
Abstract
Major advances in cancer immunotherapy have dramatically expanded the potential to manipulate immune cells in cancer patients with metastatic disease to counteract cancer spread and extend patient lifespan. One of the most successful types of immunotherapy is the immune checkpoint inhibitors, such as anti-CTLA-4 and anti-PD-1, that keep anti-tumour T cells active. However, not every patient with metastatic disease benefits from this class of drugs and patients often develop resistance to these therapies over time. Tremendous research effort is now underway to uncover new immunotherapeutic targets that can be used in patients who are refractory to anti-CTLA-4 or anti-PD-1 treatment. Here, we discuss results from experimental model systems demonstrating that modulating the immune response can negatively affect metastasis formation. We focus on molecules that boost anti-tumour immune cells and opportunities to block immunosuppression, as well as cell-based therapies with enhanced tumour recognition properties for solid tumours. We also present a list of challenges in treating metastatic disease with immunotherapy that must be considered in order to move laboratory observations into clinical practice and maximise patient benefit.

Similar content being viewed by others
Background
Cancer cells can detach from the primary tumour, intravasate into the blood or lymphatic system and migrate to distant sites where they extravasate from the blood or lymph vessels to seed secondary sites. Controlling the spread of cancer and outgrowth at these secondary sites remains the most challenging aspect of oncology. Across all cancer types, only around 20% of patients with stage IV metastatic disease survive beyond 5 years of diagnosis.1 Advances in immunotherapy have started to reverse these dire statistics and, because of their success in some types of metastatic cancer, immunotherapies have been heralded as revolutionary drugs in the treatment of metastatic disease. T-cell checkpoint inhibitors, in particular, such as those that target programmed cell death protein 1 (PD-1), its ligand PD-L1, or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), are responsible for the majority of immunotherapy successes in cancers such as melanoma and lung cancer; other cancer types exhibit lower numbers of responders with these drugs, which could be due to the lack of T-cell infiltration or (neo)antigen expression.2 Manipulating a cancer patient’s own T cells has solidified the concept that immune cells can be targeted effectively for patient benefit. However, cancer immunotherapy has a limited range. We have already learnt from clinical trials testing anti-CTLA-4 and anti-PD1 that these drugs are largely futile in eradicating secondary tumours and extending the lifespan of most cancer patients with metastasis, due to inherent or acquired drug resistance.3 Consequently, alternative immunotherapeutic approaches are required to counteract metastasis in patients that are unresponsive to CTLA-4 and/or PD-1/PD-L1 inhibitors. As invasion and metastasis rely heavily on the pro-tumour and anti-tumour functions of immune cells, understanding the cellular and molecular processes that underpin the progression of metastatic disease is likely to offer novel immunotherapy options. In this review, we discuss the advances that have been made in generating immunotherapeutic alternatives to CTLA-4 and PD-1/PD-L1 in experimental model systems of solid tumours, as well as highlighting the challenges in treating secondary tumours with immunotherapy.
Enhancing endogenous anti-tumour immunity
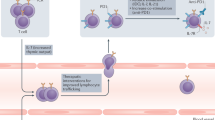
In addition to CTLA-4 and PD-1, a number of other T-cell checkpoint inhibitors that might prove extremely valuable in treating metastatic tumours have emerged over the past years, such as TIM-3 and LAG-3 (Fig. 1).4 Furthermore, the catalogue of inhibitory molecules has expanded to include receptors expressed on natural killer (NK) cells,5 several of which are shared by γδ T cells.6 Given that NK cells are critical in combating metastasis,7 blocking inhibitory molecules on NK cells, which limit their killing and cytotoxic molecule production, such as TIGIT and NKG2A, could prove very useful in treating patients with metastatic disease. Below we outline such T-cell-associated immune checkpoint inhibitors and NK cell-associated immune checkpoint inhibitors. Employing alternative immune checkpoint inhibitors to anti-CTLA-4 and anti-PD-1 that activate innate-like and/or adaptive lymphocytes could provide additional benefit or perhaps even function more effectively than the standard anti-CTLA-4/PD-1 therapies (Fig. 1).

a In metastatic progression, cancer cells detach from the primary tumour, intravasate into the blood or lymphatic system and migrate to distant sites where they extravasate from the blood or lymph vessels to seed secondary tumour sites. b Cancer cells, metastasis-associated macrophages and other cells at metastatic sites can express a plethora of immunomodulatory proteins to inhibit and activate anti-tumour T cells. The binding of these ligands to their cognate checkpoint receptors, such as programmed death-ligand 1 (PD-L1) with programmed cell death protein 1 (PD-1) and galectin-9 or carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) with TIM-3, dampens T-cell activation and effector anti-tumour T-cell responses. Checkpoint molecules, such as VISTA, LAG-3 and CTLA-4 are inhibitory receptors that deliver negative stimulation signals upon binding to MHC-II, FGL1 and the co-stimulation molecules CD80 and CD86. c Engagement of stimulatory receptors such as OX40, ICOS, CD40, B7-H3 and CD27 with their cognate ligand, or agonists that artificially provide these signals, drives T-cell activation, differentiation and effector responses (R? = unknown receptor). Dendritic cells can be activated through CD40 and CD70 to induce their maturation and antigen-presenting properties. d Natural killer (NK) cells can be manipulated by cancer cells and myeloid cells that express inhibitory ligands to dampen their cytotoxic effector responses. The inhibitory receptors T-cell immunoreceptor with Ig and ITIM domains (TIGIT) and CD96 both have affinity for CD155, which is expressed on many types of cancer cell. NKG2A binds HLA-E on human cells or Qa-1b on mouse cells to block NK-cell-mediated killing. These inhibitory and activatory checkpoint pathways can be selectively modulated by blocking or agonist antibodies to release the brake on anti-tumour immunity in order to treat or prevent metastatic disease.
TIM-3
One of the most promising immune-modulating checkpoints currently under investigation is T-cell immunoglobulin and mucin domain 3 (TIM-3), encoded by the gene HAVCR2.8 TIM-3 can be expressed by multiple subsets of T cells including CD8+ and CD4+ T cells, regulatory T cells and NK cells,9,10,11 as well as myeloid cells, such as dendritic cells and macrophages.12,13,14,15 In the absence of ligand, TIM-3 recruits the tyrosine kinase LCK to mediate T-cell activation. However, ligand engagement of TIM-3 is known to disrupt the immunological synapse, interfere with LCK signalling and induce T-cell apoptosis (reviewed in ref. 8). The two most well-studied ligands of TIM-3 include galectin-9 and carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), both of which can be produced by cancer cells and myeloid cells (reviewed in ref. 8).
The expression of TIM-3 is associated with advanced tumour stage and lymph node metastasis in lung cancer patients.10 In patients with metastatic melanoma, TIM-3 expression is also associated with dysfunctional/exhausted CD8+ T cells and NK cells, and the inhibition of TIM-3 signalling ex vivo increases the functional capacity of these cells.16,17 Interestingly, the reversion of patient-derived CD8+ T-cell dysfunction requires dual blockade of both TIM-3 and PD-1.16 After anti-PD-1 therapy, the expression of TIM-3 increases on CD4+ and CD8+ T cells in metastatic lung cancer patients and lung cancer mouse models driven by mutant epidermal growth factor receptor (EGFR) or mutant KRAS,18 suggesting that this molecule is involved in adaptive resistance to PD-1 inhibition. Indeed, the combinatorial treatment of lung-tumour-bearing CC10-rtTA;Tre-EgfrT790M/L858R mice with anti-PD-1 and anti-TIM-3 extends survival compared with anti-PD-1 blockade alone.18 Similarly, targeting both PD-1 and TIM-3 in mice bearing transplantable CT26 or MC38 colorectal cancer cells or B16 melanoma cells slows primary tumour growth.19 In the MMTV-PyMT mammary tumour model, paclitaxel and anti-TIM-3 blocking antibodies control primary tumour growth more efficiently than chemotherapy alone, while anti-TIM-3 monotherapy is ineffective.15 Mammary tumour-infiltrating CD103+ dendritic cells express high levels of TIM-3, whereas the expression of TIM-3 in CD8+ T cells is negligible in this model. These TIM-3-expressing CD103+ dendritic cells play a crucial role in the chemotherapy response, as their depletion abrogates the response to paclitaxel/anti-TIM-3 treatment. The blockade of TIM-3 or galectin-9 in MMTV-PyMT tumour-bearing mice increases the expression of CXC chemokine ligand 9 (CXCL9) in CD103+ dendritic cells, which recruits cytotoxic CXC chemokine receptor 3 (CXCR3)+ CD8+ T cells to tumours.15 Given the importance of TIM-3-expressing T cells and dendritic cells in cancer progression and chemotherapy response, TIM-3 inhibition might be useful in the metastatic setting to recruit cytotoxic T cells or to reinvigorate exhausted T cells in immunotherapy-naïve and/or anti-PD-1 refractory patients.
LAG-3
Lymphocyte activation gene 3 (LAG-3) is another checkpoint molecule expressed on T cells and NK cells; it exerts its inhibitory function by binding to MHC class II and other ligands, such as LSECtin (reviewed in ref. 20). A newly discovered LAG-3 ligand produced by liver and cancer cells—fibrinogen-like protein 1 (FGL1)—has been identified to mediate antigen-specific T-cell suppression.21 Increased LAG-3 expression on CD4+ and CD8+ T cells is associated with liver metastasis in mismatch-repair-proficient colorectal cancer. Like PD-1 and TIM-3, LAG-3 expression is associated with dysfunctional tumour-infiltrating T cells in mouse models and human metastatic tumours,22,23,24 and the combination of anti-PD-1 and anti-LAG-3 treatment delays tumour growth in mice bearing subcutaneous Sa1N fibrosarcoma cells or MC38 colorectal cancer cells.25 The anti-PD-1/anti-LAG-3 combination is also highly efficacious in the metastatic IE9mp1 transplantable ovarian cancer model.26,27 In lung colonisation models of experimental metastasis using 4T1 mammary cells, LAG-3 blockade accompanied by treatment with immunostimulatory interleukin (IL)-12 reduces tumour growth more effectively than anti-LAG-3 or IL-12 treatment alone. In this model, tumour control is mediated by targeting LAG-3-expressing NK cells with anti-LAG-3 antibodies.28
The expression of LAG-3 is regulated by glycogen synthase kinase-3 (GSK-3), and attempts at decreasing LAG-3 transcription in T cells and NK cells using a GSK-3β inhibitor have shown that this is a viable strategy to counteract B16 melanoma growth in the lung.29 In another approach, instead of blocking LAG-3 signalling, LAG-3 fused to the Fc region of IgG1 (LAG-3Ig or IMP321) can be used as an MHC-II agonist to activate dendritic cells and anti-tumour T-cell responses through so-called immunopotentiation. An early phase trial with metastatic breast cancer patients demonstrated the potency of this molecule in combination with paclitaxel, where 15 out of 30 women exhibited an objective tumour response.30 Thus, manipulating LAG-3 in patients with metastasis shows promise.
B7-CD28 superfamily members
CTLA-4 and PD-1 belong to the B7-CD28 superfamily. Some of the lesser-known members of this family play a role in primary tumour progression, suggesting that there could be benefit in targeting these molecules in the metastatic setting. For example, stimulation of inducible T-cell co-stimulator (ICOS) is required for the anti-tumour efficacy of anti-CTLA-4 in B16 and MC38 primary tumours, by generating an effector T helper 1 (TH1)-like population that plays a role in limiting tumour growth.31,32,33 Targeting V-domain Ig suppressor of T-cell activation (VISTA), which can be expressed by cancer cells or antigen-presenting cells, delays tumour growth in transplantable models and the transgenic Tyr::CreERT2;BrafV600E;PtenF/F melanoma model, and shifts the tumour microenvironment towards anti-tumour immunity.34,35 B7-H3 (also known as CD276) is expressed on cancer cells and tumour-associated endothelial cells. Targeting B7-H3 with antibody–drug conjugates reduces metastatic progression, and this effect is independent of adaptive immune cells, as nude mice were used in these experiments.36 Further experimentation of inhibitors to these molecules in metastasis models is required to understand whether their function is similar between primary and secondary tumours.
TNFR superfamily members
Some data on the importance of the tumour necrosis factor receptor (TNFR) superfamily in promoting or restricting immunity to tumours exist.37 This group of molecules comprises largely co-stimulatory molecules that synergise with T-cell receptor signalling to promote T-cell division. For example, OX40 is upregulated on activated CD4+ and CD8+ T cells, and an agonistic antibody of OX40 synergises with an inhibitor of transforming growth factor (TGF)-β to reduce the primary tumour growth of 4T1 mammary cells as well as spontaneous lung metastasis.38
CD4+ T cells activate CD40 on antigen-presenting cells, to facilitate the maturation of these cells, and agonists to CD40 have been used to stimulate CD103+ dendritic cells and prime tumour-specific T cells in genetically engineered mouse models of pancreatic ductal adenocarcinoma.39,40 CD40 agonist therapy—either as monotherapy or in combination with cytokines or anti-PD-1 or agonists of the TNFR member CD137—counteracts metastasis in transplantable melanoma, pancreatic, colon and kidney cancer models,41,42,43,44 and this immunotherapy can re-polarise myeloid cells towards an anti-tumour phenotype.41,44 Similarly, agonists of another TNFR member, CD27, reduce lung tumour burden of intravenously injected B16 melanoma cells.45 Naïve CD4+ and CD8+ T cells constitutively express CD27, and its activation by the ligand CD70 on dendritic cells supports T-cell priming. CD27 signalling has been found to be necessary to generate robust cytotoxic T cells,46,47 so CD27 agonists might further improve anti-PD-1 or CTLA-4 immunotherapy in cases where tumour-specific T cells are suboptimal. Other TNFR superfamily members, such as CD30, GITR, BTLA, are currently not well studied in the metastatic setting.
TIGIT and CD96
T-cell immunoreceptor with Ig and ITIM domains (TIGIT; a co-inhibitory receptor expressed by T cells and NK cells) and CD96 comprise a pathway analogous to CTLA-4 with CD28, where they bind the same interacting partner—CD155—to negatively regulate NK cell function [reviewed in48]. Inhibition of TIGIT with neutralising antibodies in lung colonisation experiments of 4T1 mammary cell lines or B16 melanoma cells or carrying out the experiments in TIGIT knockout mice reduces lung tumours and extends survival.49 One study found that anti-TIM-3 was required in Tigit–/– mice to reduce experimental lung metastasis of B16 cells.50 CD8+ T cells also express TIGIT,51 making it an ideal immunotherapy target to boost the anti-tumour functions of two cytotoxic cell types. Similarly, anti-CD96 therapy reduces 4T1 or B16 tumours in the lung and this effect is enhanced by the addition of anti-CTLA-4, anti-PD-1 or doxorubicin chemotherapy.52 Likewise, Cd96–/– mice develop fewer experimental lung metastases than wild-type mice after tail vein injection of B16 cells, and this result is dependent on NK cells and interferon (IFN)γ.53 As both TIGIT and CD96 bind CD155, targeting both TIGIT and CD96 might be essential to achieve maximum anti-metastatic benefit.52
NKG2A
NKG2A is another putative checkpoint inhibitor for NK cells and CD8+ T cells; its activation occurs through binding of HLA-E in humans or Qa-1b in mice.54 Blocking NKG2A using the humanised anti-NKG2A antibody monalizumab increases NK-mediated killing through antibody-dependent cell-mediated cytotoxicity (ADCC) of mouse lymphoma cells in vivo; however, the potency of monalizumab in counteracting metastases of solid tumours remains to be seen.55 Clinical trials are underway examining monalizumab with cetuximab (anti-EGFR) in patients with metastatic colorectal or head and neck cancers (NCT026435509).
IL-1R8
Interleukin 1 receptor 8 (IL-1R8, also called SIGIRR or TIR8) has been identified as a checkpoint inhibitor on NK cells. Spontaneous lung metastasis from MN/MCA1 sarcoma cells is reduced in Il1r8−/− mice when compared with wild-type mice but primary tumour growth remains unaffected. Similarly, Il1r8−/− mice are protected from liver metastasis of MC38 cells, and these events are reversed by NK cell depletion.56
Cell-based immunotherapies for metastatic disease
T cells, NK cells and dendritic cells can be harvested from either cancer patients or healthy donors, expanded ex vivo, and then transfused back into cancer patients in the process of adoptive cell therapy (ACT). Whereas ACT using αβ T cells, γδ T cells or NK cells for the treatment of haematological malignancies is well documented,5,57,58 evidence for the use of such approaches to treat the metastatic disease of solid tumours is scant. This might be because of the time it takes to generate these cells, the lack of expression of specific tumour antigens or a lack of efficacy of the transplanted cells. However, a few examples of the potential of these strategies to treat metastatic disease do exist. Thus far, ACT of CD8+ T cells into metastatic melanoma patients has been demonstrated to be the most successful regimen among these types of immunotherapy (reviewed in ref. 59). Like checkpoint inhibitors, the efficacy of ACT might be dependent on high expression of (neo)antigens; however, effective ACT in breast cancer patients, whose tumours exhibit a much lower mutational burden, has been documented.60 One advantage of the innate-like lymphocytes, γδ T cells and NK cells is that they are not restricted by MHC molecules, which bypasses the importance of (neo)antigen expression. In fact, ACT of γδ T cells has induced complete remission of lung metastasis in a patient with renal cell carcinoma.61
Moreover, αβ T cells, γδ T cells and NK cells can also be genetically modified to enhance their anti-tumour properties, and several attempts have been made at harnessing the potent killing abilities of these cells by introducing transgenic T-cell receptors or chimeric antigen receptors (CARs) (reviewed in refs. 57,62,63). CAR-T cells are gaining traction in solid tumours and these cells might be useful for the metastatic disease if the correct antigen can be identified. For example, guanylyl cyclase C (GUCY2C)-targeted CAR-T cells can reduce CT26 colorectal cancer burden in the lungs of mice and extend survival when compared with CAR-T-cell control-treated mice.64,65 Human macrophages have also been engineered with CD3ζ-based CARs similar to T-cell CARs in order to direct the phagocytic activity of these cells against tumours, and these CAR-Ms reduce the burden of lung tumours by ovarian SKOV3 cancer cells.66 These HER2-directed CAR-Ms effectively reduced tumour burden and recruited T-cells and presented antigens to them. The field of cell-based immunotherapies is rapidly evolving, with various endeavours to make these products more specific, durable and safe, so that future versions are likely to improve their benefits in the metastatic setting.
Inhibiting pro-tumour immune cells and immunosuppression at metastatic sites
Data from mouse metastasis models have highlighted a critical role for myeloid cells and some innate lymphocyte populations in metastatic progression. Here, we focus on monocytes/macrophages, neutrophils, regulatory T (TREG) cells and IL-17-producing γδ T cells. Since myeloid-derived suppressor cells (MDSCs) encompass both monocytes and neutrophils that are pathologically activated by tumour-derived factors to suppress anti-tumour immune cells,67 we refrain from using the MDSC nomenclature. Instead, we refer specifically to monocytes or neutrophils to be more precise about their individual role in metastasis formation. Information on other cell populations, such as eosinophils, basophils, mast cells and innate lymphoid cells (ILCs), is scarce in the metastatic setting, prohibiting a lengthy discussion on opportunities to target these cells. Further knowledge on these lesser-studied immune cells will expand the potential of targetable pathways. For example, a recent study found that group 2 ILCs and eosinophils support lung metastasis through suppression of NK cells, highlighting the cytokines, IL-33 and IL-5 in this process.68 While this section focuses on the pro-metastatic role of immune cells, it should be noted that the role of these cells in cancer progression is dynamic and subject to their local microenvironment. Immune cells, in particular macrophages and neutrophils, are not always pro-tumour – their function depends on their location, polarisation, maturation status and stage of the disease.
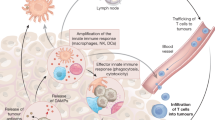
Macrophages are the best-studied group of immune cells in metastasis. We have known about their potent ability to support metastasis69 through angiogenesis70,71 for nearly 20 years, but these cells drive metastasis via multiple other mechanisms, such as providing growth factors for disseminated cancer cells as well as immune suppression of anti-tumour T cells and NK cells (reviewed in ref. 72). Neutrophils are increasingly being recognised for their pro-metastatic functions. These cells have been somewhat overlooked or avoided as they are difficult to manipulate, despite evidence of their involvement in metastasis existing more than 10 years before the data on pro-metastatic macrophages.73 Like macrophages, neutrophils can drive metastasis both from the primary tumour site or secondary locations before and after the arrival of disseminated cancer cells in distant organs (reviewed in ref. 74). Although low in number, γδ T cells can be very influential in cancer progression, where they orchestrate immune responses and modulate endothelial cells at metastatic sites primarily through the production of the pro-inflammatory cytokine IL-17.75,76 Macrophages, IL-17-producing γδ T cells and neutrophils can even work together to establish a systemic inflammatory pathway that suppresses CD8+ T cells at the pre-metastatic site and supports metastasis in p53-deficient mammary tumour models.75,77 Finally, TREG cells are known to facilitate metastatic progression by immunosuppression, and to shield cancer cells from immune detection (reviewed in ref. 78). To interfere with the activity of these pro-metastatic immune cells, three key processes that can be targeted have been identified from preclinical studies: recruitment, survival and reprogramming (Fig. 2).72

Recruitment, survival and re-programming of immune cells to a pro-tumorigenic phenotype at distant sites are key processes in the metastatic cascade. a Primary and secondary tumours release chemokines that attract aiding and abetting immune cells to encourage metastasis. In many cancers, CCR2+ bone-marrow-derived monocytes are recruited to primary and secondary tumours by the chemokine ligand CCL2, where these monocytes differentiate into tumour-associated macrophages (TAM). Pro-metastatic CXCR2+ neutrophils are recruited by CXCL1, CXCL2 or CXCL5, while pro-tumour CCR4+ regulatory T (TREG) cells require CCL17 or CCL22. Targeting these chemokine pathways can prevent the accumulation of these cell types and reduce metastasis in the liver or lung of colorectal, pancreatic and breast cancer mouse models. b Targeting colony stimulating factor (CSF)-1, granulocyte-macrophage (GM)-CSF and granulocyte (G)-CSF affects the pro-metastatic cascade. TAMs secrete interleukin (IL)-1β to activate IL-17-producing γδ T cells, which induce immunosuppressive neutrophils through G-CSF. Metastasis-associated macrophages (MAM) provide growth factors, survival signals and angiogenic factors at secondary sites to support outgrowth of cancer cells. c The cytokine IL-2 is essential for the survival of pro-tumour TREG cells as well as the activation of anti-tumour natural killer (NK) cells. Targeting selective IL-2 receptors on TREG cells might prevent their accumulation while enabling anti-tumour NK cells to remain active. d Tumour-derived factors such IL-4, vascular endothelial growth factor (VEGF) and angiopoietin 2 (ANGPT2) can induce pro-tumorigenic macrophages, while transforming growth factor (TGF)-β can enhance pro-metastatic neutrophils. CD47 functions as a ‘don’t eat me’ signal and can be upregulated on metastatic cancer cells to evade immune surveillance and phagocytosis by macrophages. Tumour-derived WNT ligands induce macrophages to secrete IL-1β, which activates IL-17-producing γδ T cells to drive pro-metastatic neutrophils. Blocking or interfering with the cytokine cascade or receptors on these pro-metastatic immune cells could reprogramme them away from a pro-tumorigenic phenotype in order to prevent metastatic disease.
Blocking recruitment of pro-metastatic immune cells
CCR2+ bone marrow-derived monocytes are readily recruited to primary and secondary tumours in multiple tumour types by the chemokine CCL2.79,80,81 Consequently, the CCL2–CCR2 axis represents one point at which the accumulation of metastasis-associated macrophages (MAM) could be prevented.82,83 CCR2 small molecule antagonists are effective in transplantable models of pancreatic cancer and hepatocellular carcinoma,80,84 suggesting that secondary tumours may also be susceptible to these drugs. As pro-metastatic γδ T cells also express CCR2,83,85,86 inhibitors of CCL2 or CCR2 could be beneficial for targeting cancer types that rely on monocytes or γδ T cells or both. In lung metastasis of MMTV-PyMT tumours, CCR2 signalling regulates monocyte retention by CCL3 activation of CCR1,87 and CCR1+ cells are also important in driving colorectal cancer liver metastasis,88 highlighting another point of intervention to thwart metastasis. CCR5 is a another promising target for macrophages (and cancer cells) in colorectal cancer liver metastasis, as its inhibition repolarises macrophages towards an anti-tumoural phenotype.89 In a Phase 1 trial called MARACON, CCR5 inhibition with the HIV drug, maraviroc, in patients with metastatic colorectal cancer was well tolerated, and tumours exhibited reduced proliferation.89 However, there are risks to targeting chemokines, and the duration of treatment is critical to avoid a rebound effect that leads to increased metastasis, as is seen in models of breast cancer lung metastasis after anti-CCL2 therapy, where interruption of treatment releases monocytes from the bone marrow and accelerates metastasis formation.90 A new mouse model in which the chemokine receptors CCR1, CCR2, CCR3 and CCR5 are deleted together has been generated.91 The use of such models in combination with metastasis models will hopefully shed light on combinatorial chemokine receptor function for pro-metastatic immune cells and will help to determine the context in which to target these receptors.
Neutrophils use a different set of chemokine receptors to monocytes/macrophages, such as CXCR1 and CXCR2. Inhibition of CXCR2 in models of spontaneous metastasis—such as the KrasG12D;Trp53R172H;Pdx1-Cre (KPC) pancreatic cancer model or the Villin-CreER;KrasG12D;Trp53F/F;Rosa26N1cd/+ (KPN) colon cancer model—reduces the occurrence of secondary tumours in the liver without affecting survival.92,93 In the context of liver metastasis as well as lung metastasis,75 neutrophils suppress CD8+ T-cell responses to help disseminated cancer cells evade anti-tumour immunity, suggesting that combining neutrophil targeting with T-cell-based immunotherapy might be better than either approach alone. Indeed, treatment of pancreatic-tumour-bearing KPC mice with CXCR2 inhibitors and anti-PD-1 antibodies extends survival beyond monotherapy controls.92
Primary 4T1 mammary tumours can induce the production of CCL17/TARC (thymus- and activation-regulated chemokine) in the pre-metastatic lung, which guides the recruitment of CCR4+ TREG cells and cancer cells to this site.94 The TREG cells then protect cancer cells by inhibiting NK cells, thereby facilitating metastasis formation. Depleting TREG cells, inhibiting CCR4 and the combined silencing of CCL17 and the TREG master transcription factor FOXP3 in CCR4+ cells reduces the number of metastatic foci in the lung.94 Another way to reduce the recruitment of TREG cells to pre-metastatic sites in liver and mammary tumour models is accomplished by reducing CCL22 secretion through miR-34 expression, as CCL22 also binds to CCR4 on TREG cells to promote their immunosuppressive effects.95 Thus, targeting chemokine receptors in patients with metastatic disease might overcome immunosuppressive barriers that are established by certain immune cell populations.
Neutralising survival factors of pro-metastatic immune cells
The colony stimulating factor (CSF) family members CSF-1, granulocyte-macrophage (GM)-CSF and granulocyte (G)-CSF are essential for the development, differentiation and survival of myeloid cells.96 It is perhaps then not surprising that cancer cells often directly or indirectly upregulate CSF molecules to promote pro-metastatic macrophages and neutrophils and thus to facilitate cancer progression. Consequently, targeting these molecules should reduce macrophage or neutrophil survival and negatively affect metastasis formation. For instance, early studies using Csf1-knockout mice, in which macrophages are severely depleted, showed that these cells are required for lung metastasis in MMTV-PyMT mammary tumour-bearing mice.69 Subsequently, antibodies and small molecules that target the CSF-1 receptor (CSF-1R) have been shown to reduce metastasis, such as in the MMTV-HER2 mammary tumour model,97 and to synergise with chemotherapy.80,98,99,100 The potency of CSF-1R inhibitors has prompted many pharmaceutical companies to trial these inhibitors in cancer patients.101 However, anti-CSF-1R therapy has been shown to lead to increased metastasis relative to controls through an increase in the number of neutrophils mediated by a compensatory increase in serum G-CSF102 or a reduction in the number of NK cells as a consequence of a decrease in the myeloid-cell derived NK survival factor IL-15.103 These data suggest that depleting macrophages completely might not be appropriate in every scenario and that it is important to understand the nuances of macrophage biology in order to manipulate pro-metastatic polarisation states. Another point to consider when using CSF-1R inhibitors is their inability to distinguish bone marrow-derived macrophages from tissue-resident macrophages, since they have different roles in tumour development and progression and these cells may function at different stages.104,105 In some cases, however, such as in pancreatic cancer where bone marrow-derived macrophages play a role in antigen presentation and tissue-resident macrophages produce and remodel extracellular matrix molecules, targeting both populations might be the best approach to prevent cancer spread.106
G-CSF is the master regulator of granulopoiesis, and several studies have shown that inhibition of G-CSF decreases neutrophil-mediated metastasis.75,107,108 GM-CSF is somewhat redundant to G-CSF in neutrophil regulation; although, its expression is dominant in certain contexts.109 Thus, data from mouse models indicate that inhibiting G-CSF and GM-CSF in metastatic cancer patients with neutrophilia to lower neutrophil numbers may reduce secondary tumour formation and/or burden. Since chemotherapy induces neutropenia, decreased neutrophil numbers is often achieved without neutralising G-CSF or GM-CSF. Indeed, chemotherapy-induced neutropenia is associated with better outcome in patients with lung, breast, gastric and colorectal cancer,110,111,112,113 supporting the notion of targeting neutrophils in patients with advanced disease. However, to offset infection and neutropenia, cancer patients on chemotherapy may be given G-CSF or GM-CSF. Whether these recombinant cytokines contribute to disease progression in this context needs further investigation.
Another controversial cytokine for the potential treatment of cancer is IL-2. IL-2 is not only important for the survival and function of TREG cells, which have high-affinity IL-2 receptors, but it is also vital for the activation of NK cells and effector T cells.114 IL-2 immunotherapy has shown limited success and severe side effects in clinical trials, which could be due to the competition of TREG cells and NK cells for cytokines. However, as TREG cells and NK cells express different IL-2 receptors (IL-2Rα and IL-2Rβ, respectively) efforts have been made to synthesise chimeric IL-2–IL-2Rβ or mutant IL-2 that preferentially binds to the IL-2Rβ in order to selectively activate NK cells; these agents show improved anti-tumour action, but have not been well studied in metastatic settings yet.115,116
Reprogramming pro-metastatic immune cells
Because of the plasticity of myeloid cells, the metastasis-promoting phenotype of monocytes, macrophages and neutrophils can be easily influenced by tumour-derived factors. In the MMTV-PyMT mammary tumour model, CD4 T-cell-derived IL-4, cancer cell-derived vascular endothelial growth factor (VEGF) and endothelial cell-derived angiopoietin 2 (ANGPT2) have all been shown to modulate the phenotype of pro-metastatic macrophages to promote lung metastasis.117,118,119 In the same model, the metastasis-promoting phenotype of macrophages can be reversed epigenetically by using the class IIa histone deacetylase (HDAC) inhibitor (TMP195), which increases their phagocytic ability, reduces primary tumour burden and prevents lung metastasis.120 These data demonstrate that class IIa HDACs acting on macrophages may enhance the efficacy of conventional therapies in breast cancer patients. WNT signalling constitutes another pathway that drives macrophage polarisation towards a pro-metastatic phenotype. Across 16 different genetically engineered mouse models of breast cancer, WNT genes were found to be upregulated in mammary tumours driven by the loss of p53. Increased expression of WNT proteins activated macrophages to secrete IL-1β, which promoted metastasis through crosstalk with γδ T cells and neutrophils in the lung; WNT inhibition by administration of LGK974 – an inhibitor of WNT ligand secretion – re-programmed these macrophages and reduced metastasis.77 In some cases, however, reprogramming macrophages might not be enough to counteract metastasis, as cancer cells can harbour alternative methods to avoid destruction by macrophages. CD47, which functions as a ‘don’t eat me’ signal, can be highly expressed by cancer cells to subvert phagocytosis by signal-regulatory protein α (SIRPα)-expressing anti-tumour macrophages.121 CD47 is upregulated on circulating colorectal cancer cells,122 and its inhibition with neutralising antibodies reduces metastasis in a variety of mouse models and patient-derived xenografts.123,124,125,126,127,128
For many years, neutrophils were thought to be short-lived cells that were unable to respond to tumour-derived factors, but several molecules, such as G-CSF75,107,129 and TGF-β,93,130,131 have been shown to repolarise these cells towards a pro-metastatic phenotype. In p53-deficient mammary tumour models, macrophage-expressed IL-1β triggers γδ T cells to express IL-17, which induces the G-CSF-dependent expansion and polarisation of neutrophils, which, in turn, suppress the activity of cytotoxic CD8+ T cells.75,77 Consequently, inhibition of TGF-β, IL-17 and G-CSF reverses the phenotype of neutrophils and promotes the activity of cytotoxic CD8+ T-cells to subvert metastasis.75,93,129,131
Finally, as TREG cells are highly abundant in tumours and suppress effector immune function, their reprogramming towards an effector phenotype could prove a fruitful strategy to increase tumour immunity and prevent metastasis. Blocking critical receptors on TREG cells, such as CD25 with antibodies and genetically altering neuropilin-1 (Nrp-1), which are required to develop and maintain the stability and function of TREG cells, changes these cells to pro-inflammatory cells that produce IFN-γ.132,133
Reprogramming any one of these immune cell populations by interfering with the cytokine cascade would, therefore, be advantageous in thwarting metastasis.
Challenges in treating metastatic disease with immunotherapy
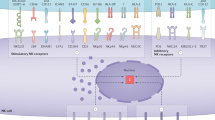
To develop successful immunotherapies for metastatic disease, a number of considerations that might affect immunotherapy efficacy need to be taken into account. The first consideration includes the type of cancer and the location of the tumour. Although it might seem obvious, genetic mutations differ significantly between cancer types, and genetic mutations affect the immune response. Metastases can differ from their primary tumour in terms of mutational and immune profiles,134,135,136 and although there are similarities between metastases of the same organ from different cancer types, there can also be cancer-specific variations (Fig. 3). For instance, CD8+ T-cell infiltration is equivalent between lung metastases from colorectal cancer and renal cell carcinoma, but NK cells are more abundant and prognostic indicators in renal cell carcinoma lung metastasis.137 The immune contexture of primary and secondary tumours can also be analogous with regards to immunologically silent or active microenvironments as, across multiple cancer types, immune active primary tumours are more likely to generate immune active metastases.138 However, the immune landscape might also very well look different between primary and secondary tumours. These differences might be dependent on the organ that harbours the secondary tumour(s) or the increased mutational burden of distant metastases.134,139,140,141,142 Adding to this complexity, the immune landscape across metastases might not be uniform, with hot and cold tumours existing within the same patient.134,140,143,144,145

Metastatic tumours differ from primary tumours in various ways. Metastases occurring in various locations must adapt to the new tissue-specific environment (coloured circles). Metastatic tumours can acquire new (epi)genetic mutations, but antigens arising from these mutations are not always presented on the surface of cancer cells, thereby preventing T-cell recognition. The immune landscape can also be very different between primary and secondary tumours, due to varying abundance of specific immune cell populations between organs. Finally, immune responses to metastatic lesions might evolve significantly over the course of time due to acquired resistance to anti-cancer therapy (chemotherapy, radiotherapy, targeted therapy, etc) by secondary tumours.
Related to this, tissue-specific immunity must also be considered, because the immune system differs between anatomical locations. Data emerging from anti-PD-1 clinical trials indicate that checkpoint inhibitors are more beneficial for patients with lung metastasis than liver metastasis, for example.146,147,148,149 In melanoma patients, liver metastases have a lower density of CD8+ T cells at the tumour margin when compared with metastases in other organs,149 which could explain the reduced response to PD-1 inhibition at this site. Likewise, in patients with metastatic prostate cancer, anti-CTLA-4 induces anti-tumour TH1 cell-type CD4+ T-cell responses in primary tumours, but this same response is absent in bone metastatic lesions. Instead, the bone marrow tumour microenvironment, which is rich in TGF-β, converts CD4+ T cells into TH17 cells to blunt anti-CTLA-4 immunotherapy at this site.150 Combining TGF-β inhibitors with anti-CTLA-4 to generate TH1 CD4+ T cell and CD8+ T-cell responses can reverse these effects in metastatic lesions. Brain and bone metastasis represent some of the most challenging tumours to treat, largely due to aberrant vascularisation at these sites or their immune-specialised status. Bone is a particularly immune-privileged site in order to protect and preserve the hematopoietic stem cell compartment. In breast cancer bone metastasis, the cycle of bone degradation and tumour growth has been shown to be a critical event in permitting the outgrowth of metastatic cells from dormancy.151 Therefore, combining immunotherapies with osteoclast inhibitors may be key in the success of treating bone metastasis. In the case of brain metastasis, the success of T cell and NK cell-based immunotherapies is heavily dependent on antibodies or small molecules being able to penetrate the blood brain barrier or reactivate T cells in highly immunosuppressive cervical lymph nodes.152 Recently, it has emerged that anti-PD-1 and anti-CTLA-4 therapy in tandem significantly increased intracranial anti-tumour activity in patients with metastatic melanoma, offering promise for the use of immune checkpoints in treating brain metastasis.153 However, modulation of T-cell trafficking molecules and/or dendritic cell assistance may be critical to overcome immunosuppression of T cells and NK cells in brain and bone metastasis.
Finally, the success of T-cell- and NK cell-based immunotherapy is dependent on the ability of these immune cells to recognise cancer cells. For example, HLA loss of heterozygosity (LOH) precludes the presentation of (neo)antigens by cancer cells, and HLA LOH can be more common in metastatic tumours than in primary tumours.134,154,155 Dormant cancer cells—non-proliferating malignant cells hiding in distant organs—are another challenge in immune-cell recognition,156 as MHC-I expression might be downregulated on these cells.157 In addition to the ability of T cells to recognise cancer cells, the other components of the cancer immunity cycle—including antigen release, antigen presentation, T-cell priming, trafficking, tumour infiltration and killing—must be intact in patients for favourable outcomes of T-cell-based therapies.158 If any of these components are missing or become inactive as a result of cancer evolution, T-cell-based immunotherapies fail or become inadequate at controlling metastatic lesions. Moreover, acquired resistance to T-cell- and NK cell-based immunotherapy may arise after initially providing a strong anti-tumour response. Some of the mechanisms of acquired resistance have been identified, including HLA loss as stated above,134,154,155 epigenetic dysfunction of T-cells,159 emergence of additional immunosuppressive pathways,160 or resistance to IFN-γ via new cancer-specific mutations.161,162 Thus, inherent and acquired resistance to T-cell- and NK cell-based immunotherapy is a major challenge.
To overcome these challenges, a deeper understanding of the immune landscape, genetic mutations, components of the cancer immunity cycle and tissue-specific immunity is needed to facilitate personalised approaches to immunotherapy in metastatic disease. What is clear from experimental models and on-going clinical trials is that the immune response to metastatic lesions can change dramatically over time, and it is not linear. Therefore, future treatment modalities will need to anticipate how pro-tumour and anti-tumour immune cells react to first line immunotherapies and mitigate roadblocks with additional immunomodulatory drugs.
Conclusions
As outlined in this article, a great many new immunotherapeutic targets are on the horizon for metastatic disease. Possibly, however, the biggest improvement for patients will come from the use of combination therapies that both boost anti-tumour immunity and attenuate immunosuppression. Biomarkers arising from the study of anti-PD-1/CTLA-4 non-responding patients or from those patients who acquire resistance might also identify suitable immunotherapy targets to re-engage anti-tumour immune activity when anti-PD-1/CTLA-4 approaches become inert. Mechanisms to induce tertiary lymphoid structures and antigen-presenting B cells—two anti-tumour features not well explored in metastasis—could support effector T-cell responses and complement immunotherapies in metastatic disease, as seen in patients with melanoma or sarcoma.163,164,165
To optimally exploit these immunotherapies, it will be important to increase our understanding of the context in which they are most efficacious. These efforts will require more knowledge regarding the interplay between specific molecules and specific cell types in the metastatic setting. Choosing the right model is paramount to address this knowledge gap. Injectable cell lines, such as the B16 melanoma cells, have been instrumental in immunotherapy discovery, but they fail to represent the full metastatic cascade. The immunotherapy field will need to adopt or create models that recapitulate the evolution of the immune response that occurs when tumours are allowed to progress from early stage to late stage metastatic disease. These models might help to uncover immunotherapy targets that specifically rewire the pre-metastatic niche, prevent cancer cell seeding or eliminate established tumours at distant sites. Combining metastasis models with humanised mice might also be useful to enhance personalised immunotherapies. It is hoped that the development of these tools will generate new insights into immunotherapeutic intervention for metastatic disease.
References
Miller, K. D., Nogueira, L., Mariotto, A. B., Rowland, J. H., Yabroff, K. R., Alfano, C. M. et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 69, 363–385 (2019).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015).
Syn, N. L., Teng, M. W. L., Mok, T. S. K. & Soo, R. A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 18, e731–e741 (2017).
Qin, S., Xu, L., Yi, M., Yu, S., Wu, K. & Luo, S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol. Cancer 18, 155 (2019).
Souza-Fonseca-Guimaraes, F., Cursons, J. & Huntington, N. D. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. 40, 142–158 (2019).
Kabelitz, D., Serrano, R., Kouakanou, L., Peters, C. & Kalyan, S. Cancer immunotherapy with gammadelta T cells: many paths ahead of us. Cell Mol. Immunol. 17, 925–939 (2020).
Lopez-Soto, A., Gonzalez, S., Smyth, M. J. & Galluzzi, L. Control of metastasis by NK Cells. Cancer Cell 32, 135–154 (2017).
Wolf, Y., Anderson, A. C. & Kuchroo, V. K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 20, 173–185 (2020).
Monney, L., Sabatos, C. A., Gaglia, J. L., Ryu, A., Waldner, H., Chernova, T. et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002).
Gao, X., Zhu, Y., Li, G., Huang, H., Zhang, G., Wang, F. et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 7, e30676 (2012).
Ndhlovu, L. C., Lopez-Verges, S., Barbour, J. D., Jones, R. B., Jha, A. R., Long, B. R. et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 119, 3734–3743 (2012).
Anderson, A. C., Anderson, D. E., Bregoli, L., Hastings, W. D., Kassam, N., Lei, C. et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318, 1141–1143 (2007).
Chiba, S., Baghdadi, M., Akiba, H., Yoshiyama, H., Kinoshita, I., Dosaka-Akita, H. et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 13, 832–842 (2012).
Ocaña-Guzman, R., Torre-Bouscoulet, L. & Sada-Ovalle, I. TIM-3 regulates distinct functions in macrophages. Front Immunol. 7, 229–229 (2016).
de Mingo Pulido, A., Gardner, A., Hiebler, S., Soliman, H., Rugo, H. S., Krummel, M. F. et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell 33, 60–74 e66 (2018).
Fourcade, J., Sun, Z., Benallaoua, M., Guillaume, P., Luescher, I. F., Sander, C. et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186 (2010).
Gallois, A., Silva, I., Osman, I. & Bhardwaj, N. Reversal of natural killer cell exhaustion by TIM-3 blockade. Oncoimmunology 3, e946365 (2014).
Koyama, S., Akbay, E. A., Li, Y. Y., Herter-Sprie, G. S., Buczkowski, K. A., Richards, W. G. et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7, 10501 (2016).
Sakuishi, K., Apetoh, L., Sullivan, J. M., Blazar, B. R., Kuchroo, V. K. & Anderson, A. C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010).
Anderson, A. C., Joller, N. & Kuchroo, V. K. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004 (2016).
Wang, J., Sanmamed, M. F., Datar, I., Su, T. T., Ji, L., Sun, J. et al. Fibrinogen-like Protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347 e312 (2019).
Zhou, G., Noordam, L., Sprengers, D., Doukas, M., Boor, P. P. C., van Beek, A. A. et al. Blockade of LAG3 enhances responses of tumor-infiltrating T cells in mismatch repair-proficient liver metastases of colorectal cancer. Oncoimmunology 7, e1448332 (2018).
Matsuzaki, J., Gnjatic, S., Mhawech-Fauceglia, P., Beck, A., Miller, A., Tsuji, T. et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl Acad. Sci. USA 107, 7875–7880 (2010).
Baitsch, L., Baumgaertner, P., Devevre, E., Raghav, S. K., Legat, A., Barba, L. et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J. Clin. Invest 121, 2350–2360 (2011).
Woo, S. R., Turnis, M. E., Goldberg, M. V., Bankoti, J., Selby, M., Nirschl, C. J. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
Huang, R. Y., Eppolito, C., Lele, S., Shrikant, P., Matsuzaki, J. & Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 6, 27359–27377 (2015).
Huang, R. Y., Francois, A., McGray, A. R., Miliotto, A. & Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 6, e1249561 (2017).
Ohs, I., Ducimetiere, L., Marinho, J., Kulig, P., Becher, B. & Tugues, S. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. 77, 7059–7071 (2017).
Rudd, C. E., Chanthong, K. & Taylor, A. Small molecule inhibition of GSK-3 specifically inhibits the transcription of inhibitory co-receptor LAG-3 for enhanced anti-tumor immunity. Cell Rep. 30, 2075–2082 e2074 (2020).
Brignone, C., Gutierrez, M., Mefti, F., Brain, E., Jarcau, R., Cvitkovic, F. et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J. Transl. Med. 8, 71 (2010).
Fu, T., He, Q. & Sharma, P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 71, 5445–5454 (2011).
Fan, X., Quezada, S. A., Sepulveda, M. A., Sharma, P. & Allison, J. P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 211, 715–725 (2014).
Wei, S. C., Anang, N. A. S., Sharma, R., Andrews, M. C., Reuben, A., Levine, J. H. et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl Acad. Sci. USA 116, 22699–22709 (2019).
Le Mercier, I., Chen, W., Lines, J. L., Day, M., Li, J., Sergent, P. et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 74, 1933–1944 (2014).
Wang, L., Rubinstein, R., Lines, J. L., Wasiuk, A., Ahonen, C., Guo, Y. et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 208, 577–592 (2011).
Seaman, S., Zhu, Z., Saha, S., Zhang, X. M., Yang, M. Y., Hilton, M. B. et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-Positive tumor cells and tumor vasculature. Cancer Cell 31, 501–515 e508 (2017).
Ward-Kavanagh, L. K., Lin, W. W., Sedy, J. R. & Ware, C. F. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity 44, 1005–1019 (2016).
Garrison, K., Hahn, T., Lee, W. C., Ling, L. E., Weinberg, A. D. & Akporiaye, E. T. The small molecule TGF-beta signaling inhibitor SM16 synergizes with agonistic OX40 antibody to suppress established mammary tumors and reduce spontaneous metastasis. Cancer Immunol. Immunother. 61, 511–521 (2012).
Lin, J. H., Huffman, A. P., Wattenberg, M. M. Walter, D. M., Carpenter, E. L., Feldser, D. M. et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J. Exp. Med. 217, e20190673 (2020).
Hegde, S., Krisnawan, V. E., Herzog, B. H., Zuo, C., Breden, M. A., Knolhoff, B. L. et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell 37, 289–307 e289 (2020).
Weiss, J. M., Ridnour, L. A., Back, T., Hussain, S. P., He, P., Maciag, A. E. et al. Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy. J. Exp. Med. 207, 2455–2467 (2010).
Zhang, M., Yao, Z., Dubois, S., Ju, W., Muller, J. R. & Waldmann, T. A. Interleukin-15 combined with an anti-CD40 antibody provides enhanced therapeutic efficacy for murine models of colon cancer. Proc. Natl Acad. Sci. USA 106, 7513–7518 (2009).
Singh, M., Vianden, C., Cantwell, M. J., Dai, Z., Xiao, Z., Sharma, M. et al. Intratumoral CD40 activation and checkpoint blockade induces T cell-mediated eradication of melanoma in the brain. Nat. Commun. 8, 1447 (2017).
Ma, H. S., Poudel, B., Torres, E. R., Sidhom, J. W., Robinson, T. M., Christmas, B. et al. A CD40 Agonist and PD-1 antagonist antibody reprogram the microenvironment of nonimmunogenic tumors to allow T-cell-mediated anticancer activity. Cancer Immunol. Res. 7, 428–442 (2019).
Roberts, D. J., Franklin, N. A., Kingeter, L. M., Yagita, H., Tutt, A. L., Glennie, M. J. et al. Control of established melanoma by CD27 stimulation is associated with enhanced effector function and persistence, and reduced PD-1 expression of tumor infiltrating CD8(+) T cells. J. Immunother. 33, 769–779 (2010).
Ahrends, T., Babala, N., Xiao, Y., Yagita, H., van Eenennaam, H. & Borst, J. CD27 agonism Plus PD-1 blockade recapitulates CD4+ T-cell help in therapeutic anticancer vaccination. Cancer Res. 76, 2921–2931 (2016).
Ahrends, T., Spanjaard, A., Pilzecker, B., Babala, N., Bovens, A., Xiao, Y. et al. CD4(+) T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity 47, 848–861 e845 (2017).
Dougall, W. C., Kurtulus, S., Smyth, M. J. & Anderson, A. C. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 276, 112–120 (2017).
Zhang, Q., Bi, J., Zheng, X., Chen, Y., Wang, H., Wu, W. et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 19, 723–732 (2018).
Kurtulus, S., Sakuishi, K., Ngiow, S. F., Joller, N., Tan, D. J., Teng, M. W. et al. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Invest 125, 4053–4062 (2015).
Johnston, R. J., Comps-Agrar, L., Hackney, J., Yu, X., Huseni, M., Yang, Y. et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937 (2014).
Blake, S. J., Stannard, K., Liu, J., Allen, S., Yong, M. C. R., Mittal, D. et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 6, 446 (2016).
Chan, C. J., Martinet, L., Gilfillan, S., Souza-Fonseca-Guimaraes, F., Chow, M. T., Town, L. et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 15, 431–438 (2014).
Vance, R. E., Jamieson, A. M. & Raulet, D. H. Recognition of the class Ib molecule Qa-1(b) by putative activating receptors CD94/NKG2C and CD94/NKG2E on mouse natural killer cells. J. Exp. Med. 190, 1801–1812 (1999).
Andre, P., Denis, C., Soulas, C., Bourbon-Caillet, C., Lopez, J., Arnoux, T. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK. Cells Cell 175, 1731–1743 e1713 (2018).
Molgora, M., Bonavita, E., Ponzetta, A., Riva, F., Barbagallo, M., Jaillon, S. et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 551, 110–114 (2017).
Sebestyen, Z., Prinz, I., Dechanet-Merville, J., Silva-Santos, B. & Kuball, J. Translating gammadelta (gammadelta) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 19, 169–184 (2020).
Yang, J. C. & Rosenberg, S. A. Adoptive T-cell therapy for cancer. Adv. Immunol. 130, 279–294 (2016).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
Zacharakis, N., Chinnasamy, H., Black, M., Xu, H., Lu, Y. C., Zheng, Z. et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24, 724–730 (2018).
Kobayashi, H., Tanaka, Y., Shimmura, H., Minato, N. & Tanabe, K. Complete remission of lung metastasis following adoptive immunotherapy using activated autologous gammadelta T-cells in a patient with renal cell carcinoma. Anticancer Res. 30, 575–579 (2010).
Silva-Santos, B., Mensurado, S. & Coffelt, S. B. gammadelta T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 19, 392–404 (2019).
Morvan, M. G. & Lanier, L. L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16, 7–19 (2016).
Magee, M. S., Kraft, C. L., Abraham, T. S., Baybutt, T. R., Marszalowicz, G. P., Li, P. et al. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. Oncoimmunology 5, e1227897 (2016).
Magee, M. S., Abraham, T. S., Baybutt, T. R., Flickinger, J. C., Ridge, N. A., Marszalowicz, G. P. et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)- expressing T cells eliminate colorectal cancer metastases. Cancer Immunol. Res. 6, 509–516 (2018).
Klichinsky, M., Ruella, M., Shestova, O., Lu, X. M., Best, A., Zeeman, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020).
Tcyganov, E., Mastio, J., Chen, E. & Gabrilovich, D. I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 51, 76–82 (2018).
Schuijs, M. J., Png, S., Richard, A. C., Tsyben, A., Hamm, G., Stockis, J. et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat. Immunol. 21, 998–1009 (2020).
Lin, E. Y., Nguyen, A. V., Russell, R. G. & Pollard, J. W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 193, 727–740 (2001).
Lin, E. Y., Li, J. F., Gnatovskiy, L., Deng, Y., Zhu, L., Grzesik, D. A. et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 66, 11238–11246 (2006).
De Palma, M., Venneri, M. A. & Galli, R. Sergi, Sergi, L., Politi, L. S., Sampaolesi, M. et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 8, 211–226 (2005)
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018).
Welch, D. R., Schissel, D. J., Howrey, R. P. & Aeed, P. A. Tumor-elicited polymorphonuclear cells, in contrast to “normal” circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proc. Natl Acad. Sci. USA 86, 5859–5863 (1989).
Coffelt, S. B., Wellenstein, M. D. & de Visser, K. E. Neutrophils in cancer: neutral no more. Nat. Rev. Cancer 16, 431–446 (2016).
Coffelt, S. B., Kersten, K., Doornebal, C. W., Weiden, J., Vrijland, K., Hau, C. S. et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 (2015).
Kulig, P., Burkhard, S., Mikita-Geoffroy, J., Croxford, A. L., Hovelmeyer, N., Gyulveszi, G. et al. IL17A-mediated endothelial breach promotes metastasis formation. Cancer Immunol. Res. 4, 26–32 (2016).
Wellenstein, M. D., Coffelt, S. B., Duits, D. E. M., van Miltenburg, M. H., Slagter, M., de Rink, I. et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 572, 538–542 (2019).
Moreno Ayala, M. A., Li, Z. & DuPage, M. Treg programming and therapeutic reprogramming in cancer. Immunology 157, 198–209 (2019).
Qian, B. Z., Li, J., Zhang, H., Kitamura, T., Zhang, J., Campion, L. R. et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011).
Mitchem, J. B., Brennan, D. J., Knolhoff, B. L., Belt, B. A., Zhu, Y., Sanford, D. E. et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73, 1128–1141 (2013).
Zhao, L., Lim, S. Y., Gordon-Weeks, A. N., Tapmeier, T. T., Im, J. H., Cao, Y. et al. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology 57, 829–839 (2013).
Kitamura, T., Doughty-Shenton, D., Cassetta, L., Fragkogianni, S., Brownlie, D., Kato, Y. et al. Monocytes differentiate to immune suppressive precursors of metastasis-associated macrophages in mouse models of metastatic breast cancer. Front Immunol. 8, 2004 (2017).
Kersten, K., Coffelt, S. B., Hoogstraat, M., Verstegen, N. J. M., Vrijland, K., Ciampricotti, M. et al. Mammary tumor-derived CCL2 enhances pro-metastatic systemic inflammation through upregulation of IL1beta in tumor-associated macrophages. Oncoimmunology 6, e1334744 (2017).
Li, X., Yao, W., Yuan, Y., Chen, P., Li, B., Li, J. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 66, 157–167 (2017).
McKenzie, D. R., Kara, E. E., Bastow, C. R., Tyllis, T. S., Fenix, K. A., Gregor, C. E. et al. IL-17-producing gammadelta T cells switch migratory patterns between resting and activated states. Nat. Commun. 8, 15632 (2017).
Akitsu, A., Ishigame, H., Kakuta, S., Chung, S. H., Ikeda, S., Shimizu, K. et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vgamma6(+)gammadelta T cells. Nat. Commun. 6, 7464 (2015).
Kitamura, T., Qian, B. Z., Soong, D., Cassetta, L., Noy, R., Sugano, G. et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 212, 1043–1059 (2015).
Kitamura, T., Fujishita, T., Loetscher, P., Revesz, L., Hashida, H., Kizaka-Kondoh, S. et al. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc. Natl Acad. Sci. USA 107, 13063–13068 (2010).
Halama, N., Zoernig, I., Berthel, A., Kahlert, C., Klupp, F., Suarez-Carmona, M. et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell 29, 587–601 (2016).
Bonapace, L., Coissieux, M. M., Wyckoff, J., Mertz, K. D., Varga, Z., Junt, T. et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133 (2014).
Dyer, D. P., Medina-Ruiz, L., Bartolini, R., Schuette, F., Hughes, C. E., Pallas, K. et al. Chemokine receptor redundancy and specificity are context dependent. Immunity 50, 378–389 e375 (2019).
Steele, C. W., Karim, S. A., Leach, J. D. G., Bailey, P., Upstill-Goddard, R., Rishi, L. et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 29, 832–845 (2016).
Jackstadt, R., van Hooff, S. R., Leach, J. D., Cortes-Lavaud, X., Lohuis, J. O., Ridgway, R. A. et al. Epithelial NOTCH signaling rewires the tumor microenvironment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell 36, 319–336 e317 (2019).
Olkhanud, P. B., Baatar, D., Bodogai, M., Hakim, F., Gress, R., Anderson, R. L. et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 69, 5996–6004 (2009).
Yang, P., Li, Q.-j, Feng, Y., Zhang, Y., Markowitz, G. J., Ning, S. et al. TGF-b-miR-34a-CCL22 signaling-induced treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 22, 291–303 (2012).
Hamilton, J. A., Cook, A. D. & Tak, P. P. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat. Rev. Drug Discov. 16, 53–70 (2016).
Linde, N., Casanova-Acebes, M., Sosa, M. S., Mortha, A., Rahman, A., Farias, E. et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 9, 21 (2018).
Zhu, Y., Knolhoff, B. L., Meyer, M. A., Nywening, T. M., West, B. L., Luo, J. et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069 (2014).
Salvagno, C., Ciampricotti, M., Tuit, S., Hau, C. S., van Weverwijk, A., Coffelt, S. B. et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat. Cell Biol. 21, 511–521 (2019).
Ruffell, B., Chang-Strachan, D., Chan, V., Rosenbusch, A., Ho, C. M., Pryer, N. et al. Macrophage IL-10 Blocks CD8(+) T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637 (2014).
Pathria, P., Louis, T. L. & Varner, J. A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 40, 310–327 (2019).
Swierczak, A., Cook, A. D., Lenzo, J. C., Restall, C. M., Doherty, J. P., Anderson, R. L. et al. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol. Res. 2, 765–776 (2014).
Beffinger, M., Tallon de Lara, P., Tugues, S., Vermeer, M., Montagnolo, Y., Ohs, I. et al. CSF1R-dependent myeloid cells are required for NKmediated control of metastasis. JCI Insight. 3, e97792 (2018).
Gomez Perdiguero, E., Klapproth, K., Schulz, C., Busch, K., Azzoni, E., Crozet, L. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551 (2015).
Kielbassa, K., Vegna, S., Ramirez, C. & Akkari, L. Understanding the origin and diversity of macrophages to tailor their targeting in solid cancers. Front Immunol. 10, 2215–2215 (2019).
Zhu, Y., Herndon, J. M., Sojka, D. K., Kim, K.-W., Knolhoff, B. L., Zuo, C. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338.e326 (2017).
Kowanetz, M., Wu, X., Lee, J., Tan, M., Hagenbeek, T., Qu, X. et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl Acad. Sci. USA 107, 21248–21255 (2010).
Wculek, S. K. & Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 (2015).
Quail, D. F., Olson, O. C., Bhardwaj, P., Walsh, L. A., Akkari, L., Quick, M. L. et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat. Cell Biol. 19, 974–987 (2017).
Di Maio, M., Gridelli, C., Gallo, C., Shepherd, F., Piantedosi, F. V., Cigolari, S. et al. Chemotherapy-induced neutropenia and treatment efficacy in advanced non-small-cell lung cancer: a pooled analysis of three randomised trials. Lancet Oncol. 6, 669–677 (2005).
Han, Y., Yu, Z., Wen, S., Zhang, B., Cao, X. & Wang, X. Prognostic value of chemotherapy-induced neutropenia in early-stage breast cancer. Breast Cancer Res. Treat. 131, 483–490 (2012).
Yamanaka, T., Matsumoto, S., Teramukai, S., Ishiwata, R., Nagai, Y. & Fukushima, M. Predictive value of chemotherapy-induced neutropenia for the efficacy of oral fluoropyrimidine S-1 in advanced gastric carcinoma. Br. J. Cancer 97, 37–42 (2007).
Shitara, K., Matsuo, K., Takahari, D., Yokota, T., Inaba, Y., Yamaura, H. et al. Neutropaenia as a prognostic factor in metastatic colorectal cancer patients undergoing chemotherapy with first-line FOLFOX. Eur. J. Cancer 45, 1757–1763 (2009).
Fan, M. Y., Low, J. S., Tanimine, N., Finn, K. K., Priyadharshini, B., Germana, S. K. et al. Differential roles of IL-2 signaling in developing versus mature Tregs. Cell Rep. 25, 1204–1213.e1204 (2018).
Jounaidi, Y., Cotten, J. F., Miller, K. W., Forman, S. A. & Hospital, M. G. Tethering IL2 to its receptor IL2Rβ enhances anti-tumor activity and expansion of natural killer NK92 cells. Cancer Res. 77, 5938–5951 (2018).
Sim, G. C., Liu, C., Wang, E., Liu, H., Creasy, C., Dai, Z. et al. IL2 Variant Circumvents ICOS+ Regulatory T-cell Expansion and Promotes NK Cell Activation. Cancer Immunol. Res. 4, 983–994 (2016).
DeNardo, D. G., Barreto, J. B., Andreu, P., Vasquez, L., Tawfik, D., Kolhatkar, N. et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 (2009).
Mazzieri, R., Pucci, F., Moi, D., Zonari, E., Ranghetti, A., Berti, A. et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19, 512–526 (2011).
Qian, B. Z., Zhang, H., Li, J., He, T., Yeo, E. J., Soong, D. Y. et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 212, 1433–1448 (2015).
Guerriero, J. L., Sotayo, A., Ponichtera, H. E., Castrillon, J. A., Pourzia, A. L., Schad, S. et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432 (2017).
Logtenberg, M. E. W., Scheeren, F. A. & Schumacher, T. N. The CD47-SIRPalpha immune checkpoint. Immunity 52, 742–752 (2020).
Steinert, G., Scholch, S., Niemietz, T., Iwata, N., Garcia, S. A., Behrens, B. et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 74, 1694–1704 (2014).
Lian, S., Xie, R., Ye, Y., Lu, Y., Cheng, Y., Xie, X. et al. Dual blockage of both PD-L1 and CD47 enhances immunotherapy against circulating tumor cells. Sci. Rep. 9, 4532 (2019).
Vaeteewoottacharn, K., Kariya, R., Pothipan, P., Fujikawa, S., Pairojkul, C., Waraasawapati, S. et al. Attenuation of CD47-SIRPalpha signal in cholangiocarcinoma potentiates tumor-associated macrophage-mediated phagocytosis and suppresses intrahepatic metastasis. Transl. Oncol. 12, 217–225 (2019).
Ngo, M., Han, A., Lakatos, A., Sahoo, D., Hachey, S. J., Weiskopf, K. et al. Antibody therapy targeting CD47 and CD271 effectively suppresses melanoma metastasis in patient-derived xenografts. Cell Rep. 16, 1701–1716 (2016).
Zhao, H., Wang, J., Kong, X., Li, E., Liu, Y., Du, X. et al. CD47 Promotes tumor invasion and metastasis in non-small cell lung cancer. Sci. Rep. 6, 29719 (2016).
Willingham, S. B., Volkmer, J. P., Gentles, A. J., Sahoo, D., Dalerba, P., Mitra, S. S. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl Acad. Sci. USA 109, 6662–6667 (2012).
Edris, B., Weiskopf, K., Volkmer, A. K., Volkmer, J. P., Willingham, S. B., Contreras-Trujillo, H. et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl Acad. Sci. USA 109, 6656–6661 (2012).
Casbon, A. J., Reynaud, D., Park, C., Khuc, E., Gan, D. D., Schepers, K. et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl Acad. Sci. USA 112, E566–E575 (2015).
Fridlender, Z. G., Sun, J., Kim, S., Kapoor, V., Cheng, G., Ling, L. et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194 (2009).
Pang, Y., Gara, S. K., Achyut, B. R., Li, Z., Yan, H. H., Day, C. P. et al. TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer Discov. 3, 936–951 (2013).
Overacre-delgoffe, A. E., Chikina, M., Dadey, R. E., Yano, H., Shayan, G., Horne, W. et al. Interferon- γ drives T reg fragility to promote anti-tumor immunity. Cell 169, 1130–1141 (2018).
Rech, A. J., Mick, R., Martin, S., Recio, A., Aqui, N. A., Jr, D. J. P. et al. CD25 Blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med. 4, 134ra62 (2015).
De Mattos-Arruda, L., Sammut, S. J., Ross, E. M., Bashford-Rogers, R., Greenstein, E., Markus, H. et al. The genomic and immune landscapes of lethal metastatic breast cancer. Cell Rep. 27, 2690–2708.e2610 (2019).
Hoadley, K. A., Siegel, M. B., Kanchi, K. L., Miller, C. A., Ding, L., Zhao, W. et al. Tumor evolution in two patients with basal-like breast cancer: a retrospective genomics study of multiple metastases. PLoS Med. 13, 1–20 (2016).
Savas, P., Teo, Z. L., Lefevre, C., Flensburg, C., Caramia, F., Alsop, K. et al. The subclonal architecture of metastatic breast cancer: results from a prospective community-based rapid autopsy program “CASCADE”. PLoS Med. 13, 1–25 (2016).
Remark, R., Alifano, M., Cremer, I., Lupo, A., Dieu-Nosjean, M. C., Riquet, M. et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin. Cancer Res. 19, 4079–4091 (2013).
Robinson, D. R., Wu, Y. M., Lonigro, R. J., Vats, P., Cobain, E., Everett, J. et al. Integrative clinical genomics of metastatic cancer. Nature 548, 297–303 (2017).
Yates, L. R., Knappskog, S., Wedge, D., Farmery, J. H. R., Gonzalez, S., Martincorena, I. et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 32, 169–184.e167 (2017).
Savas, P., Teo, Z. L., Lefevre, C., Flensburg, C., Caramia, F., Alsop, K. et al. The subclonal architecture of metastatic breast cancer: results from a prospective community-based rapid autopsy program “CASCADE”. PLoS Med. 13, e1002204 (2016).
Hoadley, K. A., Siegel, M. B., Kanchi, K. L., Miller, C. A., Ding, L., Zhao, W. et al. Tumor evolution in two patients with basal-like breast cancer: a retrospective genomics study of multiple metastases. PLoS Med. 13, e1002174 (2016).
Bertucci, F., Ng, C. K. Y., Patsouris, A., Droin, N., Piscuoglio, S., Carbuccia, N. et al. Genomic characterization of metastatic breast cancers. Nature 569, 560–564 (2019).
Jimenez-Sanchez, A., Memon, D., Pourpe, S., Veeraraghavan, H., Li, Y., Vargas, H. A. et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 170, 927–938 e920 (2017).
Angelova, M., Mlecnik, B., Vasaturo, A., Bindea, G., Fredriksen, T., Lafontaine, L. et al. Evolution of metastases in space and time under immune selection. Cell 175, 751–765 e716 (2018).
Van den Eynde, M., Mlecnik, B., Bindea, G., Fredriksen, T., Church, S. E., Lafontaine, L. et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell 34, 1012–1026 e1013 (2018).
Nosrati, A., Tsai, K. K., Goldinger, S. M., Tumeh, P., Grimes, B., Loo, K. et al. Evaluation of clinicopathological factors in PD-1 response: derivation and validation of a prediction scale for response to PD-1 monotherapy. Br. J. Cancer 116, 1141–1147 (2017).
Weide, B., Martens, A., Hassel, J. C., Berking, C., Postow, M. A., Bisschop, K. et al. Baseline biomarkers for outcome of melanoma patients treated with Pembrolizumab. Clin. Cancer Res. 22, 5487–5496 (2016).
Funazo, T., Nomizo, T. & Kim, Y. H. Liver metastasis is associated with poor progression-free survival in patients with non-small cell lung cancer treated with nivolumab. J. Thorac. Oncol. 12, e140–e141 (2017).
Tumeh, P. C., Hellmann, M. D., Hamid, O., Tsai, K. K., Loo, K. L., Gubens, M. A. et al. Liver metastasis and treatment outcome with Anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol. Res. 5, 417–424 (2017).
Jiao, S., Subudhi, S. K., Aparicio, A., Ge, Z., Guan, B., Miura, Y. et al. Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell 179, 1177–1190 e1113 (2019).
Lu, X., Mu, E., Wei, Y., Riethdorf, S., Yang, Q., Yuan, M. et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 20, 701–714 (2011).
Lorger, M., Andreou, T., Fife, C. & James, F. Immune checkpoint blockade - how does it work in brain metastases? Front. Mol. Neurosci. 12, 282–282 (2019).
Long, G. V., Atkinson, V., Menzies, A. M., Lo, S., Guminski, A. D., Brown, M. P. et al. A randomized phase II study of nivolumab or nivolumab combined with ipilimumab in patients (pts) with melanoma brain metastases (mets): The Anti-PD1 Brain Collaboration (ABC). J. Clin. Oncol. 35, 9508–9508 (2017).
McGranahan, N., Rosenthal, R., Hiley, C. T., Rowan, A. J., Watkins, T. B. K., Wilson, G. A. et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259–1271 e1211 (2017).
Turajlic, S., Xu, H., Litchfield, K., Rowan, A., Chambers, T., Lopez, J. I. et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell 173, 581–594.e512 (2018).
Boire, A., Coffelt, S. B., Quezada, S. A., Vander Heiden, M. G. & Weeraratna, A. T. Tumour dormancy and reawakening: opportunities and challenges. Trends Cancer 5, 762–765 (2019).
Pantel, K., Schlimok, G., Kutter, D., Schaller, G., Genz, T., Wiebecke, B. et al. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 51, 4712–4715 (1991).
Chen, D. S. & Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 (2013).
Philip, M., Fairchild, L., Sun, L., Horste, E. L., Camara, S., Shakiba, M. et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017).
Nakamura, K. & Smyth, M. J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol. Immunol. 17, 1–12 (2020).
Shin, D. S., Zaretsky, J. M., Escuin-Ordinas, H., Garcia-Diaz, A., Hu-Lieskovan, S., Kalbasi, A. et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7, 188–201 (2017).
Zaretsky, J. M., Garcia-Diaz, A., Shin, D. S., Escuin-Ordinas, H., Hugo, W., Hu-Lieskovan, S. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Cabrita, R., Lauss, M., Sanna, A. & Donia, M. Skaarup Larsen, M., Mitra, S. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 577, 561–565 (2020)
Petitprez, F., de Reynies, A., Keung, E. Z., Chen, T. W., Sun, C. M., Calderaro, J. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020).
Helmink, B. A., Reddy, S. M., Gao, J., Zhang, S., Basar, R., Thakur, R. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Acknowledgements
We thank members of the Coffelt lab, Leo Carlin and Ed Roberts for helpful discussions in the preparation of this article.
Author information
Authors and Affiliations
Contributions
S.C.E., W.H.M.H. and S.B.C. established the design of the article, drafted the paper and made critical revisions to the paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Data availability
Not applicable.
Competing interests
The authors declare no competing interests.
Funding information
We acknowledge funding from the Cancer Research UK Glasgow Centre (A25142), Breast Cancer Now (2018JulPR1101) and Tenovus Scotland (Project S17-17).
Additional information
Note: This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Edwards, S.C., Hoevenaar, W.H.M. & Coffelt, S.B. Emerging immunotherapies for metastasis. Br J Cancer 124, 37–48 (2021). https://doi.org/10.1038/s41416-020-01160-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-01160-5
This article is cited by
-
Locally sourced: site-specific immune barriers to metastasis
Nature Reviews Immunology (2023)
-
Immune checkpoint inhibition combined with targeted therapy using a novel virus-like drug conjugate induces complete responses in a murine model of local and distant tumors
Cancer Immunology, Immunotherapy (2023)
-
The role of immune checkpoint inhibitors in patients with intracranial metastatic disease
Journal of Neuro-Oncology (2023)
-
Dual-functional alginate and collagen–based injectable hydrogel for the treatment of cancer and its metastasis
Journal of Nanobiotechnology (2022)
-
Metastasis prevention: targeting causes and roots
Clinical & Experimental Metastasis (2022)
