
Abstract
Background
Proteasome inhibitors (PIs), including carfilzomib, potentiate the activity of selinexor, a novel, first-in-class, oral selective inhibitor of nuclear export (SINE) compound, in preclinical models of multiple myeloma (MM).
Methods
The safety, efficacy, maximum-tolerated dose (MTD) and recommended phase 2 dose (RP2D) of selinexor (80 or 100 mg) + carfilzomib (56 or 70 mg/m2) + dexamethasone (40 mg) (XKd) once weekly (QW) was evaluated in patients with relapsed refractory MM (RRMM) not refractory to carfilzomib.
Results
Thirty-two patients, median prior therapies 4 (range, 1–8), were enrolled. MM was triple-class refractory in 38% of patients and 53% of patients had high-risk cytogenetics del(17p), t(4;14), t(14;16) and/or gain 1q. Common treatment-related adverse events (all/Grade 3) were thrombocytopenia 72%/47% (G3 and G4), nausea 72%/6%, anaemia 53%/19% and fatigue 53%/9%, all expected and manageable with supportive care and dose modifications. MTD and RP2D were identified as selinexor 80 mg, carfilzomib 56 mg/m2, and dexamethasone 40 mg, all QW. The overall response rate was 78% including 14 (44%) ≥ very good partial responses. Median progression-free survival was 15 months.
Conclusions
Weekly XKd is highly effective and well-tolerated. These data support further investigation of XKd in patients with MM.
Similar content being viewed by others


Introduction
Exportin 1 (XPO1) is a critical nuclear exporter for tumour suppressor proteins (TSPs, e.g. p53, IκB and FOXO3a) [1,2,3] and eIF4E-bound oncoprotein messenger RNAs (mRNAs) (e.g. c-Myc, Bcl-xL, MDM2 and cyclin D1) [1, 2, 4, 5]. XPO1 is overexpressed in many cancers including multiple myeloma (MM), and this overexpression of XPO1 enables cancer cells to escape TSP-mediated cell cycle arrest and apoptosis [1, 2, 6, 7]. Indeed, XPO1 overexpression correlates with poor prognosis of cancer patients and drug resistance of their disease [1, 2, 6, 8, 9].
Selinexor is a novel, first-in-class oral selective inhibitor of nuclear export (SINE) compound that blocks XPO1, forcing the nuclear retention and activation of TSPs, ultimately causing cancer cell death [1]. Preclinical data demonstrate that selinexor reactivates multiple TSPs relevant to MM, inhibits nuclear factor-κB activity, reduces c-Myc levels and reactivates glucocorticoid receptor signalling in the presence of dexamethasone, all of which has the downstream effect of suppressing MM cell growth [1,2,3, 10]. Consistent with these preclinical results, in the STORM (Selinexor Treatment of Refractory Myeloma) clinical trial in patients with MM refractory to at least one proteasome inhibitor (PI), one immunomodulatory agent (IMiD) and daratumumab (triple-class refractory), selinexor 80 mg and dexamethasone 20 mg, both twice weekly, induced a 26.4% overall response rate (ORR) and 4.4-month median duration of response (DOR) [11]. Based on these results, selinexor received accelerated approval from the Food and Drug Administration (FDA) for patients with relapsed refractory MM (RRMM) [12, 13].
The combination of nuclear export and proteasome inhibition is synergistic in preclinical models, wherein PIs prevent the proteasomal degradation of TSPs and SINE compounds force their nuclear retention, leading to apoptosis of cancer cells [1, 14]. Importantly, elevated levels of XPO1 can mediate PI resistance by exporting TSPs from the nucleus [15, 16]. Thus, XPO1 inhibition can overcome PI resistance in MM cells in cell lines as well as in patient-derived MM cells ex vivo [8, 14, 17], and selinexor was shown to sensitise PI-refractory MM cells to bortezomib and carfilzomib [16]. These preclinical results were confirmed in phase 3 BOSTON (Bortezomib, Selinexor, and Dexamethasone in Patients With Multiple Myeloma) trial, where once weekly (QW) selinexor, bortezomib and dexamethasone (XVd) showed superior median progression-free survival (PFS; 13.9 vs. 9.5 months) and ORR (76.4 vs. 62.3%), reduced peripheral neuropathy and a trend to reduced mortality, as compared with standard twice weekly bortezomib and dexamethasone (Vd), despite XVd using 40% less bortezomib and 25% less dexamethasone than standard Vd [18]. The BOSTON trial led to an FDA approval of XVd for adult patients with MM who have received at least one prior therapy [12].
Carfilzomib, a second-generation PI that is more potent than bortezomib [19,20,21], is approved for the treatment of MM with dexamethasone and in combination with other agents [22]. Selinexor and carfilzomib showed marked synergistic anti-tumour activity in various preclinical models in vitro and in vivo [17, 23]. A phase I trial in patients with RRMM validated these preclinical results, demonstrating that the combination of selinexor and carfilzomib with low-dose dexamethasone (XKd) is tolerable and is effective in inducing responses in heavily pretreated MM, including in disease refractory to carfilzomib [24]. The present trial was conducted to further validate these results and to show that weekly XKd is safe and tolerable and derives durable responses in patients whose MM is not refractory to carfilzomib.
Methods
Study design and oversight
This trial is part of the multi-arm Phase 1b/2 STOMP (Selinexor and Backbone Treatments of Multiple Myeloma Patients) study evaluating the safety and efficacy of selinexor in combination with FDA-approved therapies for RRMM (ClinicalTrials.gov #NCT02343042). Here, we report data from the dose-evaluation and dose-expansion phases of varying doses of XKd. The primary objectives of the dose-evaluation phase were to determine the maximum-tolerated dose (MTD), safety and tolerability, and to identify the recommended phase 2 dose (RP2D) for the XKd regimen.
Patients with previously treated MM that was not refractory specifically to carfilzomib (but may have had prior PI treatment), and who had not received prior selinexor, were eligible for enrollment. Refractory disease and high-risk disease were defined per International Myeloma Working Group (IMWG) guidelines as lack of at least a minimal response (MR) while on therapy, or disease progression within 2 months of completing therapy [25, 26]. A full list of inclusion/exclusion criteria has been published previously [27].
The study protocol was approved by the institutional review board or an independent ethics committee at each participating centre and was in accordance with the Declaration of Helsinki, the International Conference on Harmonisation-Good Clinical Practice and local laws. All patients provided written informed consent prior to enrollment. All authors reviewed the data for accuracy and collaborated in the preparation of the manuscript.
Treatments
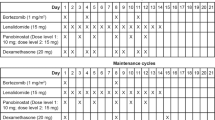
In the dose-evaluation phase, the starting selinexor dose was 100 mg QW, carfilzomib 20/56 mg/m2 QW (20 mg/m2 only on C1D1 and 56 mg/m2 thereafter, dosed on days 1, 8 and 15, of 28-day cycles) and 40 mg QW dexamethasone (Table 1). Rules for dose level evaluation vs. reduction followed standard 3 + 3 rules. As selinexor and carfilzomib antineoplastic effects synergise, and as selinexor is typically used at 60, 80 or 100 mg QW, and carfilzomib 45–70 mg/m2 weekly, optimising dosing of the combination requires modulating both agents. To be conservative, we chose either the MTD of selinexor (i.e. 100 mg—Bahlis et al. [27]) with one level down from MTD of carfilzomib QW (i.e. 56 mg/m2; Berenson et al. [28]) or one level down from MTD of selinexor (i.e. 80 mg) with the MTD of carfilzomib QW (i.e. 70 mg/m2); the step-down dose involved one level down from MTDs of both selinexor (i.e. 80 mg) and carfilzomib (i.e. 56 mg/m2). Ultimately, we will choose the dose with the highest benefit/risk ratio.
Dose-limiting toxicities (DLTs) were evaluated only in patients enrolled during the dose-evaluation phase over their first cycle of treatment. DLTs were defined as any of the following: (1) missing ≥25% of scheduled doses, a dose reduction or discontinuation due to treatment-related adverse events (TRAEs); (2) occurrence of Grade ≥3 nausea, vomiting, dehydration, diarrhoea or fatigue lasting >3 days despite optimal supportive care medications, or Grade 4 of these adverse events (AEs); (3) any other Grade 3 or 4 non-haematologic toxicity; (4) febrile neutropenia, Grade 4 neutropenia or Grade 4 thrombocytopenia lasting >7 days, and Grade ≥3 thrombocytopenia with clinically significant bleeding, petechiae or purpura.
Patients received 5-hydroxytryptamine-3 (5-HT3) antagonist (ondansetron 8 mg or equivalent, or an alternative if 5-HT3 antagonists were not tolerated) before each dose of selinexor and continued 2–3 times daily for at least 2 days. Additional supportive care was included as part of the protocol (see supplementary materials for full AE management guidelines).
Study assessments and statistics
Efficacy was assessed using modified IMWG guidelines [29]. ORR was defined as the percentage of patients who achieved a confirmed partial response (PR) or better before the progressive disease (PD) or initiating a new anti-MM treatment; clinical benefit rate was defined similarly but included MR. PFS was defined as the duration from the first dose of study treatment to the first confirmed PD or death due to any cause. DOR was defined for responders only as the duration from first PR or better to first confirmed PD or death due to any cause. For PFS and DOR, patients who discontinued treatment prior to confirmed PD or death, or who were still on treatment with no confirmed PD at the time of the data extract, were censored at the latest response assessment on or before the date of treatment discontinuation (where applicable). Overall survival (OS) was defined as the duration from the first dose of study treatment to death due to any cause. Survival endpoints were analysed using Kaplan–Meier methodology; median follow-up times were estimated using the reverse Kaplan–Meier method [30].
Safety was monitored throughout the study and severity was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events, v4.03. All patients who received at least a single dose of study medication were included in safety and efficacy analyses.
The sample size for the dose-evaluation phase of the study was based on the standard 3 + 3 dose-escalation scheme. The expansion phase was designed to test the null hypothesis that the true ORR was ≤30% against a one-sided alternative and required a sample size of 20 patients, where the first 10 patients treated at the RP2D were considered the first stage of the two-stage design. If ≤3 patients responded in Stage 1, the expansion phase would be terminated. If ≥4 patients responded, an additional ten patients were to be enrolled to include a total of 20 patients at the RP2D. If the total number of patients responding was ≥9, the treatment would be accepted as promising for further study. Assuming a true ORR of 55%, this design achieved at least 80% power at a one-sided 0.10 significance level.
Results
Patients and treatment
A total of 32 patients were enrolled between March 2018 and Feb 2021: 3 patients in the selinexor 100 mg with 56 mg/m2 carfilzomib cohort, 3 in the selinexor 80 mg with 70 mg/m2 carfilzomib cohort, 18 in the selinexor 80 mg with 56 mg/m2 carfilzomib cohort, 3 in the selinexor 60 mg with 70 mg/m2 carfilzomib cohort, and 2 and 3 in the selinexor 60 mg with 56 or 70 mg/m2 carfilzomib, respectively, cohorts in which selinexor was given on days 1, 8 and 15, but not on day 22 of the 4-week cycle (Table 1). The ‘stepped down’ selinexor 60 mg QW dose combined with 70 mg/m2 carfilzomib cohort and the cohorts in which selinexor was administered only on weeks 1–3 of the 4-week cycle were exploratory dose-evaluation cohorts initiated to test selinexor with the currently approved once-weekly carfilzomib dose (i.e. 70 mg/m2 QW) or to test the impact of 3/4-week dosing of selinexor on safety and tolerability.
The median age was 69.5 years (range 35–76 years), 63% were males and the median number of prior therapies was 4 (range 1–8). All patients had been treated with bortezomib and at least one IMiD, 3 (9%) had previously received carfilzomib, 59% had MM refractory to a PI, 75% had MM refractory to an IMiD and 66% had MM refractory to anti-CD38 monoclonal antibody (mAb). In addition, 50% of the patients had MM refractory to both a PI and an IMiD, and 38% had MM refractory to a PI, an IMiD and an anti-CD38 mAb. Fifty-three per cent of the patients had high-risk cytogenetics, defined as del 17p, t(4;14), t(14;16) or gain of 1q (3 or 4 fold). Patient demographics and disease characteristics at baseline are shown in Table 2.
As of the cut-off date of 11 May 2021, 10 (31.3%) patients were still receiving treatment, 12 (38%) patients discontinued due to PD, 5 (15.6%) discontinued due to AEs (cardiac failure; peripheral neuropathy; left ventricular failure; upper respiratory tract infection + elevated creatinine; atrial fibrillation + dyspnoea + dizziness), 1 patient discontinued due to drug toxicity, 1 patient withdrew consent, 1 patient died (respiratory failure not related to study drugs) and 1 patient discontinued to undergo autologous haematopoietic stem-cell transplantation.
Efficacy
The response was evaluated in all 32 patients. The overall ORR was 78.1% (25/32): 2 (6.3) stringent complete responses (sCRs), 3 (9.4%) CR, 9 (28.1%) very good PRs (VGPRs) and 11 (34.4%) PR. One (3.1%) additional patient achieved an MR (Table 3 and Fig. 1). PFS for individual patients is presented in Fig. 2a, and the Kaplan–Meier curve is shown in Fig. 2b. The overall median PFS was 15.0 months (95% confidence interval (CI), 12.0–NE; median follow-up 8.0 months), the median DOR was 22.7 months (95% CI, 11.8–NE; median follow-up 5.6 months) and the median OS (mOS) was not reached (95% CI, NE–NE; median follow-up 15.1 months). In the nine patients with ≤2 prior therapies, the ORR was 88.9%.

Waterfall plot depicts the best % changes in the IgA, primary myeloma marker (serum M-protein, urine M-protein or difference between involved and uninvolved serum-free light-chain levels) from baseline. F free light chain, S SPEP, U UPEP, A IgA.

a Time to PFS event or censor for all dosed patients. PFS event is confirmed disease progression or death before documented treatment discontinuation or the start of a new anti-MM treatment. Y-axis labels indicate which drugs each patient was refractory to (V bortezomib, K carfilzomib, R lenalidomide, P pomalidomide, D daratumumab) and whether they had high-risk cytogenetics (* = Yes), defined as any of del(17p), t(4;14), t(14;16) or gain 1q at initial diagnosis or screening. TX toxicity to study drug, AE adverse event, CP disease progression (clinical progression), WC withdrawal of consent, OT other, event PFS event. b Kaplan-Meier analysis of progression-free survival (PFS) for all dosed patients (n = 32). Median PFS for all patients was 15.0 months (95% CI, 12.0–NE) and 23.7 months (95% CI, 3.9–NE) patients with triple-class refractory MM.
XKd induced responses in patients with triple-class refractory MM (n = 12): ORR was 66.7% (8/12), the median PFS was 23.7 months (95% CI, 3.9–NE), the median DOR was 22.7 months (95% CI, NE–NE) and the mOS was 20.4 months (95% CI, 20.4–NE). Likewise, the presence of high-risk genetics [i.e. del(17p), t(4;14), t(14;16) or gain 1q] at initial diagnosis or screening did not compromise the efficacy of XKd: In the 17 patients with high-risk cytogenetics, the ORR was 82.4% (14/17), the median PFS was 15.0 months (95% CI, 4.9–NE), the median DOR was 22.7 months (95% CI, 13.1–NE) and the mOS was not reached (95% CI, 20.4–NE).
XKd induced rapid responses, 16 of the 25 responders achieved PR or better within the first cycle of treatment and 23 had ≥PR within the second cycle.
Discussion
The aim of the present research was to determine the MTD, RP2D, safety and efficacy of weekly XKd for the treatment of RRMM. Although numbers were relatively small in this phase 2 study, across all doses tested, weekly XKd demonstrated high rates of anti-MM activity against heavily pretreated RRMM (1–8 prior therapies, median = 4), with an ORR of 78.1% including 2 (6.3%) sCR, 3 (9.4%) CR, 9 VGPR (28.1%) and 11 PR (34.4%). Moreover, XKd induced long-lasting responses with median DOR of 22.7 months, median PFS of 15.0 months and median OS not reached (median follow-up 15.1 months). No clinically significant and unexpected cumulative toxicities have been observed. Importantly, efficacy was preserved in genetically high-risk disease; ORR was 82.4%, median PFS 15.0 months, median DOR 22.7 months and median OS not reached. Similar results were observed in triple-class refractory MM where ORR was 66.7%, median PFS 23.7 months, median DOR 22.7 months and median OS 20.4 months. Anti-MM activity was not affected by any specific prior therapy consistent with the lack of cross-reactivity of selinexor with other anti-MM agents.
The high efficacy demonstrated here by XKd is on par with the efficacy demonstrated by currently recommended triplet combinations utilising carfilzomib and dexamethasone to treat RRMM. In RRMM patients treated with a median of 2 prior therapies (range 1–4), none of whom had previous treatment with daratumumab or had triple-class refractory MM, daratumumab, carfilzomib and dexamethasone (DKd) induced an ORR of 84% [31]. At a median follow-up of 16.6 months, DKd-induced median PFS was not reached [31]. In patients with RRMM with a median of six prior therapies (range 2–12), carfilzomib, pomalidomide and dexamethasone (KPd) induced an ORR of 50% [32]. At a median follow-up of 26.3 months, KPd induced a median PFS of 7.2 months and a median OS of 20.6 months [32]. While the current sample size is relatively small, these findings suggest that XKd is at least as active, and potentially more active in patients with anti-CD38 mAb refractory disease than other triplet combinations in heavily pretreated MM. These data position XKd as an optimal combination to address the unmet clinical need in the growing population of MM patients with prior therapy with an anti-CD38 mAb for which there is a scarcity of data and limited clinical efficacy with established therapy regimens. We do also note that most physicians treating progressing MM prefer to utilise agents in novel therapeutic classes, rather than switching to a second-generation compound in the same class, and hence XKd may provide a preferred regimen.
Two dose levels tested, 100 mg selinexor with 56 mg/m2 carfilzomib and 80 mg selinexor with 70 mg/m2 carfilzomib, both with 40 mg dexamethasone QW, were found to be intolerable in our patient cohort. However, the next lower level, 80 mg selinexor with 56 mg/m2 carfilzomib and 40 mg dexamethasone all QW, was identified as MTD and determined to be the RP2D. Additional dose levels including 60 mg selinexor (on days 1, 8, 15 and 22) with 70 mg/m2 carfilzomib and 80/100 mg selinexor with carfilzomib 70/56 mg/m2, both only on days 1, 8 and 15 of the 28-day cycles, are being evaluated in eight patients.
Mechanistically, the notable potency of the XKd regimen is well supported by preclinical studies [8, 14,15,16,17]: while PIs prevent the proteasome-mediated destruction of TSPs and other key regulatory/anti-cancer proteins in both the cytoplasm and the nucleus, they cannot control the geographic distribution of these molecules within the cell [33, 34]. Since TSPs and many other regulatory proteins only function in the nuclear compartment, resistance to PIs often occurs in the setting of elevated levels of XPO1—mediating the rapid nuclear export and functional inactivation of these key anti-cancer proteins. When XPO1 inhibitors are combined with PIs, levels of TSPs and other proteins are greatly increased, and they are restricted to the nuclear compartment leading to greatly enhanced anti-cancer activity [14, 17, 23].
The XKd safety profile was similar to that observed previously for each of the components [11, 18], with the most common non-haematologic TRAEs being nausea and fatigue, mostly Grades 1 and 2, reversible and manageable with dose modifications and/or supportive care. Prophylaxis with antivirals and intravenous (IV) hydration for carfilzomib, along with anti-nausea agents for selinexor, are important to optimise therapy. The most common haematological TRAEs were thrombocytopenia (with no associated bleeding) and anaemia, which were also reversible and manageable with dose modifications and/or supportive care. Discontinuation due to TRAEs occurred in 15.6% of all patients in the current study (median prior therapies = 4), and in 11.1% of patients at the RP2D, a rate comparable to that reported for other triplet combinations containing carfilzomib and dexamethasone with half the number of prior therapies (i.e. KRd = 16.8% [35], DKd = 22% [36]). Moreover, some of the most common AEs associated with carfilzomib, but not selinexor, were lower or similar in the current trial compared to other pivotal clinical trials testing carfilzomib in triplet combinations [35, 36]. Importantly, the XKd triplet is one of the simplest regimens to administer, requiring only the “backbone” weekly carfilzomib infusions (30 min of hydration, followed by 30 min of carfilzomib) with appropriate monitoring; the oral selinexor (and dexamethasone) can be given during the infusions. This is considerably simpler than double parenteral infusions (e.g. DKd) or parenteral-daily oral regimens (e.g. KPd).
In conclusion, the QW combination of oral selinexor 80 mg, dexamethasone 40 mg and IV carfilzomib 56 mg/m2 provided deep and durable responses in patients with heavily pretreated RRMM, of whom 37.5% had triple-class refractory MM and 53.1% had high-risk cytogenetics. Based on the preliminary results presented in this study in a more heavily pretreated population than those previously reported with carfilzomib-based triplets, QW XKd has compelling, durable activity, including in high-risk and triple-class refractory MM, and additional studies in both previously treated, as well as newly diagnosed MM, are warranted.
Safety
Two of the first three patients enrolled into the selinexor 100 mg with 56 mg/m2 carfilzomib cohort experienced a DLT (Grade 3 thrombocytopenia and Grade 3 vomiting) (Table 1). Likewise, two of the first three patients enrolled into the selinexor 80 mg with 70 mg/m2 carfilzomib cohort experienced a DLT (Grade 4 thrombocytopenia ± pneumonia) (Table 1). No DLTs were observed in the first three patients enrolled into the 80 mg selinexor with 56 mg/m2 carfilzomib and 40 mg dexamethasone; three additional patients were enrolled in this cohort to confirm the tolerability and no DLTs occurred; therefore, this dose level and schedule were determined to be the MTD and the RP2D for the expansion phase. Assessment of DLTs in the dose-evaluation cohorts that were initiated after the RP2D is still ongoing.
All 32 patients received at least one dose of selinexor and were therefore included in safety analyses (Table 4). Among all patients, the most common (>50%) non-haematologic TRAEs (all; Grade 3 [no Grade 4 non-haematologic TRAEs were reported]) were nausea (71.9%; 6.3%) and fatigue (53.1%; 9.4%), were mostly grade 1/2 and manageable by dose modification and/or supportive care. Grade 3/4 non-haematological TRAEs that occurred in at least two patients were fatigue (9.4%, Grade 3), pneumonia (6.3%, Grade 3), nausea (6.3%, Grade 3) and hyperglycaemia (6.3%, one Grade 3 and one Grade 4). The most common haematologic TRAEs (all, Grade 3/4) were thrombocytopenia (71.9%, 46.9%) and anaemia (53.1%, 18.8% all Grade 3). Grade 4 thrombocytopenia occurred in seven patients (21.9%) and was not accompanied by bleeding in any patient. No cases of febrile neutropenia were reported. Overall, 17 (53.1%) patients had TRAEs leading to dose interruptions and 21 (65.6%) had TRAEs leading to dose reductions.
Among patients at the RP2D (N = 18), the most common (≥50%) non-haematologic TRAEs (all; Grade 3) were nausea (77.8%; 11.1%), fatigue (55.6%; 5.6%) and decreased appetite (50.0%; 5.6%), were mostly Grade 1/2 and manageable by dose modification and/or supportive care. Nausea was the only Grade ≥3 non-haematological TRAEs that occurred in at least two patients (11.1%, G3). Most common haematologic TRAEs (all, Grade 3/4) were thrombocytopenia (77.8%, 50.0%) and anaemia (61.1%, 11.1% all G3). Grade 4 thrombocytopenia occurred in three patients (16.7%) and again was not accompanied by bleeding in any patient.
No patient had nausea or vomiting leading to discontinuation of any study drug. All patients received at least one prophylactic anti-nausea agent. Twenty-four (75%) received a 5-HT3 antagonist, and of those, 19 (79.2%) received at least one additional antiemetic medication. Ten patients (31.3%) received two or more additional antiemetic medications. Four (12.5%) patients received an additional appetite stimulant (i.e. megesterol, mirtazapine) and 11 (34.4%) patients received potassium chloride tablets. Six (18.8%) patients received transfusions (either red blood cell or platelet transfusions). Six (18.8%) patients received the thrombopoietin receptor agonists eltrombopag or romiplostim, one (3.1%) patient received filgrastim and two patients (6.3%) received an erythropoietin-stimulating agent.
Six patients had at least one serious AE) attributed to any of the study drugs: 2 pneumonia (6.3% of patients, Grade 3), 1 anaemia (Grade 3), 1 coronavirus disease 2019 (Grade 3), 1 cardiac failure (Grade 1), 1 fatigue (Grade 3), 1 influenza (Grade 3), 1 left ventricular failure (Grade 3), 1 encephalopathy (Grade 3), 1 Pneumocystis jirovecii pneumonia (Grade 3), 1 viral pneumonia (Grade 3), 1 pulmonary embolism (Grade 3), and 1 thrombocytopenia (Grade 4, no concurrent bleeding).
Data availability
Karyopharm Therapeutics agrees to share the individual participant data that underlie the results reported in this Article (after de-identification), including the study protocol. To gain access, data requestors should submit a request to medicalinformation@karyopharm.com.
References
Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M. et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28:155–65.
Fung HYJ, Chook YM. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol. 2014;27:52–61.
Parikh K, Cang S, Sekhri A, Liu D. Selective inhibitors of nuclear export (SINE)—a novel class of anti-cancer agents. J Hematol Oncol. 2014;7:78.
Gravina GL, Senapedis W, McCauley D, Baloglu E, Shacham S, Festuccia C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol. 2014;7:85.
Li S, Fu J, Mapara M, Landesman Y, Lentzsch S. Combined inhibition of XPO1 and eIF4E prevents protein translation and has synergistic anti-myeloma effects. EHA. 2020;294827:EP910.
Schmidt J, Braggio E, Kortuem KM, Egan JB, Zhu YX, Xin CS, et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia. 2013;27:2357–65.
Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–32.
Azmi AS, Uddin MH, Mohammad RM. The nuclear export protein XPO1—from biology to targeted therapy. Nat Rev Clin Oncol. 2021;18:152–69.
Chanukuppa V, Paul D, Taunk K, Chatterjee T, Sharma S, Kumar S, et al. XPO1 is a critical player for bortezomib resistance in multiple myeloma: a quantitative proteomic approach. J Proteom. 2019;209:103504.
Argueta C, Kashyap T, Klebanov B, Unger TJ, Guo C, Harrington S, et al. Selinexor synergizes with dexamethasone to repress mTORC1 signaling and induce multiple myeloma cell death. Oncotarget. 2018;9:25529–44.
Chari A, Vogl DT, Gavriatopoulou M, Nooka AK, Yee AJ, Huff CA, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N. Engl J Med. 2019;381:727–38.
US FDA. XPOVIO™ (selinexor). Prescribing information. Reference ID: 4719344. 2020, Silver Spring: US FDA.
Syed YY. Selinexor: FIRST global approval. Drugs. 2019;79:1485–94.
Kashyap T, Argueta C, Aboukameel A, Unger TJ, Klebanov B, Mohammad RM, et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-κB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget. 2016;7:78883–95.
Turner JG, Dawson J, Cubitt CL, Baz R, Sullivan DM. Inhibition of CRM1-dependent nuclear export sensitizes malignant cells to cytotoxic and targeted agents. Semin Cancer Biol. 2014;27:62–73.
Turner JG, Dawson J, Emmons MF, Cubitt CL, Kauffman M, Shacham S, et al. CRM1 inhibition sensitizes drug resistant human myeloma cells to topoisomerase II and proteasome inhibitors both in vitro and ex Vivo. J Cancer. 2013;4:614–25.
Turner JG, Kashyap T, Dawson JL, Gomez J, Bauer AA, Grant S, et al. XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IκBα and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget. 2016;7:78896–909.
Grosicki S, Simonova M, Spicka I, Pour L, Kriachok I, Gavriatopoulou M, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. Lancet. 2020;396:1563–73.
Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hájek R, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17:27–38.
Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110:3281–90.
Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res. 2011;17:2734–43.
US FDA. KYPROLIS® (carfilzomib). Prescribing information. Reference ID: 4659713. 2020, Silver Spring: US FDA.
Rosebeck S, Alonge MM, Kandarpa M, Mayampurath A, Volchenboum SL, Jasielec J, et al. Synergistic myeloma cell death via novel intracellular activation of caspase-10-dependent apoptosis by carfilzomib and selinexor. Mol Cancer Ther. 2016;15:60–71.
Jakubowiak AJ, Jasielec JK, Rosenbaum CA, Cole CE, Chari A, Mikhael J, et al. Phase 1 study of selinexor plus carfilzomib and dexamethasone for the treatment of relapsed/refractory multiple myeloma. Br J Haematol. 2019;186:549–60.
Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e46.
Rajkumar SV, Harousseau J-L, Durie B, Anderson KC, Dimopoulos M, Kyle R, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117:4691–5.
Bahlis NJ, Sutherland H, White D, Sebag M, Lentzsch S, Kotb R, et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood. 2018;132:2546–54.
Berenson JR, Cartmell A, Bessudo A, Lyons RM, Harb W, Tzachanis D, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood. 2016;127:3360–8.
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos M-V, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538–48.
Shuster JJ. Median follow-up in clinical trials. J Clin Oncol. 1991;9:191–2.
Chari A, Martinez-Lopez J, Mateos MV, Blade J, Benboubker L, Oriol A, et al. Daratumumab plus carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood. 2019;134:421–31.
Shah JJ, Stadtmauer EA, Abonour R, Cohen AD, Bensinger WI, Gasparetto C, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood. 2015;126:2284–90.
Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–60.
Aliabadi F, Sohrabi B, Mostafavi E, Pazoki-Toroudi H, Webster TJ. Ubiquitin-proteasome system and the role of its inhibitors in cancer therapy. Open Biol. 2021;11:200390.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N Engl J Med. 2015;372:142–52.
Dimopoulos M, Quach H, Mateos MV, Landgren O, Leleu X, Siegel D, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396:186–97.
Gasparetto C, Lentzsch S, Schiller G, Callander N, Chen C, White D, et al. A phase 1b/2 study of selinexor, carfilzomib, and dexamethasone (SKd) in relapsed/refractory multiple myeloma (RRMM). EHA. 2019;PS141:abstract 3423.
Gasparetto C, Schiller GJ, Callander NS, Lentzsch S, Tuchman SA, Bahlis N, et al. A Phase 1b/2 study of selinexor, carfilzomib, and dexamethasone (SKd) in relapsed/ refractory multiple myeloma (RRMM). Blood J. 2019;134:3157.
Gasparetto C, Lipe B, Tuchman S, Callander NS, Lentzsch S, Baljevic M, et al. Once weekly selinexor, carfilzomib, and dexamethasone (SKd) in patients with relapsed/refractory multiple myeloma. J Clin Oncol. 2020;38:8530–8530.
Gasparetto C, Lipe B, Tuchman SA, Callander NS, Lentzsch S, Baljevic M, et al. Selinexor in combination with carfilzomib and dexamethasone, all once weekly (SKd), for patients with relapsed/refractory multiple myeloma. Blood. 2020;136 Suppl 1:14–15.
Gasparetto C, Schiller GJ, Tuchman S, Callander NS, Baljevic M, Lentzsch S, et al. Once weekly selinexor, carfilzomib, and dexamethasone (XKd) in carfilzomib nonrefractory multiple myeloma (MM) patients. J Clin Oncol. 2021;39 Suppl 15:abstract 8038.
Gasparetto C, Schiller GJ, Tuchman S, Callander N, Baljevic M, Lentzsch S, et al. Once weekly selinexor, carfilzomib, and dexamethasone (XKd) in carfilzomib nonrefractory multiple myeloma (MM) patients. EHA. 2021;abstract S188.
Acknowledgements
We want to acknowledge patients, their families, and caregivers, investigators, co-investigators and study teams at each participating centre. Subsets of these data were previously presented at: EHA annual meeting 2019 [37], ASH annual meeting 2019 [38], ASCO annual meeting 2020 [39], ASH annual meeting 2020 [40], ASCO annual meeting 2021 [41] and EHA annual meeting 2021 [42].
Funding
This study was supported by research funding from Karyopharm Therapeutics, Inc.
Author information
Authors and Affiliations
Contributions
Conception and design: SS and MK. Provision of study material or patients: CG, GJS, SAT, NSC, MB, SL, ACR, RK, DW, NJB, CIC, HJS, SM, RL, MS, CPV, WIB, NB and BL. Collection and assembly of data: all authors. Data analysis and interpretation: all authors. Manuscript writing: all authors. Final approval of manuscript: all authors. Accountable for all aspects of the work: all authors.
Corresponding author
Ethics declarations
Competing interests
CG—advisory board: Karyopharm, Abbvie, Janssen, GSK, Sanofi, Oncopeptide; speaker: Karyopharm, GSK, Sanofi. GJS—research funding from Karyopharm. SAT—honoraria/advisory board: Karyopharm, Caelum, Sanofi; research funding: Karyopharm, Sanofi, Caelum; consultancy: Oncopeptides. NSC—nothing to disclose. MB—Independent Review Committee member and consultant: Karyopharm; advisory role and consultant: BMS/Celgene; advisory role: Oncopeptides, Johnson & Johnson; research support: Amgen, Exelixis. SL—consultant advisory role: Caelum Bioscience, Sorrento, Janssen, Celularity, Abbvie, GSK, Takeda, Karyopharm, Sanofi, Oncopetide; stock ownership: Caelum Bioscience, Magenta; research grant: Karyopharm, Sanofi. ACR—consultant: BMS, Celgene, Amgen, Janssen; research support: BMS, Karyopharm, Abbvie. RK—research funding: Merck, Sanofi; patents, royalties, other intellectual property: none; stock and other ownership interests: Karyopharm; consulting or advisory role: Celgene/BMS; Janssen; Amgen; Takeda; Sanofi; Merck; honoraria: none; speakers’ bureau: none; travel, accommodations, expenses: none. DW—honoraria: Amgen, Antengene, Celgene/BMS, GSK, Janssen, Karyopharm, Sanofi, Takeda. NJB—honoraria: Janssen, Celgene, Amgen, Takeda, AbbVie, GlaxoSmithKline, Sanofi, Genentech and Karyopharm; consultant/advisor role: Janssen, Celgene, Amgen, Takeda, AbbVie, GlaxoSmithKline, Karyopharm and Genentech. CIC—research funding: Gilead, BMS; consultant or advisory: BMS, Novartis, Gilead, Abbvie, Beigene. HJS—honoraria and consultant or advisory role: Amgen, Takeda, Celgene, BMS, Janssen, Sanofi-Genzyme. SM—consultant or advisory role: ad hoc advisory board for BMS, Takeda, Sanofi, Janssen, Amgen; GSK; speakers’ bureau: BMS, GSK, Takeda, Janssen, Karyopharm. RL—research funding: BMS/Celgene; consultant or advisory role: Janssen, BMS/Celgene, Amgen, Sanofi, Takeda. MS—advisory boards: Janssen, Karyopharm, Amgen, Novartis, Bristol-Myers Squibb, Takeda; no other disclosures. CPV—honoraria from BMS/Celgene, Janssen, Sanofi, GSK, Takeda. WIB—research funding: BMS, Karyopharm, Poseida, Harpoon, Regeneron, Janssen, Sanofi; consulting: Janssen, Regeneron, BMS; honoraria/speakers’ bureau: Takeda, BMS, Amgen, Janssen; no travel in the past year. NB—consultant: Karyopharm, BMS/Celgene, Sanofi, Janssen, Oncopeptides; research support: Karyopharm, BMS/Celgene, Janssen, Merck; speakers’ bureau: Oncopeptides, Janssen; advisory board: BMS/Celgene, Sanofi. SA—employee and stock holder in Karyopharm Therapeutics. OB-S—employee and stockholder in Karyopharm Therapeutics. AD—employee and stockholder in Karyopharm Therapeutics. DVD—employee and stockholder in Karyopharm Therapeutics. TZ—a former employee and stockholder in Karyopharm Therapeutics. CZ—employee and stockholder in Karyopharm Therapeutics. OSB—employee and stockholder in Karyopharm Therapeutics. JS—CMO, employee and stockholder in Karyopharm Therapeutics. SS—President, employee and stockholder in Karyopharm Therapeutics; patents and royalties: (8999996, 9079865, 9714226, PCT/US12/048319, and I574957) on hydrazide-containing nuclear transport modulators and uses, and pending patents (PCT/US12/048319, 499/2012, PI20102724, and 2012000928). MGK—CEO, employee and stockholder in Karyopharm Therapeutics. BL—advisory role: BMS, GSK, Janssen, Karyopharm.
Ethics approval and consent to participate
The study protocol was approved by the institutional review board or an independent ethics committee at each participating centre and was in accordance with the Declaration of Helsinki, the International Conference on Harmonisation-Good Clinical Practice and local laws. All patients provided written informed consent prior to enrollment.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gasparetto, C., Schiller, G.J., Tuchman, S.A. et al. Once weekly selinexor, carfilzomib and dexamethasone in carfilzomib non-refractory multiple myeloma patients. Br J Cancer 126, 718–725 (2022). https://doi.org/10.1038/s41416-021-01608-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01608-2
This article is cited by
-
Co-expression analysis of transcriptomic data from cancer and healthy specimens reveals rewiring of proteasome genes and an interaction with the XPO1 gene across several tumour types
Translational Medicine Communications (2024)
-
Targeting Nuclear Export Proteins in Multiple Myeloma Therapy
BioDrugs (2022)
-
Once weekly selinexor, carfilzomib and dexamethasone in carfilzomib non-refractory multiple myeloma patients
British Journal of Cancer (2022)
