesley J, Kar S, Lindstrom S, et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet. 2017;49:1767–78.
Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013;45:392–8. 8e1-2.
Fachal L, Aschard H, Beesley J, Barnes DR, Allen J, Kar S, et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat Genet. 2020;52:56–73.
Zhang H, Ahearn TU, Lecarpentier J, Barnes D, Beesley J, Qi G, et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat Genet. 2020;52:572–81.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–7.
Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5.
Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet. 2009;41:359–64.
Chandra V, Bhattacharyya S, Schmiedel BJ, Madrigal A, Gonzalez-Colin C, Fotsing S, et al. Promoter-interacting expression quantitative trait loci are enriched for functional genetic variants. Nat Genet. 2021;53:110–9.
Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–93.
Ghoussaini M, Edwards SL, Michailidou K, Nord S, Cowper-Sal Lari R, Desai K, et al. Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat Commun. 2014;4:4999.
Wyszynski A, Hong CC, Lam K, Michailidou K, Lytle C, Yao S, et al. An intergenic risk locus containing an enhancer deletion in 2q35 modulates breast cancer risk by deregulating IGFBP5 expression. Hum Mol Genet. 2016;25:3863–76.
Guo X, Long J, Zeng C, Michailidou K, Ghoussaini M, Bolla MK, et al. Fine-scale mapping of the 4q24 locus identifies two independent loci associated with breast cancer risk. Cancer Epidemiol Biomark Prev. 2015;24:1680–91.
Glubb DM, Maranian MJ, Michailidou K, Pooley KA, Meyer KB, Kar S, et al. Fine-scale mapping of the 5q11.2 breast cancer locus reveals at least three independent risk variants regulating MAP3K1. Am J Hum Genet. 2015;96:5–20.
Dunning AM, Michailidou K, Kuchenbaecker KB, Thompson D, French JD, Beesley J, et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat Genet. 2016;48:374–86.
Shi J, Zhang Y, Zheng W, Michailidou K, Ghoussaini M, Bolla MK, et al. Fine-scale mapping of 8q24 locus identifies multiple independent risk variants for breast cancer. Int J Cancer. 2016;139:1303–17.
Orr N, Dudbridge F, Dryden N, Maguire S, Novo D, Perrakis E, et al. Fine-mapping identifies two additional breast cancer susceptibility loci at 9q31.2. Hum Mol Genet. 2015;24:2966–84.
Darabi H, McCue K, Beesley J, Michailidou K, Nord S, Kar S, et al. Polymorphisms in a putative enhancer at the 10q21.2 breast cancer risk locus regulate NRBF2 expression. Am J Hum Genet. 2015;97:22–34.
Darabi H, Beesley J, Droit A, Kar S, Nord S, Moradi Marjaneh M, et al. Fine scale mapping of the 17q22 breast cancer locus using dense SNPs, genotyped within the Collaborative Oncological Gene-Environment Study (COGs). Sci Rep. 2016;6:32512.
Meyer KB, O’Reilly M, Michailidou K, Carlebur S, Edwards SL, French JD, et al. Fine-scale mapping of the FGFR2 breast cancer risk locus: putative functional variants differentially bind FOXA1 and E2F1. Am J Hum Genet. 2013;93:1046–60.
Betts JA, Moradi Marjaneh M, Al-Ejeh F, Lim YC, Shi W, Sivakumaran H, et al. Long noncoding RNAs CUPID1 and CUPID2 mediate breast cancer risk at 11q13 by modulating the response to DNA damage. Am J Hum Genet. 2017;101:255–66.
French JD, Ghoussaini M, Edwards SL, Meyer KB, Michailidou K, Ahmed S, et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am J Hum Genet. 2013;92:489–503.
Ghoussaini M, French JD, Michailidou K, Nord S, Beesley J, Canisus S, et al. Evidence that the 5p12 variant rs10941679 confers susceptibility to estrogen-receptor-positive breast cancer through FGF10 and MRPS30 regulation. Am J Hum Genet. 2016;99:903–11.
Horne HN, Chung CC, Zhang H, Yu K, Prokunina-Olsson L, Michailidou K, et al. Fine-mapping of the 1p11.2 breast cancer susceptibility locus. PLoS ONE. 2016;11:e0160316.
Zeng C, Guo X, Long J, Kuchenbaecker KB, Droit A, Michailidou K, et al. Identification of independent association signals and putative functional variants for breast cancer risk through fine-scale mapping of the 12p11 locus. Breast Cancer Res. 2016;18:64.
Lin WY, Camp NJ, Ghoussaini M, Beesley J, Michailidou K, Hopper JL, et al. Identification and characterization of novel associations in the CASP8/ALS2CR12 region on chromosome 2 with breast cancer risk. Hum Mol Genet. 2015;24:285–98.
Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–84. 84e1-2.
Lawrenson K, Kar S, McCue K, Kuchenbaeker K, Michailidou K, Tyrer J, et al. Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast-ovarian cancer susceptibility locus. Nat Commun. 2016;7:12675.
Udler MS, Tyrer J, Easton DF. Evaluating the power to discriminate between highly correlated SNPs in genetic association studies. Genet Epidemiol. 2010;34:463–8.
Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43.
Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–97.
Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93.
Cowper-Sal lari R, Zhang X, Wright JB, Bailey SD, Cole MD, Eeckhoute J, et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet. 2012;44:1191–8.
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30.
Cazaly E, Saad J, Wang W, Heckman C, Ollikainen M, Tang J. Making sense of the epigenome using data integration approaches. Front Pharm. 2019;10:126.
Vierstra J, Lazar J, Sandstrom R, Halow J, Lee K, Bates D, et al. Global reference mapping of human transcription factor footprints. Nature. 2020;583:729–36.
Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, et al. A human cell atlas of fetal chromatin accessibility. Science. 2020;370:6518.
Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–83.
de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506.
Schoenfelder S, Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet. 2019;20:437–55.
Inoue F, Kreimer A, Ashuach T, Ahituv N, Yosef N. Identification and massively parallel characterization of regulatory elements driving neural induction. Cell Stem Cell. 2019;25:713–27. e10.
Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8.
Inoue F, Ahituv N. Decoding enhancers using massively parallel reporter assays. Genomics. 2015;106:159–64.
Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30:271–7.
Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–7.
Gardini A. Global run-on sequencing (GRO-Seq). Methods Mol Biol. 2017;1468:111–20.
Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LT, Fernandez N, et al. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat Commun. 2015;6:6905.
Gordon MG, Inoue F, Martin B, Schubach M, Agarwal V, Whalen S, et al. lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements. Nat Protoc. 2020;15:2387–412.
Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–54.
Gariglio P, Bellard M, Chambon P. Clustering of RNA polymerase B molecules in the 5’ moiety of the adult beta-globin gene of hen erythrocytes. Nucleic Acids Res. 1981;9:2589–98.
Gariglio P, Buss J, Green MH. Sarkosyl activation of RNA polymerase activity in mitotic mouse cells. FEBS Lett. 1974;44:330–3.
Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet. 2014;46:1311–20.
Nojima T, Tellier M, Foxwell J, Ribeiro de Almeida C, Tan-Wong SM, Dhir S, et al. Deregulated expression of mammalian lncRNA through loss of SPT6 induces r-loop formation, replication stress, and cellular senescence. Mol Cell. 2018;72:970–84 e7.
Barbieri E, Hill C, Quesnel-Vallieres M, Zucco AJ, Barash Y, Gardini A. Rapid and scalable profiling of nascent RNA with fastGRO. Cell Rep. 2020;33:108373.
Gregersen LH, Mitter R, Svejstrup JQ. Using TTchem-seq for profiling nascent transcription and measuring transcript elongation. Nat Protoc. 2020;15:604–27.
Franco HL, Nagari A, Malladi VS, Li W, Xi Y, Richardson D, et al. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2018;28:159–70.
Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–94.
Li Q, Seo JH, Stranger B, McKenna A, Pe’er I, Laframboise T, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–41.
Seo JH, Li Q, Fatima A, Eklund A, Szallasi Z, Polyak K, et al. Deconvoluting complex tissues for expression quantitative trait locus-based analyses. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120363.
Baxter JS, Leavy OC, Dryden NH, Maguire S, Johnson N, Fedele V, et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat Commun. 2018;9:1028.
Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52.
Consortium ITP-CAoWG. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93.
Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB. Abundant associations with gene expression complicate GWAS follow-up. Nat Genet. 2019;51:768–9.
Beesley J, Sivakumaran H, Moradi Marjaneh M, Shi W, Hillman KM, Kaufmann S, et al. eQTL colocalization analyses identify NTN4 as a candidate breast cancer risk gene. Am J Hum Genet. 2020;107:778–87.
Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–8.
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–52.
Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51:592–9.
Hoffman JD, Graff RE, Emami NC, Tai CG, Passarelli MN, Hu D, et al. Cis-eQTL-based trans-ethnic meta-analysis reveals novel genes associated with breast cancer risk. PLoS Genet. 2017;13:e1006690.
Gao G, Pierce BL, Olopade OI, Im HK, Huo D. Trans-ethnic predicted expression genome-wide association analysis identifies a gene for estrogen receptor-negative breast cancer. PLoS Genet. 2017;13:e1006727.
Wu L, Shi W, Long J, Guo X, Michailidou K, Beesley J, et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat Genet. 2018;50:968–78.
Ferreira MA, Gamazon ER, Al-Ejeh F, Aittomaki K, Andrulis IL, Anton-Culver H, et al. Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nat Commun. 2019;10:1741.
Feng H, Gusev A, Pasaniuc B, Wu L, Long J, Abu-Full Z, et al. Transcriptome-wide association study of breast cancer risk by estrogen-receptor status. Genet Epidemiol. 2020;44:442–68.
Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet. 2018;50:621–9.
Jiang L, Xue C, Dai S, Chen S, Chen P, Sham PC, et al. DESE: estimating driver tissues by selective expression of genes associated with complex diseases or traits. Genome Biol. 2019;20:233.
Shang L, Smith JA, Zhou X. Leveraging gene co-expression patterns to infer trait-relevant tissues in genome-wide association studies. PLoS Genet. 2020;16:e1008734.
Cano-Gamez E, Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front Genet. 2020;11:424.
Howard BA, Gusterson BA. Human breast development. J Mammary Gland Biol Neoplasia. 2000;5:119–37.
Wang Z, Cao S, Morris JS, Ahn J, Liu R, Tyekucheva S, et al. Transcriptome deconvolution of heterogeneous tumor samples with immune infiltration. iScience. 2018;9:451–60.
Quon G, Haider S, Deshwar AG, Cui A, Boutros PC, Morris Q. Computational purification of individual tumor gene expression profiles leads to significant improvements in prognostic prediction. Genome Med. 2013;5:29.
Onuchic V, Hartmaier RJ, Boone DN, Samuels ML, Patel RY, White WM, et al. Epigenomic deconvolution of breast tumors reveals metabolic coupling between constituent cell types. Cell Rep. 2016;17:2075–86.
Fox NS, Haider S, Harris AL, Boutros PC. Landscape of transcriptomic interactions between breast cancer and its microenvironment. Nat Commun. 2019;10:3116.
Quiroz-Zarate A, Harshfield BJ, Hu R, Knoblauch N, Beck AH, Hankinson SE, et al. Expression quantitative trait loci (QTL) in tumor adjacent normal breast tissue and breast tumor tissue. PLoS ONE. 2017;12:e0170181.
Gascard P, Bilenky M, Sigaroudinia M, Zhao J, Li L, Carles A, et al. Epigenetic and transcriptional determinants of the human breast. Nat Commun. 2015;6:6351.
Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–84. e19.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
International Cancer Genome C, Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, et al. International network of cancer genome projects. Nature. 2010;464:993–8.
Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54.
Low SK, Chin YM, Ito H, Matsuo K, Tanikawa C, Matsuda K, et al. Identification of two novel breast cancer loci through large-scale genome-wide association study in the Japanese population. Sci Rep. 2019;9:17332.
Beesley J, Sivakumaran H, Moradi Marjaneh M, Lima LG, Hillman KM, Kaufmann S, et al. Chromatin interactome mapping at 139 independent breast cancer risk signals. Genome Biol. 2020;21:8.
Chen Z, Wen W, Beeghly-Fadiel A, Shu XO, Diez-Obrero V, Long J, et al. Identifying putative susceptibility genes and evaluating their associations with somatic mutations in human cancers. Am J Hum Genet. 2019;105:477–92.
Meyer KB, Maia AT, O’Reilly M, Teschendorff AE, Chin SF, Caldas C, et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108.
Schmitt AD, Hu M, Ren B. Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol. 2016;17:743–55.
Dryden NH, Broome LR, Dudbridge F, Johnson N, Orr N, Schoenfelder S, et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014;24:1854–68.
Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre BM, et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015;25:582–97.
Fullwood MJ, Ruan Y. ChIP-based methods for the identification of long-range chromatin interactions. J Cell Biochem. 2009;107:30–9.
Baxter JS, Johnson N, Tomczyk K, Gillespie A, Maguire S, Brough R, et al. Functional annotation of the 2q35 breast cancer risk locus implicates a structural variant in influencing activity of a long-range enhancer element. Am J Hum Genet. 2021;108:1190–203.
Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell. 2019;176:1516.
Kichaev G, Yang WY, Lindstrom S, Hormozdiari F, Eskin E, Price AL, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10:e1004722.
Lichou F, Trynka G. Functional studies of GWAS variants are gaining momentum. Nat Commun. 2020;11:6283.
Klein, DC. & Hainer, SJ. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 2020;28:69-85.
Montalbano, A, Canver, MC. & Sanjana, NE. High-Throughput Approaches to Pinpoint Function within the Noncoding Genome. Mol Cell. 2017;68:44-59.
Gilbert, LA, Larson, MH, Morsut, L, Liu, Z, Brar, GA, Torres, SE. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442-51.
Li, S, Garay, JP, Tubbs, CA & Franco, HL. CRISPR-based knock-in mutagenesis of the pioneer transcription factor FOXA1: optimization of strategies for multi-allelic proteins in cancer cells. FEBS Open Bio. 2021;11:1537-1551.
Nunez, JK, Chen, J, Pommier, GC, Cogan, JZ, Replogle, JM, Adriaens, C. et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184:2503–2519 e2517.
Funding
This work was supported by Programme Grants from Breast Cancer Now as part of Programme Funding to the Breast Cancer Now Toby Robins Research Centre to OF and SH. We thank SH and AG in the Breast Cancer Now Toby Robins Research Centre Bioinformatics Core for Bioinformatics Support and thank Breast Cancer Now, working in partnership with Walk the Walk for supporting the work of this team.
Author information
Authors and Affiliations
Contributions
All authors contributed to the identification of data sources and methods and to the writing and reviewing of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Romualdo Cardoso, S., Gillespie, A., Haider, S. et al. Functional annotation of breast cancer risk loci: current progress and future directions. Br J Cancer 126, 981–993 (2022). https://doi.org/10.1038/s41416-021-01612-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01612-6
This article is cited by
-
Evaluation of circulating plasma proteins in breast cancer using Mendelian randomisation
Nature Communications (2023)
