
Abstract
To enable survival in adverse conditions, cancer cells undergo global metabolic adaptations. The amino acid cysteine actively contributes to cancer metabolic remodelling on three different levels: first, in its free form, in redox control, as a component of the antioxidant glutathione or its involvement in protein s-cysteinylation, a reversible post-translational modification; second, as a substrate for the production of hydrogen sulphide (H2S), which feeds the mitochondrial electron transfer chain and mediates per-sulphidation of ATPase and glycolytic enzymes, thereby stimulating cellular bioenergetics; and, finally, as a carbon source for epigenetic regulation, biomass production and energy production. This review will provide a systematic portrayal of the role of cysteine in cancer biology as a source of carbon and sulphur atoms, the pivotal role of cysteine in different metabolic pathways and the importance of H2S as an energetic substrate and signalling molecule. The different pools of cysteine in the cell and within the body, and their putative use as prognostic cancer markers will be also addressed. Finally, we will discuss the pharmacological means and potential of targeting cysteine metabolism for the treatment of cancer.
Similar content being viewed by others

Background
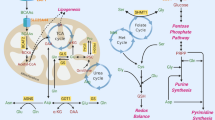
Cysteine is a sulphur-containing proteinogenic amino acid; it has a free thiol group, which is likely to confer particular properties on functional sites of proteins that contain this highly conserved residue. As a multifaceted precursor, cysteine contributes to the survival and proliferation of cancer cells. Besides being a component of proteins and glutathione, cysteine is an important source of energy and biomass (Fig. 1).

Cysteine has different fates, including the synthesis of glutathione or proteins, oxidative or non-oxidative catabolism and reversible post-translational protein modification (protein cysteinylation, production of reactive sulphide species and oxidation to cysteine disulphides).
Cancer cells face a range of intrinsic and extrinsic adverse/stressful conditions, such as nutrient and oxygen deficiency, and have consequently developed means to adapt their metabolism in order to survive. Cysteine has three main roles in the metabolic rewiring of cancer cells: as a precursor of glutathione under the action of glutamyl-cysteine ligase (GCL), contributing to oxidative stress control; as a substrate for the production of hydrogen sulphide (H2S), which stimulates cellular bioenergetics; and as a carbon source for biomass and energy production (Fig. 2a). Cysteine is also essential for the ability of cancer cells to evade drug exposure and cell injury and adapt to other stressful conditions such as hypoxia. As cysteine and glutathione are scavengers of free radicals (mainly reactive oxygen species (ROS)), they can abrogate the effects of the majority of oxidative or alkylating drugs used in cancer therapy, affording an important resistance mechanism.1,2,3,4,5,6,7 Glutathione is also a highly important component in detoxification, allowing the physiological and pathophysiological maintenance of cell metabolism.8,9,10,11 Moreover, the relevance of cysteine in the production of other organic compounds and H2S (Fig. 2a, b) highlights the importance of the bioavailability of cysteine to enable cells to adapt to metabolically challenging conditions, as well as mediating cancer cell survival, tumour growth, metastases formation, resistance to therapy and disease recurrence.

a Cysteine can be taken up in the form of cystine, through the cystine–glutamate antiporter transport system (xCT), or as cysteine through the excitatory amino acid transporter 3 (EAAT3) or the alanine-serine-cysteine-transporter 2 (ASCT2). Cysteine metabolism is tightly linked to that of glutamine, forming a network of amino acids capable of supplying the core metabolic pathways that underlie pivotal processes in cancer: reactive oxygen species (ROS) scavenging and chemoresistance dependent on glutathione synthesis; carbon and energy metabolism through fatty acid synthesis and the tricarboxylic acid (TCA) cycle, one-carbon metabolism and the production of ATP by the mitochondrial electron transfer chain (mETC), and sulphur and energy production as a generator of hydrogen sulphide (H2S), an electron (e−) donor for the mETC. b Cysteine (Cys) is a precursor of other organic compounds, such as homocysteine (Hcy), 3-mercaptopyruvate (3-MP), cystathionine, cystine, cysteine sulphinic acids (CSA), 3-sulphopyruvate (3-SP), hypotaurine (hT), taurine, glutathione (reduced, GSH), γ-glutamyl-cysteine (Glu-Cys) and cysteinylglycine (Cys-Gly).
In this article, the cellular and systemic roles of cysteine in the metabolic remodelling that occurs in cancer cells will be outlined. Emphasis will be placed on the metabolic pathways and relevant players (Table 1) that interfere with cysteine anabolism and catabolism, and the determinants that affect cysteine bioavailability, thereby influencing cancer development. Finally, insights into the pharmacological targeting of cysteine metabolism will be presented.
Cysteine bioavailability: main players and regulators
As mentioned above, the bioavailability of cysteine in a cancer cell can influence metabolic fitness and the development of therapy resistance. Although cysteine can be derived from the catabolism of extracellular glutathione, protein catabolism or de novo synthesis from methionine,12,13,14 the major source of cellular cysteine is the dietary intake of cystine, the oxidised form of cysteine.12,15 The oxidative environment of the plasma favours free cysteine dimerisation into cystine, which then becomes the predominantly available form for cells to take up from the surrounding milieu.
Cystine transporters
A number of cystine transporters have been described and studied in the context of cancer; however, as some of these transporters are cystine–glutamate antiporters (Fig. 2),16,17 many of these studies have focused on the relevance of glutamate, mainly in the central nervous system, describing a correlation between increased glutamate efflux and increased cancer aggressiveness and invasive capacity (and, consequently, poor prognosis).16,18,19,20,21,22 As cystine influx through the cystine–glutamate xCT antiporter occurs concomitantly with glutamate efflux, increased cystine import can also be related to poor prognosis. Thus, cancer cells in metabolic and phenotypic equilibrium can be disturbed by blocking cyst(e)ine/glutamate transporters.16,23,24
Moreover, in cancer cell lines (Table 1), increased expression levels of xCT were found to be associated with increased intracellular levels of glutathione and cisplatin resistance.7 In fact, xCT is considered to be the main transporter of cystine in cancer (Table 1)25,26,27 and its expression is controlled by nuclear factor erythroid 2-related factor 2 (NRF2), a master regulator of the cellular redox state,28 highlighting the importance of xCT in an oxidative stress-resistant cancer cell phenotype. In addition, xCT expression can be regulated by the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/PKB/mTOR)29,30,31 and mitogen-activated protein kinase (MAPK) pathways in synergy with activating transcription factor 4 (ATF4), which is activated by endoplasmic reticulum stress.27 Decreased expression of xCT leads to an increase in the rate of sulphur transfer from homocysteine to cysteine (through the trans-sulphuration branch of the methionine cycle), supporting the pivotal role of xCT in cyst(e)ine transport and cysteine provision.29,32 The neutral and basic amino acid transporter (RBAT, also designated by SLC3A1) is another cystine transporter with possible implications in cancer development. Its expression has been linked to the capacity of breast cancer cells to control their redox state by promoting the accumulation of reduced glutathione, thereby decreasing ROS levels and increasing cell survival (Table 1).33
Non-specific cysteine transporters
While influx transporters are the main means by which cells acquire cystine, cysteine can also be taken up from the extracellular milieu into cells directly by excitatory amino acid transporter 3 (EAAT3) and the alanine-serine cysteine transporters 1 and 2 (ASCT1/2), all of which are known to be overexpressed in different cancer types34 (Table 1). However, the association between cysteine transport and the overexpression of these transporters in cancer metabolic remodelling has not yet been established, as these transporters are not specific for cysteine and so the focus of most studies is the transport of other amino acids (e.g. glutamine and glutamate).
A limited number of studies have investigated EAAT3 in cancer. For example, the role of EAAT3 was explored in brain tumour models, but the studies focused on glutamate transport and the central nervous system-specific glutamatergic cycle,35,36 which is essential for the ultimate production of neurotransmitters.37 Nonetheless, EAAT3 has been associated with increased chemoresistance in colorectal cancer models38 and reported to be highly expressed in prostate cancer.39 ASCT1 and ASCT2, which have mainly been studied in the context of glutamine dependence, are expressed at high levels in different cancer types,40,41,42 prompting these transporters to be considered putative therapeutic targets in cancer, with different inhibitors currently under investigation.40,43,44,45 Furthermore, glutamine and cysteine metabolism are deeply linked,46,47 because serine and glycine can derive from glutamine and, by entering the one-carbon metabolism pathway, they will contribute to homocysteine and cysteine syntheses.1 This fact makes ASCT1, ASCT2 and EAAT3 pivotal in the reliance of cancer cells on cysteine uptake and anabolism.
Cysteine catabolism and its interplay with other metabolic pathways in cancer
Cysteine plays a central role in cellular metabolism as a key component of carbon and sulphur metabolism (Fig. 2). There are two main pathways for cysteine catabolism: one is via its enzymatic breakdown to produce H2S and organic intermediates that will serve as carbon sources, and the second is via oxidative metabolism through cysteine dioxygenase (CDO). The production of H2S and its role in bioenergetics and signalling will be addressed later in the article.
Cysteine degradation and energy metabolism
Although the role of cysteine degradation in cancer development has predominantly been explored regarding H2S production, the usefulness of cysteine as a carbon source is also evident along its catabolic pathways, as its degradation gives rise to other organic compounds relevant for carbon and energy metabolism (Fig. 2). These compounds include pyruvate, which can be converted into acetyl-CoA and enter the tricarboxylic acid (TCA) cycle or be used for fatty acid synthesis, and α-ketoglutarate, a precursor of glutamate and an intermediate of the TCA cycle.48,49,50,51 Moreover, through the action of two of the enzymes involved in the production of H2S—cysteine aminotransferase (CAT) and 3-mercaptopyruvate sulphurtransferase (3-MST)—cysteine is sequentially converted into 3-mercaptopyruvate (3-MP) with the release of an amino group that will react with α-ketoglutarate, ending with the formation of glutamate and pyruvate (Fig. 3a), again connecting cysteine metabolism with the TCA cycle. Given the increased expression of cysteine catabolic enzymes, such as cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE) and 3-MST in different cancer cell types, it is likely that their relevance for cancer development is shared between the production of H2S (detailed later in the article) and the accompanying generation of metabolites that constitute carbon sources.52,53,54,55,56,57,58,59,60

Cysteine can be a substrate for hydrogen sulphide (H2S) synthesis, be oxidatively catabolised to taurine or be a substrate for glutathione production. a The trans-sulphuration pathway. The enzymes cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) catalyse the conversion of homocysteine into cysteine, generate hydrogen sulphide through several alternative reactions or cysteine per/polysulphide (CysSS(n)H) with cystine as substrate. Cysteine is converted by cysteine aminotransferase (CAT) into 3-mercaptopyruvate (3-MP), which is a substrate of 3-MP sulphurtransferase (3-MST), of which there is a cytosolic and a mitochondrial isoform. 3-MST can also generate CysS(n)SH and glutathione per-/polysulphide (GS(n)SH), respectively, using Cys and GSH as sulphur acceptors. H2S is catabolised by a mitochondrial sulphide oxidation pathway. H2S is a substrate of sulphide:quinone oxidoreductase (SQR), which preferentially uses glutathione as a sulphur acceptor, to generate oxidised glutathione (GSSH) that is converted back into GSH by persulphide dioxygenase (PDO) using oxygen as co-substrate, yielding sulphite (SO32−) as co-product. Oxidised glutathione is also a substrate of rhodanese (Rhod), which uses SO32− as co-substrate to yield thiosulphate (S2O32−) and glutathione. SO32− oxidation by sulphite oxidase yields sulphate (SO42−). b Cysteine oxidation. Cysteine can be oxidised by cysteine dioxygenase (CDO) to cysteinesulphinate (CSA), which is converted by cysteinesulphinate decarboxylase (CSD) into hypotaurine (hT), which is further oxidised to taurine. Alternatively, cysteinesulphinate can be transaminated by aspartate aminotransferase (AAT) to 3-sulphinopyruvate (3-SP), which decomposes to form pyruvate and sulphite/sulphate. c Cysteine is a substrate for glutathione generation. Cysteine is converted into γ-glutamyl-cysteine (Glu-Cys) by glutamate-cysteine ligase (GCL) and subsequently to glutathione by glutathione synthase (GS). Conversely, GSH is converted into cysteinylglycine (Cys-Gly) by γ-glutamyl transpeptidase (γGT) and finally back to cysteine by dipeptidase (dPP).
Cysteine-oxidative catabolism
In the second pathway for cysteine catabolism, CDO catalyses the conversion of cysteine into cysteine sulphinic acid (CSA; Figs. 2b and 3b), leading to the cellular production of taurine, pyruvate (Fig. 2b) and sulphate.61,62,63 CDO is highly regulated at the level of protein turnover, as oxidative degradation of cysteine by CDO is highly inducible,64 and high cysteine levels inhibit CDO ubiquitinylation and reduce its proteasomal degradation.65 Deficient CDO activity has been related to lower sulphate levels in plasma, elevated fasting plasma cysteine concentrations, and lower sulphate-conjugate:glucuronide-conjugate ratios for paracetamol detoxification products, consistent with impaired cysteine catabolism.66 CDO competes with GCL (Fig. 3c) for cysteine and thereby contributes to the regulation of the intracellular availability of cysteine and glutathione67 and to H2S production.68
In non-transformed cells, NRF2 promotes the entry of cysteine into the taurine synthesis pathway via CDO1,69 and modestly limits the accumulation of cysteine and glutathione synthesis. In cancer, CDO has been shown to divert the flow of cysteine away from glutathione synthesis, thereby promoting the production of ROS.70 As cells use NADPH to reduce cystine to cysteine, the increased cysteine catabolism by CDO implies the increased consumption of NADPH, which will affect other cell metabolic pathways and the antioxidant capacity as well. Cysteine starvation or xCT blockade confers the opposite result and increases cellular NADPH pools.69
The important role of taurine as a cellular osmoregulator will not be described in detail in this paper. However, it should be mentioned that taurine levels contribute to cellular homeostasis, and that this role is particularly important in the brain71 and kidney.72 The overexpression of components of cysteine catabolic pathways in cancer cells is likely to result in increased pyruvate production. This link between cysteine-oxidative catabolism-derived pyruvate and cancer has been described in pancreatic cancer73 and non-small lung cancer cells,69 although additional studies that focus on other cancer types are required to strengthen this association.
CDO1: a tumour suppressor?
The predominant notion, supported by a number of reports in different cancer types (detailed below), is that CDO1 is a tumour suppressor gene simply by virtue of its silencing in cancer; however, it is ought to be confirmed. In cancer (Table 1), CDO1 is usually silenced by promoter methylation and this is associated with poor prognosis,70,74 implying that the shutting down of CDO1 expression favours cancer progression. The CDO products sulphite and CSA are toxic to lung cancer cells. As cysteine stabilises CDO levels, cells that express high levels of NRF2 are sensitive to CDO-related toxicity.69 Taurine is also a marker of CDO activation, and lower levels of taurine in lung cancer cells are related to higher intracellular cysteine concentrations.69 In gastric cancer cells, the absence of CDO1 contributed to restore cellular glutathione levels, to prevent ROS and lipid peroxidation, and to promote resistance to ferroptosis, a type of programmed cell death dependent on iron and associated with lipid peroxide accumulation.75 Conversely, forced overexpression of CDO1 in breast cancer cells shifted the flux from glutathione synthesis towards cysteine catabolism, consequently increasing ROS levels and leading to reduced cell viability and growth.70 In breast cancer, hypermethylation of the CDO1 promoter, which is frequently observed, is a predictor of poor outcome in anthracycline-treated, oestrogen receptor-positive and lymph-node-positive patients.70 CDO1 expression might therefore be a useful indicator for the prediction of drug resistance, and hypomethylating agents (e.g. 5-azacytidine) might have a role in the treatment of breast cancer cells with epigenetically silenced CDO1—for example, by inducing sensitisation to anthracycline. As anthracyclines are ROS-generating drugs, one of the resistance mechanisms might be related to the ability of cancer cells to evade this insult.
However, in contrast to the tumour-suppressive effects described above, there is evidence that CDO1 can have a tumorigenic effect associated with aggressive glioblastoma, since its overexpression was detected in these tumours.70,74 Patient-derived glioblastoma specimens exhibit overexpression of CDO1 and consequent accumulation of CSA.76 Altogether, although many data point towards CDO1 as being a tumour suppressor gene and a putative prognostic marker for cancer outcome, divergent evidence from glioblastoma patient samples highlight the need for more studies to disclose the actual impact of cysteine-derived organic compounds in cancer metabolic rewiring.
Cysteine anabolism and its interplay with other metabolic pathways in cancer
De novo cysteine synthesis
The de novo synthesis of cysteine occurs through the trans-sulphuration pathway of the methionine cycle, which also involves serine (Fig. 3a), and renders the synthesis of cysteine dependent on the activity of one-carbon metabolism (methionine and folate cycles).77,78,79 Dietary methionine is converted into homocysteine in two consecutive steps: the first involves the condensation of homocysteine with serine, catalysed by CBS to yield cystathionine (Fig. 2b), which is subsequently hydrolysed by CSE in the second step to generate cysteine and other compounds (e.g. ammonia, α-ketoglutarate or propionate), thus establishing a link between the trans-sulphuration pathway and the TCA cycle (as reviewed in Combs and DeNicola.80) Cancer cells express higher levels of the trans-sulphuration pathway enzymes CBS and CSE than normal cells (reviewed e.g. in refs.,81,82) highlighting this pathway as a crucial metabolic pathway in cancer cells.83 A very recent study presents the trans-sulphuration pathway as being directly beneficial for cancer cells to maintain the redox equilibrium and evade ferroptosis, as, upon inhibition of cysteine import, glutathione synthesis is supplied by cysteine derived from the trans-sulphuration pathway.84 In the same study, the concept was proven in cancer cell lines by reverting lipid oxidation through the addition of homocysteine. The relevance of the trans-sulphuration pathway in the survival of cancer cells and reversion of ferroptosis was also proven by silencing CBS, thus inhibiting cysteine synthesis.85
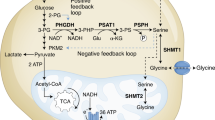
One-carbon metabolism and cysteine bioavailability
The de novo synthesis of cysteine cannot be addressed without mentioning the involvement of serine and glycine in the one-carbon pathway. The serine synthesis pathway is mainly supplied by glucose-derived 3-phosphoglycerate, which is converted into 3-phosphohydroxypyruvate by the action of phosphoglycerate dehydrogenase, which also converts NADH into NAD+.86 Subsequently, 3-phosphohydroxypyruvate is converted into serine by two sequential reactions catalysed by phosphoserine aminotransferase 1 and phosphoserine phosphatase. Then, serine hydroxymethyltransferase converts serine into glycine, which will enter one-carbon metabolism.78 In cancer, c-Myc, a pivotal and well-known oncogene, is the main regulator of the serine synthesis pathway.79,87
Activation of the serine synthesis pathway correlates directly with glutathione synthesis,87 as serine-derived glycine is one of the components of glutathione. Serine and glycine are needed for cysteine synthesis in one-carbon metabolism, and serine-derived glycine is preferentially used by cancer cells over exogenous glycine.88 Therefore, in the context of cancer metabolic remodelling, the import of serine is a key control point of the one-carbon pathway.
One-carbon metabolism also plays a role in the regulation of gene expression and epigenetic modulation. Several types of cancer have their carcinogenesis associated with the silencing of physiologically beneficial genes, such as tumour suppressor genes, through DNA methylation,89,90 as described earlier in the article for CDO1. Furthermore, in some tumours, the need for methyl groups for epigenetic regulation prevents activation of the trans-sulphuration pathway for cysteine synthesis, instead prioritising one-carbon metabolism.83 The folate cycle uses glycine and tetrahydrofolate (converted from folic acid) and produces intermediates (5,10-methylene-tetrahydrofolate and 5-methylene-tetrahydrofolate) to supply methyl groups for purine synthesis. After the folate cycle, through the interconnection with the methionine cycle involving the entry of cobalamin (vitamin B12), folic acid is again synthesised.91 In the methionine cycle, methionine is sequentially converted into s-adenosyl-l-methionine (the methyl donor for all methylation reactions in the cell) and s-adenosyl-l-homocysteine.92 s-Adenosyl-l-homocysteine can be used for pyrimidine synthesis or converted into homocysteine, which can be re-methylated to methionine or enter the trans-sulphuration pathway to generate cysteine, through CBS and CSE.50,93 Cysteine catabolism depends on the methionine cycle, belonging to the one-carbon metabolism, which supplies different pathways relevant in carcinogenesis and cancer metabolism.
Glutathione metabolism
Together with cysteine anabolism, the extracellular catabolism of glutathione constitutes a relevant source of cysteine.12 Following ROS scavenging, the degradation of oxidised glutathione through the γ-glutamyl cycle (Fig. 3c) facilitates recycling of cysteine, glycine and glutamate. Upon its efflux from cells, oxidised glutathione is exposed to the sequential action of enzymes located on the external face of the cell membrane: γ-glutamyl transpeptidase (GGT) generates glutamate94 and the cysteinylglycine dipeptide, which is degraded by dipeptidases such as aminopeptidase N (APN), thereby releasing cysteine and glycine.95 Glutamate, cysteine and glycine can then be imported by the cell using specific transporters. The cysteinylglycine dipeptide can also be taken up by cells in a process mediated by the peptide transporter 2, before being degraded in the cytoplasm by unspecific dipeptidases,96 although the circumstances under which this occurs remain undetermined.
Glutathione catabolism can also be carried out by CHAC1 and CHAC2 isoenzymes, which belong to the γ-glutamylcyclotransferase family.97,98 These enzymes catalyse the degradation of glutathione and the release of cysteinylglycine in the cytosol, rather than outside the cell as for GGT. Although not much is known about these enzymes in cancer, the CHAC2 isoform has already been classified as a tumour suppressor gene in gastric and colorectal cancer, on the basis of its downregulation being associated with more aggressive cancer variants and its activation-inducing apoptosis in cancer cells.99
Cysteine-derived reactive sulphide species
Cysteine is a key player in cancer as a source of reactive sulphide species (RSS), particularly as a substrate for endogenous and gut-microbe-derived H2S-synthesising and -catabolising enzymes. Tight and intricate regulatory processes maintain physiological H2S levels, while imbalances either in its production or breakdown have deleterious consequences, with high H2S levels eventually becoming toxic and/or pathogenic. At low intracellular concentrations (0.01–1 μM), H2S injects electron equivalents into the mitochondrial electron transport chain (mETC) by reducing quinone to quinol via sulphide:quinone oxidoreductase (SQR), ultimately stimulating ATP production.100 However, at 3–30-fold higher concentrations, H2S becomes toxic essentially by inhibiting cytochrome c oxidase (KI 2.6 μM at mitochondrial pH 8.05; reviewed e.g. in refs.100,101). The implications of this effect of H2S on cellular bioenergetics in the context of cancer are discussed below.
The H2S-related RSS per-sulphides and poly-sulphides can be generated by the same enzymatic systems that are involved in H2S metabolism, and play a possibly as-yet-underestimated role in signalling in human health and disease,102,103,104 which is also briefly discussed below.
Biosynthesis of H2S
H2S can be generated in mammalian physiology via dedicated endogenous enzymatic systems, gut microbiota metabolism and the breakdown of dietary polysulphide sources such as, for example, garlic and onion (reviewed e.g. in refs.82,105,106,107) (Fig. 3a). H2S is mainly generated by three endogenous enzymes with different organ/tissue distributions, all of which are related to cysteine metabolism: the trans-sulphuration pathway pyridoxal-5′-phosphate-dependent-CBS and CSE, and 3-MST.105 Historically considered to be cytosolic, CBS and CSE can also—under certain (patho)physiological conditions—translocate to mitochondria108,109 or nuclei,110 or even be secreted.111 3-MST can be detected in mitochondria and in the cytosol.112 While H2S-mediated protein per-sulphidation is relevant in any cellular compartment, mitochondrial H2S generation is particularly relevant for cellular bioenergetics, as detailed later in the article. Secreted CBS and CSE have been proposed to contribute for the maintenance of homocysteine levels in the plasma.111 3-MST uses 3-MP, derived from cysteine through the action of CAT, as an activating substrate to generate a persulphide at the catalytic Cys248 residue.113 H2S is then released upon the reaction of activated 3-MST with a sulphane sulphur acceptor such as thioredoxin, cysteine, homocysteine, glutathione, or even n-acetylcysteine, thus generating the corresponding per-sulphides.114,115,116 Whereas the canonical reactions catalysed by CBS and CSE within the trans-sulphuration pathway sequentially convert homocysteine into cysteine, both enzymes catalyse a number of alternative H2S-yielding reactions, using as substrates different combinations of these very same sulphur amino acids (reviewed e.g. in refs.82,117).
Regulation of H2S and RSS levels
The reactivity of H2S and related RSS demands a tight control of their levels, which is ensured both by an intricate regulation of their synthesising enzymes and through an efficient sulphide detoxification pathway located in mitochondria. Whereas CSE and 3-MST are mostly regulated at the transcriptional level, CBS is functionally controlled by a number of post-translational modifications and interactions. The activity of CBS is increased by ~2–3-fold upon s-adenosyl-l-methionine binding to its C-terminal domain.118 Redox reactions at key cysteine residues—namely, s-glutathionylation at Cys346119 and reduction of the Cys272-X-X-Cys275 disulphide—also enhance CBS enzymatic activity.119 Conversely, at the N-terminal domain, a non-catalytic regulatory haem sensor negatively impacts the enzymatic activity of CBS upon reduction and completely inactivates the enzyme upon binding of nitric oxide or carbon monoxide.100,120,121,122 An N-terminal intrinsically disordered peptide sequence has been shown to bind another haem moiety, although its function remains to be determined.123 As CBS and CSE use the same substrates to generate H2S, their function is interdependently regulated through substrate/product accumulation/depletion, which will favour one biochemical pathway over the other. Indeed, inhibition of CBS can yield overall higher H2S production through CSE, which presents a higher catalytic efficiency.124
Sulphide levels are enzymatically controlled through the catabolic sulphide-oxidising pathway (SOP) located in mitochondria. The first (irreversible and committing) step is catalysed by SQR, which oxidises H2S and transfers the electron equivalents to coenzyme Q and the sulphur atom to an acceptor molecule that becomes persulphidated.100,125,126 The preferred sulphur-accepting substrate is still a matter of debate, but glutathione appears to be the most effective and plausible in physiological conditions.127,128,129,130 The resulting glutathione persulphide is a substrate for either persulphide dioxygenase, which oxidises the sulphane sulphur yielding sulphite and glutathione, or rhodanese, which generates thiosulphate. Finally, sulphite oxidase converts sulphite into sulphate.125 The expression and activity of the SOP enzymes appear to be strongly related to the H2S levels to which the corresponding cells or tissues are exposed. For example, whereas sulphide-oxidising activity is virtually undetectable in nervous system cells, colonocytes display high expression and activity of SOP enzymes,131,132,133 consistent with the high H2S concentrations that result from gut microbial metabolism. Indeed, the apical localisation of SOP enzymes in human colonic crypts places this pathway optimally at the host–microbiome interface.134
As well as H2S, several other RSS are synthesised in human physiology, and similarly fulfil a number of signalling functions, which result essentially from modification of, or interaction with, target proteins. H2S, for example, can bind to protein metal centres, such as haem moieties in mitochondrial cytochrome c oxidase or haemoglobin (reviewed e.g. in ref.135). Probably the most prevalent H2S-mediated modification of target proteins, however, involves per-sulphidation (or polysulphidation) of cysteine residues (i.e. the addition of sulphane sulphur) with the concomitant functional and/or structural consequences (reviewed e.g. in refs.82,102,103), some of which are described below in relation to cancer. The enzymatic pathways involved in H2S metabolism are per se sources of low molecular weight per-sulphides and poly-sulphides. Indeed, both CBS and CSE can synthesise cysteine persulphide (CysSSH) and poly-sulphides (CysS(n)SH) from cystine,104,136 whereas 3-MP-activated 3-MST can generate from cysteine, glutathione or n-acetylcysteine, the corresponding per-sulphides (respectively, CysSSH, GSSH and NACSSH).114,115 Within the sulphide oxidation pathway, GSSH is also generated mostly as a product of SQR. The mitochondrial cysteinyl-tRNA synthase (CARS2) has been identified as the main cellular source of CysSSH, yielding free CysSSH and co-translationally inserting persulphidated cysteine into nascent polypeptides.137 The intrinsic reactivity and peculiar chemistry of per-sulphides and poly-sulphides indicate that they might carry out numerous signalling functions that could currently be underestimated mainly owing to the technical difficulties in studying these metabolites in biological milieu.
NFS1 and iron–sulphur clusters
Another enzyme capable of metabolising cysteine and producing a sulphur carrier is the mitochondrial cysteine desulphurase (NFS1). NFS1 degrades cysteine and releases sulphide, which can be used to generate iron–sulphur (Fe–S) clusters,138 versatile co-factors that carry out electron transfer, catalysis and afford structural stability. These Fe–S clusters are synthesised in the mitochondrion before being exported out by chaperones and channels to participate in the maturation of Fe–S proteins (reviewed in ref.139) The association between NFS1 activity and cancer relates to temporary abrupt increases in oxygen tensions experienced by cancer cells that differently affect tumours according to their tissue and organ localisation. Metastatic or primary lung tumours were shown to rely on enhanced expression of NFS1 to maintain the supply of Fe–S clusters as co-factors of multiple essential proteins and enzymes in the cell that are exposed to (damaging) high oxygen concentrations.140 Whereas the continuous supply of Fe–S clusters by NFS1 also prevented the iron-starvation response from being triggered in lung adenocarcinomas, NFS1 suppression was shown to predispose cancer cells to ferroptosis.140 Thus, cysteine contributes to the inhibition of ferroptosis both as a component of glutathione, which facilitates the scavenging of lipid peroxides by the phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4), and as a substrate of NFS1. Accordingly, dysregulation of the Fe–S clusters biogenesis, including decreased expression of NFS1, has also been described as being relevant in mechanisms of resistance to cancer therapy.138 Therefore, the activation of ferroptosis seems to be an important goal in cancer therapy, and triggering NFS1 might be a suitable strategy.
Cysteine-derived H2S in cancer development
A role for cysteine in cancer associated with disturbed metabolism and signalling of H2S, per-sulphides and poly-sulphides has been established on the basis of a range of evidence. In different cancer types, higher expression and activity, and changes in the localisation, of H2S-synthesising and -breakdown enzymes have been observed in cancer specimens or cell models (Table 1), as compared with tumour-adjacent normal tissue or non-tumorigenic cells, and associated with different aspects of cancer development (e.g. refs.81,134,141) In addition to perturbations in endogenous RSS metabolism, excessive H2S generated via bacterial cysteine desulphydrase from gut microbiota species such as Fusobacterium nucleatum has been linked to the development of colorectal cancer.106,142 The microbial influence in systemic and cellular metabolism is a new, controversial and developing field in cancer research. However, in colorectal cancer—the most explored cancer model in this matter—it seems that both microbially derived and endogenous H2S play a role in cancer progression and colon health.106
Following pioneering studies that linked CBS overexpression with colorectal and ovarian cancer (Table 1), an increase in the expression of all enzymes involved—individually or jointly—in the synthesis of H2S has also been reported for breast, gastric, lung, liver, bladder, kidney and prostate carcinomas, melanoma, neuroblastoma, glioma and astrocytoma.81,143,144,145 Even though the overexpression of enzymes involved in both the synthesis and breakdown of H2S has thus far only been reported for colorectal cancer,134 it can be envisaged that overexpression of both enzymatic pathways might be a common trait for any cancer type where H2S production is increased, as excess H2S can become toxic even for the most robust cancer cells (see below).
The exact manner by which cysteine-derived enhanced H2S metabolism contributes to cancer development is still to be fully clarified, although some common trends can be established. Different lines of evidence show that H2S modulates several aspects related to cancer cell adaptation within the tumour microenvironment.
H2S in cancer: metabolism and bioenergetics
Perhaps the most well-established role of H2S in cancer cells is its contribution to stimulating cell bioenergetics and glycolytic metabolism. At subtoxic concentrations, H2S has been shown to stimulate ATP production at the level of oxidative phosphorylation, both as a source of electron equivalents for the mETC (via SQR-mediated quinone reduction, Table 1), and through per-sulphidation of ATPase, which maintains the enzyme in its activated state.146 Szabo et al.52 reported that increased oxygen consumption by mitochondria isolated from colorectal cancer HCT116 cells treated with cysteine was suppressed upon CBS inhibition. Similarly, in ovarian cancer cells, CBS inhibition resulted in mitochondrial dysfunction and ROS overproduction (Table 1), consistent with a malfunctioning mETC.58 Mitochondrial bioenergetics are also stimulated by the H2S-generating 3-MST substrate 3-MP (Table 1) both in the murine colon cancer CT26 cell line147 and in mouse hepatoma cells,148 where silencing of 3-MST or SQR decreased basal cellular bioenergetics, further suggesting that mitochondrial bioenergetics are partially sustained by SQR-mediated H2S oxidation (Table 1). In line with these observations, exposure of the SW480 colorectal cancer cell line to n-acetylcysteine yielded a synchronous upregulation of 3-MST and SQR expression and activity.114
In contrast to the stimulation of ATP production by low H2S levels, higher H2S concentrations inhibit complex IV of the mETC and thereby impair ATP production. Libiad et al.134 established a link between the increased expression, and changes in the localisation, of SOP enzymes and suppression of the growth-restricting effects of excess H2S in colorectal carcinoma cells. Indeed, given that colorectal cancer cell lines display increased expression of H2S-synthesising enzymes, increased expression of SOP enzymes affords a higher capacity of these cells to dispose of excess H2S while stimulating cell bioenergetics.
Stimulating effects of H2S on glycolytic metabolism in cancer cells have been demonstrated to result from per-sulphidation of lactate dehydrogenase (LDH, particularly the LDH-A isoform)149,150 and glyceraldehyde-3-phosphate dehydrogenase, although the functional consequences of per-sulphidation of the latter appear to be controversial.151,152
H2S and cancer: beyond disturbed metabolism
In addition to the energetic stimulus afforded by low H2S concentrations, the link between the adaptability of cancer cells to an evolving and challenging environment and enhanced H2S metabolism extends to adaptation to hypoxia, antioxidant capacity, neoangiogenesis, cell cycle regulation, apoptosis evasion and chemoresistance. CBS and CSE have been reported to re-localise to mitochondria in response to hypoxia, a common feature of the tumour microenvironment, resulting in targeted H2S delivery that can stimulate ATP production and protect mitochondria from oxidative stress.109,153 Accordingly, exposure of SW480 colorectal cancer cells to hypoxia resulted in an enrichment in mitochondria of SQR protein levels and H2S detoxification activity.154 Besides the bioenergetics stimulation, the antioxidant nature of H2S is expected to protect cancer cells from oxidative damage. Indeed, protein per-sulphidation is posited to confer a protection mechanism to prevent irreversible oxidation of protein cysteine residues.155
The dysregulated proliferation of cancer cells leads to nutrient and oxygen deprivation, underlying the need for the formation of new blood vessels. The roles of CBS and CSE in neoangiogenesis have been shown for different cancer types, and possibly involve per-sulphidation of KATP channels and the activation of MAPK signalling pathways,156,157,158,159,160 which have been demonstrated in endothelial cells. Several studies have shown that H2S itself activates angiogenesis in cancer,161,162,163,164 evidence that was used to develop strategies to promote the release of H2S and the activation of angiogenesis under certain pathological circumstances, such as wound healing165,166 and inflammatory diseases.167
Evasion of apoptosis brought about by H2S has been attributed mostly to CSE-generated H2S mediating the per-sulphidation of key proteins in different signalling pathways: nuclear factor-κB (NF-κB) in hepatoma cells,168 the Keap1-transcriptional regulator of NRF2,169 and the extracellular-signal-regulated kinase (ERK)-activating protein kinase 1. CBS has been implicated in colorectal cancer carcinogenesis, its overexpression being detected even in precancerous lesions, such as hyperplastic polyps. Therefore, despite being only a partial effector of carcinogenesis, CBS belongs to a panel of intervening players, including NF-κB, KRAS, p53 and Wnt components, that contribute to metabolic rewiring and increased proliferative and invasive potential.170 Emerging evidence implies a role for CBS in the resistance of cancer cells to ferroptosis,85,171 although the mechanistic details of this observation remain to be unravelled (detailed below).
Cysteine plasma pools and bioavailability
In plasma, cysteine is the major thiol that contributes to glutathione levels and protein synthesis.172 Under normal conditions, protein synthesis prevails over the other cysteine-dependent pathways. Although the degradation of glutathione contributes to the cysteine pool, the resulting levels are not sufficient for ‘normal’ metabolism upon cystine scarcity.173
In healthy volunteers, the total cysteine availability in plasma is 200–300 μM, distributed across three pools—free reduced, free oxidised and protein bound. Up to 65% of cysteine is bound to proteins (protein s-cysteinylation; Fig. 4)174,175 and this pool increases with age.176 The remaining cysteine circulates mostly as cystine (25–30%, 40–50 μM) and the low abundance pool constituted by reduced cysteine. The concentration of cystine in blood is higher in women than in men and also increases with age.177 Cystine bioavailability across various tissues is ensured by different strategies, including drug-transporter-dependent mechanisms. The ability of NRF2 to regulate xCT coupled with the decline of NRF2 with age178 might account for the increased levels of plasma cystine seen with increasing age. Plasma from xCT-knockout mice contains a higher proportion of oxidised cysteine179 and xCT expression is increased in many tumours,180 pointing out its relevance in the context of cancer and eventually contributing for cancer metabolic rewiring. As many cysteine-containing proteins (transporters, receptors and enzymes) at extracellular surfaces or in extracellular fluids are prone to oxidation, their activity might be influenced by the thiol/disulphide redox microenvironment.181

The oxidation of a cysteine residue within a protein can result in the formation of a cysteinyl radical. l-Cystine is reduced to l-cysteine under the action of l-cystine reductase. Reaction between protein cysteinyl residues and low molecular weight thiols such as free cysteine can yield s-cysteinylated proteins.
Total plasma cysteine as a marker of cancer risk
The level of total plasma cysteine (free reduced, free oxidised and protein bound) has been associated with the risk of developing cancer. For instance, increased cysteine availability in plasma is related to an enhanced risk of breast cancer, particularly oestrogen receptor-positive (ER+) and/or progesterone receptor-positive (PR+) cancer and in combination with low folate availability in plasma.182 In breast cancer, higher cysteine plasma concentrations are found in patients with higher body weight,182,183 which is a known risk factor for breast cancer, progression and mortality,184 further linking cysteine, metabolic dysregulation and cancer. However, a correlation between higher plasma cysteine levels and increased breast cancer risk was not observed in another prospective nested case–control study, wherein an inverse cysteine–cancer risk relationship was particularly evident in leaner women183 or related to the catechol-O-methyltransferase (COMT) genotype compatible with high enzymatic activity.185 Inconsistencies in the outcome of these studies might be attributed to the heterogeneity of the populations studied and/or different conditions of analysis and concentrations of plasma cysteine used. On the other hand, the association with the COMT genotype185 and ER+ breast cancer182 places cysteine metabolism as a relevant feature of oestrogen-dependent hormonal breast cancer. COMT o-methylates catechol oestrogen metabolites and mediates their detoxification, and it is plausible that the activity of this enzyme is inhibited by the accumulation of the cysteine precursor homocysteine.185 These toxic oestrogen metabolites are also detoxified by glutathione, which is therefore dependent on cysteine availability. Conjugates formed between glutathione and toxic metabolites are further metabolised into cysteinyl-s-conjugates, which have long half-lives in circulation.186 In fact, increased levels of cysteinyl-s-conjugates in biological fluids have been associated with several cancers, including melanoma, non-Hodgkin lymphoma, breast, ovarian and thyroid carcinomas, and with poor prognosis, recurrence and survival (see below).186
On the other hand, plasma cysteine levels have been reported to show an inverse correlation with the risk of cervical dysplasia, showing a weak negative association between the levels of cysteine and the development of low-grade, but not high-grade, squamous intraepithelial lesions in a large case–control study.187 Similarly, higher serum concentrations of cysteine were associated with a significantly reduced risk of oesophageal squamous cell carcinomas and gastric adenocarcinomas.188 In another study conducted in male smokers, high serum cysteine levels were associated with gastric adenocarcinomas but not with oesophageal squamous cell carcinomas189 or head and neck squamous cell carcinoma.189 The plasma cysteine concentration was inversely related to the incidence of colorectal cancer in postmenopausal women, for rectal and proximal tumours (P = 0.06), but not for distal tumours.190 Furthermore, the association was significant for localised tumours, but not for metastases, and was not observed in a study conducted only in men191 or other cohorts of non-postmenopausal women or women at different physiological stages.192,193 In a metabolomics study, the levels of cysteine were inversely related to overall glioma risk, being lower in the circulation of glioma patients compared with controls years in advance of diagnosis.194 This result was consistent with the accumulation of CSA in patient-derived low-grade glioma and with the intra-tumoural expression of CDO1,76 since the decreased level of cysteine can be due to the overexpression and activity of CDO1 found in high-grade glioblastomas.
Interestingly, higher plasma levels of cystine indicated a high probability of response in patients with non-small cell lung cancer before and after treatment with the immune checkpoint inhibitor nivolumab, which targets programmed cell death protein 1.195 Considering everything stated before implying cysteine as relevant to sustain the high performance (survival and proliferation) of cancer cells, acting as a detoxifying component or as an energy or biomass source; this observation can be a clue to use cysteine levels to predict the therapy response and the behaviour of tumours.
Cysteine fractions and protein s-cysteinylation
Sullivan et al.196 showed that cystine levels are lower in tumour interstitial fluid in murine pancreatic adenocarcinomas (PDACs) than in plasma, which may indicate that cancer cells actively uptake cysteine. In the same work, cystine availability was lower in autochthonous PDAC tumours compared with subcutaneous tumours, supporting the relevance of anatomical location for the metabolic microenvironment. Nunes et al.197 reported increased total cysteine availability in the serum of women with ovarian tumours, regardless of whether the tumours were benign or malignant. The distinction between malignant and benign phenotypes was established by the presence of lower levels of free cysteine in the plasma of women with benign tumours. In addition, protein s-cysteinylation levels distinguish healthy controls from those with neoplasms, suggesting that discriminating plasma cysteine pools might be valuable for early diagnosis and outcome prediction. Patients ascites fluid, which is representative of the ovarian tumour microenvironment, was also rich in cysteine, derived predominantly from the s-cysteinylated form of albumin in plasma.176 S-cysteinylated proteins were found to be abundant in the ascites fluid and plasma of patients with ovarian cancer.197
Micropinocytosis as an entry mode for cysteine and cysteine
Cysteine, in addition to other amino acids, is also made available to tumour cells though micropinocytosis of extracellular proteins.198 Several pathways have been implicated in micropinocytosis events, such as the ERK/MAPK pathway activated by oncogenic RAS, and PI3K/mTOR signalling pathways.199 As previously mentioned, due to its reactivity, cysteine is not an abundant core residue in protein sequences. However, in plasma most proteins are reversibly post-translationally modified by cysteinylation114 and, as mentioned above, s-cysteinylated albumin represents a major source of cysteine for cells, including cancer cells.13 Although the cellular pathways required for protein s-cysteinylation are not fully elucidated (Fig. 4), evidence shows that CBS might have a relevant role in this setting, as no s-cysteinylated albumin was detected in CBS-deficient mice.200
Disulphide-containing proteins have long been reported to be the major source of cystine in lysosomes via endoproteolysis.201 This evidence reinforces the role of micropinocytosis as a route for the import of different compounds (proteins), to be used as nutrients or signalling molecules in cancer promotion.202,203 Essentially, the imported disulphide-containing proteins undergo cathepsin-catalysed degradation in the lysosome, leading to the formation of cystine, which is then effluxed by the cystinosin transporter into the cytosol, where it is reduced to cysteine in a process that requires NADPH, leading to the formation of oxidised glutathione.204,205 Both thiol availability and redox status have been shown to influence the expression of cystinosin, and a shift towards a more oxidised status of cysteine and glutathione with a progressive increase in the mRNA levels of cystinosin has been reported. So, cysteinylated proteins taken up from the plasma by micropinocytosis can constitute a source of cysteine that once in the cell will be used in the different cysteine metabolic pathways, even in the s-cysteinylation of other proteins.
Protein s-cysteinylation in cancer
Although the role of protein s-cysteinylation has not been fully explored in humans, some insights into its role in cancer, relating to the s-cysteinylation of certain proteins with cancer features, have emerged. For example, heparinase, an enzyme that degrades heparan sulphate and enhances the invasive and metastatic capacity of cancer cells, was reported to be activated by s-cysteinylation.206 In addition, enzymes involved in the control of oxidative stress can be modulated by s-cysteinylation, as is the case for the metalloenzyme Cu/Zn-superoxide dismutase 1 (SOD1), which catalyses the dismutation of superoxide anions into oxygen and hydrogen peroxide. s-Cysteinylation of SOD1 prevents the inhibitory action of hydrogen peroxide on SOD1,207,208 which contributes to an increased antioxidant potential of cancer cells and thereby protects them from oxidative damage. In fact, the reactive species interactome has been the subject of a very interesting review stating that the adaptive, selective and evolutionary cellular mechanisms that are required to overcome oxidative stress are largely dependent on cysteine redox switches, leading to enzyme modification and improved antioxidant capacity.209 Furthermore, s-cysteinylation of proteins and peptides from major histocompatibility complex classes I and II has been outlined as a means of reducing the immune effectiveness of T-lymphocytes,210 which is important in cancer as it can promote immune evasion and reduce the elimination of cancer cells by the immune system.
Therapeutic strategies targeting cysteine metabolism in cancer
Throughout this review, the trans-sulphuration pathway has been presented as a metabolic pathway that involves the interconversion (and de novo synthesis) of cysteine and homocysteine through the intermediate cystathionine, cysteine being a critical component of glutathione (Fig. 5b),211 and both cysteine and homocysteine contributing to the production of H2S. As stated before, glutathione is the most abundant antioxidant in the cell, with a key role in ROS scavenging in cancer cells.212 However, clinical trials aimed at inhibiting glutathione synthesis were unsuccessful, despite some strategies having provided promising results.213 Instead, inhibition of the H2S-synthesising trans-sulphuration pathway enzymes CBS and CSE, together with 3-MST, still holds great promise for cancer treatment,81,143,214 as outlined below. Another strategy outlined below relies on inhibition of the xCT system, which mediates the entry of cystine that fuels the trans-sulphuration pathway. Other therapeutic approaches outlined below include systemically decreasing cysteine levels and inhibiting GPX4.

a Drugs targeting cysteine metabolism. b Key inhibition targets and interplay between the trans-sulphuration pathway, the xCT antiporter and ferroptosis. c Inhibition of glutathione- and thioredoxin-dependent antioxidant pathways by sulphasalazine or auranofin combined with L-BSO. d Novel selenium-chrysin nanoformulations acting as inhibitors of glutathione bioavailability/synthesis and CBS activity. CBS cystathionine β-synthase, CSE cystathionine γ-lyase, GCS γ-glutamyl cysteine synthetase, GSS glutathione synthetase, Pyr pyruvate, α-KB α-ketobutyrate, GSH glutathione reduced form, GSSG glutathione oxidised form, ROS reactive oxygen species, FIN ferroptosis-inducing compounds.
Systemic depletion of cysteine
Strategies to bring about the systemic decrease of cysteine levels are advanced in the management of cancer, and some studies have presented promising results in breast, prostate and leukaemia models in vivo. These studies report a significant reduction in tumour burden upon the systemic depletion of cysteine using cyst(e)inase.215 Cyst(e)inase degrades extracellular cysteine and cystine and leads to decreased levels of intracellular cysteine and glutathione, which profoundly affects the redox potential of cancer cells216 and counteracts the essential role of cysteine in cancer cells.80 Cystine starvation has been reported to play an important role in cell death in different types of cancer. The mechanism is not fully understood but it is believed that the increased intracellular glutathione levels and increased ROS217 are closely coupled with the build-up of toxic lipid peroxides and consequent ferroptosis.218,219
Targeting H2S biosynthesis
The inhibition of H2S biosynthesis has been shown to sensitise cancer cells to chemotherapy, as it disturbs the mitochondrial equilibrium by impairing bioenergetics.220,221,222 Thus, the blockade of enzymes that contribute to the production of H2S constitutes an attractive anti-cancer strategy. As such, drug screening targeting CBS, CSE or 3-MST has been conducted; however, as yet, no inhibitor with pharmacological potential has been identified.114,220,223,224,225 Different CBS inhibitors have been designed and tested over the past years, starting with amino-oxyacetic acid (AOAA)52,58,60 and related prodrugs;226 unfortunately, however, AOAA was shown to also inhibit CSE activity.227 Subsequently, several compounds (e. g. hexachlorophene and benserazide) that were already clinically available were tested and repurposed as CBS inhibitors (with the knowledge that they were not specific for CBS).225 In 2018, Wang et al.171 designed and synthesised a novel, potent and bioactive inhibitor of CBS. This pharmacological probe (CH004, Fig. 5a) allows selective inhibition of CBS (raising cellular homocysteine levels and suppressing the production of H2S in a dose-dependent manner) over CSE; it can suppress cell proliferation with cell cycle arrest at S phase and, notably, it can reduce tumour growth in a xenograft mouse model. Importantly, the underlying cell death mechanism is triggered by CBS inhibition in HepG2 cells via ferroptosis, suggesting that CBS has a previously unreported function in this process. Nevertheless, further studies are required to elucidate the exact role and underlying molecular mechanisms of the trans-sulphuration pathway in cancer cell death.
In contrast to inhibiting H2S biosynthesis, some studies have addressed the advantage of ‘poisoning’ cancer cells with high levels of H2S, by using donors that release H2S in the tumour and somehow block the most important signalling pathways sustaining cancer survival, such as PI3K and MAPK.228,229,230
Inhibiting xCT
The search for strategies and drugs aimed at inducing oxidative stress has revealed xCT to be a suitable target—increased glutamate efflux through this antiporter is an important process for cells to generate sufficient glutathione to cope with high intracellular ROS levels.
Wangpaichitr et al.231 found that riluzole (Fig. 5a), approved for the treatment of amyotrophic lateral sclerosis, increases ROS levels by multiple mechanisms, including decreasing LDH-A and NAD+ levels to increase oxidative stress, as well as interfering with the xCT antiporter to block the cystine–glutamate pump. Together, these two mechanisms seem to work jointly to enhance ROS levels and lead to cell death in cisplatin-resistant lung cancer cells; this is an important achievement as no drugs are available to overcome cisplatin resistance or kill cisplatin-resistant cells.
Sulphasalazine (Fig. 5a), a pro-drug that combines sulphapyridine (an antibiotic) and 5-aminosalicylic (an anti-inflammatory agent) linked by an azo bridge, has huge therapeutic potential, but is, unfortunately, labile under physiological conditions (70% degradation by colonic bacteria via azo cleavage; Fig. 5c). Nevertheless, sulphasalazine has been demonstrated to be an effective treatment in cancer models, by inhibiting xCT to activate ferroptosis232,233 and restore sensitivity to chemotherapy.234 Gout et al.235 also reported that targeting xCT with sulphasalazine potently suppresses lymphoma growth. The effect of sulphasalazine on reducing the ROS defence capacity of cancer cells and sensitising them to available chemotherapeutic drugs (e.g. cisplatin, docetaxel) is associated with the activation of p38 MAPK-mediated growth suppression.236
As well as triggering cell death, xCT inhibition also induces the metabolic rewiring of cancer cells. Timmerman et al.237 detected metabolic responses related to perturbations in glutamine metabolism in 47 independent breast cancer-derived cell lines, meaning that the inhibition of xCT promotes an adjustment in glutamine metabolism, which indirectly contributes to the import of cysteine through the exchange with glutamine-derived glutamate through xCT. This metabolic adaptation can be relevant in triple-negative breast cancers (those lacking ERs, PRs and HER2, which constitute approximately a quarter of breast tumours) that express xCT, which exhibit increased levels of ROS induced by decreased glutamate levels upon glutamine scarcity. The authors hypothesised that xCT inhibition might be further potentiated by limiting glutamate or glutamine availability, and show that xCT is a compelling therapeutic target for triple-negative tumours. Okazaki et al.238 studied genes related to glutaminolysis in order to determine the sensitivity of glutamine metabolism to xCT-targeted therapy in head and neck squamous cell carcinoma. A metabolome analysis disclosed that sulphasalazine triggers an increase in the glutamate-derived TCA cycle intermediate α-ketoglutarate in addition to a decrease in cysteine and glutathione. This observation means that xCT blockage induces the accumulation of glutamate that is converted into α-ketoglutarate instead of being used in glutamate-cysteine exchange and consequently that is why cysteine does not enter the cell and give rise to glutathione.
GPX4 inhibition
Through the metabolomic profiling of 177 cancer cell lines, Yang et al.239 showed that glutathione depletion constitutes one mechanism of ferroptosis. Two classes of ferroptosis-inducing compounds were investigated (Fig. 5b), based on different approaches for inhibiting GPX4 (Fig. 4b). One of these classes (class I ferroptosis-inducing compounds, erastin derivatives) inhibits GPX4 through the depletion of glutathione, and the other (class II ferroptosis-inducing compounds, RSL3 derivatives) inhibits GPX4 directly, without glutathione depletion. Indirect inhibition of GPX4 using l-buthionine sulphoximine (L-BSO; an inhibitor of GCL and consequently of glutathione synthesis) enhanced ferroptotic cell death induced by all ferroptosis-inducing compounds, and its modulation effect is specific to ferroptosis-inducing compounds. Later, Viswanathan et al.240 identified ML210 and ML162 as two new class II ferroptosis-inducing compounds. In this study, it was also demonstrated that treatment with the inhibitor of cholesterol synthesis fluvastatin (Fig. 5a) led to a decrease in the expression of GPX4 in a time- and concentration-dependent manner, and a cumulative effect was observed when using fluvastatin and RSL3, showing that both contribute to ferroptosis.
Drug combinations
Combinations of agents that target cysteine metabolism have also been explored. Harris et al.5 combined L-BSO with sulphasalazine or auranofin (a gold salt used in the treatment of rheumatoid arthritis) to inhibit both glutathione- and thioredoxin-dependent antioxidant pathways, triggering synergistic cancer cell death (Fig. 5c). Ye et al.241 investigated the effect of the combined administration of paclitaxel and RSL3: alone and at low concentrations, these agents do not cause substantial cell death. Low-concentration paclitaxel (3–6 nM) is reported to interfere with glutaminolysis, a process that is essential for ferroptosis. Remarkably, the combination of these drugs induced ferroptosis and significant cell death in p53-mutated hypopharyngeal squamous cell carcinoma. Also, low-concentration paclitaxel (2 nM) enhanced RSL3-induced ferroptosis by upregulating the expression of p53 variants
Nanoformulations
Nanoformulation using targeted nanoparticles such as dendrimers is an emerging strategy in cancer therapeutics. Precise drug delivery by dendrimer nanoparticles is easily achieved by surface targeting, and folate-targeted dendrimers in particular are a relevant choice in cancer therapeutics, because cancer cells overexpress the folate receptor.242 Mota et al.243 developed folate-targeted dendrimers that are able to load and release L-BSO, whereas Santos et al.244 also used nanoparticles to produce selenium-chrysin (SeChry) nanoformulations to target glutathione bioavailability and CBS, aiming at novel ovarian cancer therapeutics (Fig. 5d). SeChry was chosen as a plausible competitive inhibitor of xCT as xCT is also able to take up selenium. Interestingly, this nanoformulation increased the specificity for SeChry delivery to ovarian cancer cells, as the nanoparticles were functionalised with folate and cancer cells express more folate receptor, and therefore significantly reduced the toxicity against non-malignant cells. Although SeChry did not affect the uptake of cysteine, it did increase glutathione depletion, indicating that it might induce oxidative stress, which will be scavenged by GPX4, a selenium-dependent enzyme.245 Also, in vitro enzymatic assays revealed an inhibitory effect of SeChry towards CBS, thus inhibiting H2S production.244
Conclusions and future perspectives
Once inside a cell, cysteine can have different fates, including the synthesis of glutathione or proteins, oxidative or non-oxidative catabolism, and reversible post-translational protein modification, each of which can contribute to the development and progression of cancer (Fig. 1). Having such a wide relevance in cancer, cysteine metabolism is undoubtedly a relevant target.
As mentioned above, a number of compounds or drug formulations have been investigated with regard to inhibiting cysteine metabolism, either by targeting H2S-synthesising trans-sulphuration pathway enzymes (e.g. CBS) or via xCT inhibition. However, there are many more studies that need to be carried out, and the description of ferroptosis—a relatively unexplored cancer cell death mechanism that is inhibited by glutathione-dependent GPX4—reinforces the importance of cysteine in cancer and points to new perspectives regarding the ways whereby cysteine import and synthesis and glutathione synthesis can be efficiently targeted in order to trigger cell death in cancer. Targeted nanoformulation is a very efficient and attractive platform that might solve the issues of low solubility and metabolic stability of some therapeutic agents. Selenium-containing drugs are expected to lead to important advances in novel cancer therapeutics, due the involvement of GPX4 in the elimination of lipid peroxides and in ferroptosis prevention. As well as chemical targeting, enzymatic targeting is another challenging strategy and cyst(e)inase-mediated depletion of serum cysteine and cystine pools has already been demonstrated to suppress the growth of tumours in different animal models.215
Herein, we describe the multiple possibilities whereby an enhanced cysteine transport and metabolism enables various types of cancer cells to adapt to the challenging tumour microenvironment, and to thrive, proliferate and acquire chemoresistance. This cysteine-centred view of cancer biology offers various possibilities to develop new and improved diagnostics tools and pharmacological strategies to target cancer.
References
Lopes-Coelho, F., Gouveia-Fernandes, S., Gonçalves, L. G., Nunes, C., Faustino, I., Silva, F. et al. HNF1β drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumor Biol. 37, 4813–4829 (2016).
Colla, R., Izzotti, A., De Ciucis, C., Fenoglio, D., Ravera, S., Speciale, A. et al. Glutathione-mediated antioxidant response and aerobic metabolism: two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 7, 70715–70737 (2016).
Zanotto-Filho, A., Masamsetti, V. P., Loranc, E., Tonapi, S. S., Gorthi, A., Bernard, X. et al. Alkylating agent-induced NRF2 blocks endoplasmic reticulum stress-mediated apoptosis via control of glutathione pools and protein thiol homeostasis. Mol. Cancer Ther. 15, 3000–3014 (2016).
Lien, E. C., Lyssiotis, C. A., Juvekar, A., Hu, H., Asara, J. M., Cantley, L. C. et al. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 18, 572–578 (2016).
Harris, I., Treloar, A., Inoue, S., Sasaki, M., Gorrini, C., Lee, K. et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015).
Traverso, N., Ricciarelli, R., Nitti, M., Marengo, B., Furfaro, A. L., Pronzato, M. A. et al. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013, 972913 (2013).
Okuno, S., Sato, H., Kuriyama-Matsumura, K., Tamba, M., Wang, H., Sohda, S. et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br. J. Cancer 88, 951–956 (2003).
Ballatori, N., Krance, S. M., Notenboom, S., Shi, S., Tieu, K. & Hammond, C. L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 390, 191–214 (2009).
Wu, G., Fang, Y.-Z., Yang, S., Lupton, J. R. & Turner, N. D. Glutathione metabolism and its implications for health. J. Nutr. 134, 489–492 (2004).
Wang, W. & Ballatori, N. Endogenous glutathione conjugates: occurrence and biological functions. Pharmacol. Rev. 50, 335–356 (1998).
Kalinina, E. V., Chernov, N. N. & Novichkova, M. D. Role of glutathione, glutathione transferase, and glutaredoxin in regulation of redox-dependent processes. Biochem. Biokhimiia 79, 1562–1583 (2014).
Hanigan, M. H. & Ricketts, W. A. Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Biochemistry 32, 6302–6306 (1993).
Davidson, S. M., Jonas, O., Keibler, M. A., Hou, H. W., Luengo, A., Mayers, J. R. et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 23, 235–241 (2017).
Mosharov, E., Cranford, M. R. & Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 39, 13005–13011 (2000).
Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 261, 2256–2263 (1986).
Lo, M., Wang, Y.-Z. & Gout, P. W. The x cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J. Cell. Physiol. 215, 593–602 (2008).
Bianchi, M. G., Bardelli, D., Chiu, M. & Bussolati, O. Changes in the expression of the glutamate transporter EAAT3/EAAC1 in health and disease. Cell. Mol. Life Sci. 71, 2001–2015 (2014).
Fazzari, J., Lin, H., Murphy, C., Ungard, R. & Singh, G. Inhibitors of glutamate release from breast cancer cells; new targets for cancer-induced bone-pain. Sci. Rep. 5, 8380 (2015).
Shiozaki, A., Iitaka, D., Ichikawa, D., Nakashima, S., Fujiwara, H., Okamoto, K. et al. xCT, component of cysteine/glutamate transporter, as an independent prognostic factor in human esophageal squamous cell carcinoma. J. Gastroenterol. 49, 853–863 (2014).
Stepulak, A., Rola, R., Polberg, K. & Ikonomidou, C. Glutamate and its receptors in cancer. J. Neural Transm. 121, 933–944 (2014).
Koochekpour, S., Majumdar, S., Azabdaftari, G., Attwood, K., Scioneaux, R., Subramani, D. et al. Serum glutamate levels correlate with Gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clin. Cancer Res. 18, 5888–5901 (2012).
Dornier, E., Rabas, N., Mitchell, L., Novo, D., Dhayade, S., Marco, S. et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun. 8, 2255 (2017).
Drayton, R. M., Dudziec, E., Peter, S., Bertz, S., Hartmann, A., Bryant, H. E. et al. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin. Cancer Res. 20, 1990–2000 (2014).
Doxsee, D. W., Gout, P. W., Kurita, T., Lo, M., Buckley, A. R., Wang, Y. et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate 67, 162–171 (2007).
Ji, X., Qian, J., Rahman, S. M. J., Siska, P. J., Zou, Y., Harris, B. K. et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 37, 5007–5019 (2018).
Koppula, P., Zhang, Y., Shi, J., Li, W. & Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 292, 14240–14249 (2017).
Lim, J. K. M., Delaidelli, A., Minaker, S. W., Zhang, H. F., Colovic, M., Yang, H. et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl Acad. Sci. USA 116, 9433–9442 (2019).
Habib, E., Linher-Melville, K., Lin, H.-X. & Singh, G. Expression of xCT and activity of system xc(−) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 5, 33–42 (2015).
Lien, E. C., Ghisolfi, L., Geck, R. C., Asara, J. M. & Toker, A. Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci. Signal. 10, eaao6604 (2017).
Mossmann, D., Park, S. & Hall, M. N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757 (2018).
Gu, Y., Albuquerque, C. P., Braas, D., Zhang, W., Villa, G. R., Bi, J. et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol. Cell 67, 128–38.e7 (2017).
Kang, E. S., Lee, J., Homma, T., Kurahashi, T., Kobayashi, S., Nabeshima, A. et al. xCT deficiency aggravates acetaminophen-induced hepatotoxicity under inhibition of the transsulfuration pathway. Free Radic. Res. 51, 80–90 (2017).
Jiang, Y., Cao, Y., Wang, Y., Li, W., Liu, X., Lv, Y. et al. Cysteine transporter SLC3A1 promotes breast cancer tumorigenesis. Theranostics 7, 1036–1046 (2017).
Zhang, W., Trachootham, D., Liu, J., Chen, G., Pelicano, H., Garcia-Prieto, C. et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 14, 276–286 (2012).
Bianchi, M. G., Franchi-Gazzola, R., Reia, L., Allegri, M., Uggeri, J., Chiu, M. et al. Valproic acid induces the glutamate transporter excitatory amino acid transporter-3 in human oligodendroglioma cells. Neuroscience 227, 260–270 (2012).
Aronica, E., Gorter, J. A., Ijlst-Keizers, H., Rozemuller, A. J., Yankaya, B., Leenstra, S. et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur. J. Neurosci. 17, 2106–2118 (2003).
McKenna, M. C., Stridh, M. H., McNair, L. F., Sonnewald, U., Waagepetersen, H. S. & Schousboe, A. Glutamate oxidation in astrocytes: roles of glutamate dehydrogenase and aminotransferases. J. Neurosci. Res. 94, 1561–1571 (2016).
Pedraz-Cuesta, E., Christensen, S., Jensen, A. A., Jensen, N. F., Bunch, L., Romer, M. U. et al. The glutamate transport inhibitor dl-Threo-β-benzyloxyaspartic acid (DL-TBOA) differentially affects SN38- and oxaliplatin-induced death of drug-resistant colorectal cancer cells. BMC Cancer 15, 411 (2015).
Pissimissis, N., Papageorgiou, E., Lembessis, P., Armakolas, A. & Koutsilieris, M. The glutamatergic system expression in human PC-3 and LNCaP prostate cancer cells. Anticancer Res. 29, 371–377 (2009).
White, M. A., Lin, C., Rajapakshe, K., Dong, J., Shi, Y., Tsouko, E. et al. Glutamine transporters are targets of multiple oncogenic signaling pathways in prostate cancer. Mol. Cancer Res. 15, 1017–1028 (2017).
Vanhove, K., Derveaux, E., Graulus, G.-J., Mesotten, L., Thomeer, M., Noben, J.-P. et al. Glutamine addiction and therapeutic strategies in lung cancer. Int. J. Mol. Sci. 20, 252 (2019).
Biancur, D. E., Paulo, J. A., Małachowska, B., Quiles Del Rey, M., Sousa, C. M., Wang, X. et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 8, 15965 (2017).
Wahi, K. & Holst, J. ASCT2: a potential cancer drug target. Expert Opin. Therap. Targets 23, 555–558 (2019).
Schulte, M. L., Fu, A., Zhao, P., Li, J., Geng, L., Smith, S. T. et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 24, 194–202 (2018).
Wang, Q., Hardie, R.-A., Hoy, A. J., van Geldermalsen, M., Gao, D., Fazli, L. et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 236, 278–289 (2015).
Muir, A., Danai, L. V., Gui, D. Y., Waingarten, C. Y., Lewis, C. A., Vander & Heiden, M. G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, e27713 (2017).
Lian, G., Gnanaprakasam, J. R., Wang, T., Wu, R., Chen, X., Liu, L. et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. Elife 7, e36158 (2018).
Zou, S., Shimizu, T., Shimizu, S., Higashi, Y., Nakamura, K., Ono, H. et al. Possible role of hydrogen sulfide as an endogenous relaxation factor in the rat bladder and prostate. Neurourol. Urodyn. 37, 2519–2526 (2018).
Huang, C. W. & Moore, P. K. in Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide (eds Moore, P. K. & Whiteman, M.) 3–25 (Springer International Publishing, Cham 2015).
Pascale, R. M., Peitta, G., Simile, M. M. & Feo, F. Alterations of methionine metabolism as potential targets for the prevention and therapy of hepatocellular carcinoma. Medicina 55, 296 (2019).
Adeva-Andany, M. M., López-Maside, L., Donapetry-García, C., Fernández-Fernández, C. & Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids 49, 1005–1028 (2017).
Szabo, C., Coletta, C., Chao, C., Módis, K., Szczesny, B., Papapetropoulos, A. et al. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl Acad. Sci. USA 110, 12474–12479 (2013).
Wang, L., Shi, H., Zhang, X., Zhang, X., Liu, Y., Kang, W. et al. I157172, a novel inhibitor of cystathionine gamma-lyase, inhibits growth and migration of breast cancer cells via SIRT1-mediated deacetylation of STAT3. Oncol. Rep. 41, 427–436 (2019).
You, J., Shi, X., Liang, H., Ye, J., Wang, L., Han, H. et al. Cystathionine- γ-lyase promotes process of breast cancer in association with STAT3 signaling pathway. Oncotarget 8, 65677–65686 (2017).
Turbat-Herrera, E. A., Kilpatrick, M. J., Chen, J., Meram, A. T., Cotelingam, J., Ghali, G. et al. Cystathione β-synthase is increased in thyroid malignancies. Anticancer Res. 38, 6085–6090 (2018).
Alix-Panabières, C., Cayrefourcq, L., Mazard, T., Maudelonde, T., Assenat, E. & Assou, S. Molecular portrait of metastasis-competent circulating tumor cells in colon cancer reveals the crucial role of genes regulating energy metabolism and DNA repair. Clin. Chem. 63, 700–713 (2017).
Sekiguchi, F., Sekimoto, T., Ogura, A. & Kawabata, A. Endogenous hydrogen sulfide enhances cell proliferation of human gastric cancer AGS cells. Biol. Pharm. Bull. 39, 887–890 (2016).
Bhattacharyya, S., Saha, S., Giri, K., Lanza, I. R., Nair, K. S., Jennings, N. B. et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE 8, e79167–e79167 (2013).
Poisson, L. M., Munkarah, A., Madi, H., Datta, I., Hensley-Alford, S., Tebbe, C. et al. A metabolomic approach to identifying platinum resistance in ovarian cancer. J. Ovarian Res. 8, 13 (2015).
Sen, S., Kawahara, B., Gupta, D., Tsai, R., Khachatryan, M., Roy-Chowdhuri, S. et al. Role of cystathionine β-synthase in human breast cancer. Free Radic. Biol. Med. 86, 228–238 (2015).
Bella, D. L., Hahn, C. & Stipanuk, M. H. Effects of nonsulfur and sulfur amino acids on the regulation of hepatic enzymes of cysteine metabolism. Am. J. Physiol. 277, E144–E153 (1999).
Bella, D. L., Hirschberger, L. L., Hosokawa, Y. & Stipanuk, M. H. Mechanisms involved in the regulation of key enzymes of cysteine metabolism in rat liver in vivo. Am. J. Physiol. 276, E326–E335 (1999).
Lee, J. I., Londono, M., Hirschberger, L. L. & Stipanuk, M. H. Regulation of cysteine dioxygenase and gamma-glutamylcysteine synthetase is associated with hepatic cysteine level. J. Nutr. Biochem. 15, 112–122 (2004).
Stipanuk, M. H. Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr. 24, 539–577 (2004).
Kwon, Y. H. & Stipanuk, M. H. Cysteine regulates expression of cysteine dioxygenase and gamma-glutamylcysteine synthetase in cultured rat hepatocytes. Am. J. Physiol. Endocrinol. Metab. 280, E804–E815 (2001).
Davies, M. H., Ngong, J. M., Pean, A., Vickers, C. R., Waring, R. H. & Elias, E. Sulphoxidation and sulphation capacity in patients with primary biliary cirrhosis. J. Hepatol. 22, 551–560 (1995).
Dominy, J. E. Jr., Hwang, J. & Stipanuk, M. H. Overexpression of cysteine dioxygenase reduces intracellular cysteine and glutathione pools in HepG2/C3A cells. Am. J. Physiol. Endocrinol. Metab. 293, E62–E69 (2007).
Jurkowska, H., Roman, H. B., Hirschberger, L. L., Sasakura, K., Nagano, T., Hanaoka, K. et al. Primary hepatocytes from mice lacking cysteine dioxygenase show increased cysteine concentrations and higher rates of metabolism of cysteine to hydrogen sulfide and thiosulfate. Amino Acids 46, 1353–1365 (2014).
Kang, Y. P., Torrente, L., Falzone, A., Elkins, C. M., Liu, M., Asara, J. M. et al. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 8, e45572 (2019).
Jeschke, J., O’Hagan, H. M., Zhang, W., Vatapalli, R., Calmon, M. F., Danilova, L. et al. Frequent inactivation of cysteine dioxygenase type 1 contributes to survival of breast cancer cells and resistance to anthracyclines. Clin. Cancer Res. 19, 3201–3211 (2013).
Pasantes-Morales, H. Taurine homeostasis and volume control. Adv. Neurobiol. 16, 33–53 (2017).
Khan, S., Bano, Z., Singh, L. R., Hassan, M. I., Islam, A. & Ahmad, F. Testing the ability of non-methylamine osmolytes present in kidney cells to counteract the deleterious effects of urea on structure, stability and function of proteins. PLoS ONE 8, e72533 (2013).
Yu, L., Teoh, S. T., Ensink, E., Ogrodzinski, M. P., Yang, C., Vazquez, A. I. et al. Cysteine catabolism and the serine biosynthesis pathway support pyruvate production during pyruvate kinase knockdown in pancreatic cancer cells. Cancer Metab. 7, 13 (2019).
Brait, M., Ling, S., Nagpal, J. K., Chang, X., Park, H. L., Lee, J. et al. Cysteine dioxygenase 1 is a tumor suppressor gene silenced by promoter methylation in multiple human cancers. PLoS ONE 7, e44951 (2012).
Hao, S., Yu, J., He, W., Huang, Q., Zhao, Y., Liang, B. et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia 19, 1022–1032 (2017).
Prabhu, A., Sarcar, B., Kahali, S., Yuan, Z., Johnson, J. J., Adam, K. P. et al. Cysteine catabolism: a novel metabolic pathway contributing to glioblastoma growth. Cancer Res. 74, 787–796 (2014).
Pérez-Miguelsanz, J., Vallecillo, N., Garrido, F., Reytor, E., Pérez-Sala, D. & Pajares, M. A. Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim. Biophys. Acta 1864, 1165–1182 (2017).
Amelio, I., Cutruzzolá, F., Antonov, A., Agostini, M. & Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 39, 191–198 (2014).
Nikiforov, M. A., Chandriani, S., O’Connell, B., Petrenko, O., Kotenko, I., Beavis, A. et al. A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol. Cell. Biol. 22, 5793–5800 (2002).
Combs, J. A. & DeNicola, G. M. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers 11, 678 (2019).
Hellmich, M. R. & Szabo, C. Hydrogen sulfide and cancer. Handb. Exp. Pharmacol. 230, 233–41. (2015).
Giuffrè, A. & Vicente, J. B. Hydrogen sulfide biochemistry and interplay with other gaseous mediators in mammalian physiology. Oxid. Med. Cell Longev. 2018, 6290931 (2018).
Zhu, J., Berisa, M., Schworer, S., Qin, W., Cross, J. R. & Thompson, C. B. Transsulfuration activity can support cell growth upon extracellular cysteine limitation. Cell Metab. 30, 865–76 e5 (2019).
Cao, J., Chen, X., Jiang, L., Lu, B., Yuan, M., Zhu, D. et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 11, 1251 (2020).
Liu, N., Lin, X. & Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 122, pages279–292 (2020).
DeBerardinis, R. J. Serine metabolism: some tumors take the road less traveled. Cell Metab. 14, 285–286 (2011).
Sun, L., Song, L., Wan, Q., Wu, G., Li, X., Wang, Y. et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 25, 429–444 (2015).
Labuschagne, C., van den Broek, N., Mackay, G., Vousden, K. & Maddocks, O. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7, 1248–1258 (2014).
Liang, Y., Zhang, C. & Dai, D.-Q. Identification of differentially expressed genes regulated by methylation in colon cancer based on bioinformatics analysis. World J. Gastroenterol. 25, 3392–3407 (2019).
Liu, B., Song, J., Luan, J., Sun, X., Bai, J., Wang, H. et al. Promoter methylation status of tumor suppressor genes and inhibition of expression of DNA methyltransferase 1 in non-small cell lung cancer. Exp. Biol. Med. 241, 1531–1539 (2016).
Nazki, F. H., Sameer, A. S. & Ganaie, B. A. Folate: metabolism, genes, polymorphisms and the associated diseases. Gene 533, 11–20 (2014).
Nilsson, R., Nicolaidou, V. & Koufaris, C. Mitochondrial MTHFD isozymes display distinct expression, regulation, and association with cancer. Gene 144032 (2019).
Kulkarni, A., Dangat, K., Kale, A., Sable, P., Chavan-Gautam, P. & Joshi, S. Effects of altered maternal folic acid, vitamin B12 and docosahexaenoic acid on placental global DNA methylation patterns in Wistar rats. PLoS ONE 6, e17706–e17706 (2011).
Umapathy, A., Li, B., Donaldson, P. J. & Lim, J. C. Functional characterisation of glutathione export from the rat lens. Exp. Eye Res. 166, 151–159 (2018).
Hausheer, F. H., Parker, A. R., Petluru, P. N., Jair, K. W., Chen, S., Huang, Q. et al. Mechanistic study of BNP7787-mediated cisplatin nephroprotection: modulation of human aminopeptidase N. Cancer Chemother. Pharmacol. 67, 381–391 (2011).
Frey, I. M., Rubio-Aliaga, I., Siewert, A., Sailer, D., Drobyshev, A., Beckers, J. et al. Profiling at mRNA, protein, and metabolite levels reveals alterations in renal amino acid handling and glutathione metabolism in kidney tissue of Pept2−/− mice. Physiol. Genomics 28, 301–310 (2007).
Nguyen, Y. T. K., Park, J. S., Jang, J. Y., Kim, K. R., Vo, T. T. L., Kim, K. W. et al. Structural and functional analyses of human ChaC2 in glutathione metabolism. Biomolecules 10, 31 (2019).
Kaur, A., Gautam, R., Srivastava, R., Chandel, A., Kumar, A., Karthikeyan, S. et al. ChaC2, an enzyme for slow turnover of cytosolic glutathione. J. Biol. Chem. 292, 638–651 (2017).
Liu, S., Fei, W., Shi, Q., Li, Q., Kuang, Y., Wang, C. et al. CHAC2, downregulated in gastric and colorectal cancers, acted as a tumor suppressor inducing apoptosis and autophagy through unfolded protein response. Cell Death Dis. 8, e3009 (2017).
Vicente, J. B., Malagrino, F., Arese, M., Forte, E., Sarti, P. & Giuffre, A. Bioenergetic relevance of hydrogen sulfide and the interplay between gasotransmitters at human cystathionine beta-synthase. Biochim. Biophys. Acta 1857, 1127–1138 (2016).
Jiang, J., Chan, A., Ali, S., Saha, A., Haushalter, K. J., Lam, W. L. et al. Hydrogen sulfide-mechanisms of toxicity and development of an antidote. Sci. Rep. 6, 20831 (2016).
Cuevasanta, E., Moller, M. N. & Alvarez, B. Biological chemistry of hydrogen sulfide and persulfides. Arch. Biochem. Biophys. 617, 9–25 (2017).
Filipovic, M. R., Zivanovic, J., Alvarez, B. & Banerjee, R. Chemical biology of H2S signaling through persulfidation. Chem. Rev. 118, 1253–1337 (2018).
Yadav, P. K., Martinov, M., Vitvitsky, V., Seravalli, J., Wedmann, R., Filipovic, M. R. et al. Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc. 138, 289–299 (2016).
Kabil, O. & Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 20, 770–782 (2014).
Blachier, F., Beaumont, M. & Kim, E. Cysteine-derived hydrogen sulfide and gut health: a matter of endogenous or bacterial origin. Curr. Opin. Clin. Nutr. Metab. Care 22, 68–75 (2019).
Rose, P., Moore, P. K. & Zhu, Y. Z. H2S biosynthesis and catabolism: new insights from molecular studies. Cell. Mol. Life Sci. 74, 1391–1412 (2017).
Fu, M., Zhang, W., Wu, L., Yang, G., Li, H. & Wang, R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl Acad. Sci. USA 109, 2943–2948 (2012).
Teng, H., Wu, B., Zhao, K., Yang, G., Wu, L. & Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc. Natl Acad. Sci. USA 110, 12679–12684 (2013).
Kabil, O., Zhou, Y. & Banerjee, R. Human cystathionine beta-synthase is a target for sumoylation. Biochemistry 45, 13528–13536 (2006).
Bearden, S. E., Beard, R. S. Jr. & Pfau, J. C. Extracellular transsulfuration generates hydrogen sulfide from homocysteine and protects endothelium from redox stress. Am. J. Physiol. Heart Circ. Physiol. 299, H1568–H1576 (2010).
Frasdorf, B., Radon, C. & Leimkuhler, S. Characterization and interaction studies of two isoforms of the dual localized 3-mercaptopyruvate sulfurtransferase TUM1 from humans. J. Biol. Chem. 289, 34543–34556 (2014).
Yadav, P. K., Yamada, K., Chiku, T., Koutmos, M. & Banerjee, R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013 (2013).
Zuhra, K., Tomé, C. S., Masi, L., Giardina, G., Paulini, G., Malagrinò, F. et al. N-acetylcysteine serves as substrate of 3-mercaptopyruvate sulfurtransferase and stimulates sulfide metabolism in colon cancer cells. Cells 8, 828 (2019).
Kimura, Y., Koike, S., Shibuya, N., Lefer, D., Ogasawara, Y. & Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H2S2, H2S3 and H2S. Sci. Rep. 7, 10459 (2017).
Yadav, P. K., Vitvitsky, V., Carballal, S., Seravalli, J. & Banerjee, R. Thioredoxin regulates human mercaptopyruvate sulfurtransferase at physiologically relevant concentrations. J. Biol. Chem. 295, 6299–6311 (2020).
Kabil, O., Vitvitsky, V., Xie, P. & Banerjee, R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 15, 363–372 (2011).
McCorvie, T. J., Kopec, J., Hyung, S. J., Fitzpatrick, F., Feng, X., Termine, D. et al. Inter-domain communication of human cystathionine beta-synthase: structural basis of S-adenosyl-l-methionine activation. J. Biol. Chem. 289, 36018–36030 (2014). 2014.
Niu, W. N., Yadav, P. K., Adamec, J. & Banerjee, R. S-glutathionylation enhances human cystathionine beta-synthase activity under oxidative stress conditions. Antioxid. Redox Signal. 22, 350–361 (2015).
Banerjee, R. & Zou, C. G. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 433, 144–156 (2005).
Vicente, J. B., Colaco, H. G., Mendes, M. I., Sarti, P., Leandro, P. & Giuffre, A. NO* binds human cystathionine beta-synthase quickly and tightly. J. Biol. Chem. 289, 8579–8587 (2014).
Vicente, J. B., Colaco, H. G., Sarti, P., Leandro, P. & Giuffre, A. S-Adenosyl-l-methionine modulates CO and NO* binding to the human H2S-generating enzyme cystathionine beta-synthase. J. Biol. Chem. 291, 572–581 (2016).
Kumar, A., Wissbrock, A., Goradia, N., Bellstedt, P., Ramachandran, R., Imhof, D. et al. Heme interaction of the intrinsically disordered N-terminal peptide segment of human cystathionine-beta-synthase. Sci. Rep. 8, 2474 (2018).
Banerjee, R. Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Curr. Opin. Chem. Biol. 37, 115–121 (2017).
Libiad, M., Yadav, P. K., Vitvitsky, V., Martinov, M. & Banerjee, R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 (2014).
Landry, A. P., Moon, S., Kim, H., Yadav, P. K., Guha, A., Cho, U. S. et al. A catalytic trisulfide in human sulfide quinone oxidoreductase catalyzes coenzyme a persulfide synthesis and inhibits butyrate oxidation. Cell Chem. Biol. 26, 1515–25 e4 (2019).
Augustyn, K. D., Jackson, M. R. & Jorns, M. S. Use of tissue metabolite analysis and enzyme kinetics to discriminate between alternate pathways for hydrogen sulfide metabolism. Biochemistry 56, 986–996 (2017).
Jackson, M. R., Melideo, S. L. & Jorns, M. S. Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 (2012).
Landry, A. P., Ballou, D. P. & Banerjee, R. H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. J. Biol. Chem. 292, 11641–11649 (2017).
Mishanina, T. V., Yadav, P. K., Ballou, D. P. & Banerjee, R. Transient kinetic analysis of hydrogen sulfide oxidation catalyzed by human sulfide quinone oxidoreductase. J. Biol. Chem. 290, 25072–25080 (2015).
Linden, D. R., Furne, J., Stoltz, G. J., Abdel-Rehim, M. S., Levitt, M. D. & Szurszewski, J. H. Sulphide quinone reductase contributes to hydrogen sulphide metabolism in murine peripheral tissues but not in the CNS. Br. J. Pharmacol. 165, 2178–2190 (2012).
Vitvitsky, V., Kabil, O. & Banerjee, R. High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox Signal. 17, 22–31 (2012).
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 (2014).
Libiad, M., Vitvitsky, V., Bostelaar, T., Bak, D. W., Lee, H. J., Sakamoto, N. et al. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 294, 12077–12090 (2019).
Pietri, R., Roman-Morales, E. & Lopez-Garriga, J. Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxid. Redox Signal. 15, 393–404 (2011).
Ida, T., Sawa, T., Ihara, H., Tsuchiya, Y., Watanabe, Y., Kumagai, Y. et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl Acad. Sci. USA 111, 7606–7611 (2014).
Akaike, T., Ida, T., Wei, F. Y., Nishida, M., Kumagai, Y., Alam, M. M. et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 8, 1177 (2017).
Ferecatu, I., Canal, F., Fabbri, L., Mazure, N. M., Bouton, C. & Golinelli-Cohen, M. P. Dysfunction in the mitochondrial Fe-S assembly machinery leads to formation of the chemoresistant truncated VDAC1 isoform without HIF-1alpha activation. PLoS ONE 13, e0194782 (2018).
Melber, A. & Winge, D. R. Steps toward understanding mitochondrial Fe/S cluster biogenesis. Methods Enzymol. 599, 265–292 (2018).
Alvarez, S. W., Sviderskiy, V. O., Terzi, E. M., Papagiannakopoulos, T., Moreira, A. L., Adams, S. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017).
Augsburger, F. & Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)—hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 104083 (2018).
Hale, V. L., Jeraldo, P., Mundy, M., Yao, J., Keeney, G., Scott, N. et al. Synthesis of multi-omic data and community metabolic models reveals insights into the role of hydrogen sulfide in colon cancer. Methods 149, 59–68 (2018).
Szabo, C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 15, 185–203 (2016).
Zhu, H., Blake, S., Chan, K. T., Pearson, R. B. & Kang, J. Cystathionine β-synthase in physiology and cancer. Biomed. Res. Int. 2018, 3205125 (2018).
Akbari, M., Sogutdelen, E., Juriasingani, S. & Sener, A. Hydrogen sulfide: emerging role in bladder, kidney, and prostate malignancies. Oxid. Med. Cell Longev. 2019, 2360945 (2019).
Modis, K., Ju, Y., Ahmad, A., Untereiner, A. A., Altaany, Z., Wu, L. et al. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharm. Res. 113, 116–124 (2016).
Augsburger, F., Randi, E. B., Jendly, M., Ascencao, K., Dilek, N., Szabo, C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation, migration, and bioenergetics in murine colon cancer cells. Biomolecules 10, 447 (2020).
Modis, K., Coletta, C., Erdelyi, K., Papapetropoulos, A. & Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 27, 601–611 (2013).
Untereiner, A. A., Pavlidou, A., Druzhyna, N., Papapetropoulos, A., Hellmich, M. R. & Szabo, C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 149, 174–85. (2018).
Valvona, C. J., Fillmore, H. L., Nunn, P. B. & Pilkington, G. J. The regulation and function of lactate dehydrogenase a: therapeutic potential in brain tumor. Brain Pathol. 26, 3–17 (2016).
Jarosz, A. P., Wei, W., Gauld, J. W., Auld, J., Ozcan, F., Aslan, M. et al. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is inactivated by S-sulfuration in vitro. Free Radic. Biol. Med. 89, 512–521 (2015).
Mustafa, A. K., Gadalla, M. M., Sen, N., Kim, S., Mu, W., Gazi, S. K. et al. H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72 (2009).
Wang, M., Guo, Z. & Wang, S. Regulation of cystathionine gamma-lyase in mammalian cells by hypoxia. Biochem. Genet. 52, 29–37 (2014).
Malagrino, F., Zuhra, K., Mascolo, L., Mastronicola, D., Vicente, J. B., Forte, E. et al. Hydrogen sulfide oxidation: adaptive changes in mitochondria of sw480 colorectal cancer cells upon exposure to hypoxia. Oxid. Med. Cell Longev. 2019, 8102936 (2019).
Dóka, É., Ida, T., Dagnell, M., Abiko, Y., Luong, N. C., Balog, N. et al. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 6, eaax8358 (2020).
Mustafa, A. K., Sikka, G., Gazi, S. K., Steppan, J., Jung, S. M., Bhunia, A. K. et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 109, 1259–1268 (2011).
Tang, G., Wu, L., Liang, W. & Wang, R. Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol. Pharmacol. 68, 1757–1764 (2005).
Cai, W. J., Wang, M. J., Moore, P. K., Jin, H. M., Yao, T. & Zhu, Y. C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 76, 29–40 (2007).
Papapetropoulos, A., Pyriochou, A., Altaany, Z., Yang, G., Marazioti, A., Zhou, Z. et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl Acad. Sci. USA 106, 21972–21977 (2009).
Zhao, K., Ju, Y., Li, S., Altaany, Z., Wang, R. & Yang, G. S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 15, 792–800 (2014).
Wu, D., Li, J., Zhang, Q., Tian, W., Zhong, P., Liu, Z. et al. Exogenous hydrogen sulfide regulates the growth of human thyroid carcinoma cells. Oxid. Med. Cell Longev. 2019, 6927298 (2019).
Wang, M., Yan, J., Cao, X., Hua, P. & Li, Z. Hydrogen sulfide modulates epithelial-mesenchymal transition and angiogenesis in non-small cell lung cancer via HIF-1alpha activation. Biochem. Pharmacol. 172, 113775 (2020).
Tao, B. B., Cai, W. J. & Zhu, Y. C. H2S is a promoter of angiogenesis: identification of H2S “receptors” and its molecular switches in vascular endothelial cells. Handb. Exp. Pharmacol. 230, 137–152 (2015).
Wu, D., Li, M., Tian, W., Wang, S., Cui, L., Li, H. et al. Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways. Sci. Rep. 7, 5134 (2017).
Kaur, K., Carrazzone, R. J. & Matson, J. B. The benefits of macromolecular/supramolecular approaches in hydrogen sulfide delivery: a review of polymeric and self-assembled hydrogen sulfide donors. Antioxid. Redox Signal. 32, 79–95 (2020).
Zhao, X., Liu, L., An, T., Xian, M., Luckanagul, J. A., Su, Z. et al. A hydrogen sulfide-releasing alginate dressing for effective wound healing. Acta Biomater. 104, 85–94 (2020).
Zhao, A. S., Zou, D., Wang, H. H., Han, X., Yang, P. & Huang, N. Hydrogen sulphide-releasing aspirin enhances cell capabilities of anti-oxidative lesions and anti-inflammation. Med. Gas Res. 9, 145–152 (2019).
Zhen, Y., Pan, W., Hu, F., Wu, H., Feng, J., Zhang, Y. et al. Exogenous hydrogen sulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-kappaB pathway in PLC/PRF/5 hepatoma cells. Int. J. Oncol. 46, 2194–2204 (2015).
Yang, G., Zhao, K., Ju, Y., Mani, S., Cao, Q., Puukila, S. et al. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 18, 1906–1919 (2013).
Phillips, C. M., Zatarain, J. R., Nicholls, M. E., Porter, C., Widen, S. G., Thanki, K. et al. Upregulation of cystathionine-beta-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res. 77, 5741–5754 (2017).
Wang, L., Cai, H., Hu, Y., Liu, F., Huang, S., Zhou, Y. et al. A pharmacological probe identifies cystathionine beta-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 9, 1005 (2018).
Ookhtens, M. & Kaplowitz, N. Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine. Semin. Liver Dis. 18, 313–329 (1998).
Yu, X. & Long, Y. C. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci. Rep. 6, 30033 (2016).
Oliveira, P. V. S. & Laurindo, F. R. M. Implications of plasma thiol redox in disease. Clin. Sci. 132, 1257–1280 (2018).
Mansoor, M. A., Svardal, A. M. & Ueland, P. M. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Anal. Biochem. 200, 218–229 (1992).
Rossi, R., Giustarini, D., Milzani, A. & Dalle-Donne, I. Cysteinylation and homocysteinylation of plasma protein thiols during ageing of healthy human beings. J. Cell Mol. Med. 13, 3131–3140 (2009).
Blanco, R. A., Ziegler, T. R., Carlson, B. A., Cheng, P. Y., Park, Y., Cotsonis, G. A. et al. Diurnal variation in glutathione and cysteine redox states in human plasma. Am. J. Clin. Nutr. 86, 1016–1023 (2007).
Suh, J. H., Shenvi, S. V., Dixon, B. M., Liu, H., Jaiswal, A. K., Liu, R. M. et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl Acad. Sci. USA 101, 3381–3386 (2004).
Sato, H., Shiiya, A., Kimata, M., Maebara, K., Tamba, M., Sakakura, Y. et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 280, 37423–37429 (2005).
Polewski, M. D., Reveron-Thornton, R. F., Cherryholmes, G. A., Marinov, G. K., Cassady, K. & Aboody, K. S. Increased expression of system xc− in glioblastoma confers an altered metabolic state and temozolomide resistance. Mol. Cancer Res. 14, 1229–1242 (2016).
Moriarty-Craige, S. E. & Jones, D. P. Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 24, 481–509 (2004).
Lin, J., Lee, I. M., Song, Y., Cook, N. R., Selhub, J., Manson, J. E. et al. Plasma homocysteine and cysteine and risk of breast cancer in women. Cancer Res. 70, 2397–2405 (2010).
Zhang, S. M., Willett, W. C., Selhub, J., Manson, J. E., Colditz, G. A. & Hankinson, S. E. A prospective study of plasma total cysteine and risk of breast cancer. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive. Oncology 12, 1188–1193 (2003).
Weiderpass, E., Braaten, T., Magnusson, C., Kumle, M., Vainio, H., Lund, E. et al. A prospective study of body size in different periods of life and risk of premenopausal breast cancer. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive. Oncology 13, 1121–1127 (2004).
Goodman, J. E., Lavigne, J. A., Wu, K., Helzlsouer, K. J., Strickland, P. T., Selhub, J. et al. COMT genotype, micronutrients in the folate metabolic pathway and breast cancer risk. Carcinogenesis 22, 1661–1665 (2001).
Goncalves-Dias, C., Morello, J., Semedo, V., Correia, M. J., Coelho, N. R., Monteiro, E. C. et al. The mercapturomic profile of health and non-communicable diseases. High-throughput 8 (2019).
Goodman, M. T., McDuffie, K., Hernandez, B., Wilkens, L. R. & Selhub, J. Case–control study of plasma folate, homocysteine, vitamin B(12), and cysteine as markers of cervical dysplasia. Cancer 89, 376–382 (2000).
Murphy, G., Fan, J. H., Mark, S. D., Dawsey, S. M., Selhub, J., Wang, J. et al. Prospective study of serum cysteine levels and oesophageal and gastric cancers in China. Gut 60, 618–623 (2011).
Miranti, E. H., Freedman, N. D., Weinstein, S. J., Abnet, C. C., Selhub, J., Murphy, G. et al. Prospective study of serum cysteine and cysteinylglycine and cancer of the head and neck, esophagus, and stomach in a cohort of male smokers. Am. J. Clin. Nutr. 104, 686–693 (2016).
Miller, J. W., Beresford, S. A., Neuhouser, M. L., Cheng, T. Y., Song, X., Brown, E. C. et al. Homocysteine, cysteine, and risk of incident colorectal cancer in the Women’s Health Initiative observational cohort. Am. J. Clin. Nutr. 97, 827–834 (2013).
Lee, J. E., Li, H., Giovannucci, E., Lee, I. M., Selhub, J., Stampfer, M. et al. Prospective study of plasma vitamin B6 and risk of colorectal cancer in men. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive. Oncology 18, 1197–1202 (2009).
Chiang, F. F., Wang, H. M., Lan, Y. C., Yang, M. H., Huang, S. C. & Huang, Y. C. High homocysteine is associated with increased risk of colorectal cancer independently of oxidative stress and antioxidant capacities. Clin. Nutr. 33, 1054–1060 (2014).
Myte, R., Gylling, B., Haggstrom, J., Schneede, J., Lofgren-Burstrom, A., Huyghe, J. R. et al. One-carbon metabolism biomarkers and genetic variants in relation to colorectal cancer risk by KRAS and BRAF mutation status. PLoS ONE 13, e0196233 (2018).
Huang, J., Weinstein, S. J., Kitahara, C. M., Karoly, E. D., Sampson, J. N. & Albanes, D. A prospective study of serum metabolites and glioma risk. Oncotarget 8, 70366–70377 (2017).
Hatae, R., Chamoto, K., Kim, Y. H., Sonomura, K., Taneishi, K., Kawaguchi, S. et al. Combination of host immune metabolic biomarkers for the PD-1 blockade cancer immunotherapy. JCI Insight 5, e133501 (2020).
Sullivan, M. R., Danai, L. V., Lewis, C. A., Chan, S. H., Gui, D. Y., Kunchok, T. et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235 (2019).
Nunes, S. C., Ramos, C., Lopes-Coelho, F., Sequeira, C. O., Silva, F., Gouveia-Fernandes, S. et al. Cysteine allows ovarian cancer cells to adapt to hypoxia and to escape from carboplatin cytotoxicity. Sci. Rep. 8, 9513 (2018).
Kamphorst, J. J., Nofal, M., Commisso, C., Hackett, S. R., Lu, W., Grabocka, E. et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015).
Commisso, C., Davidson, S. M., Soydaner-Azeloglu, R. G., Parker, S. J., Kamphorst, J. J., Hackett, S. et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013).
Bar-Or, D., Curtis, C. G., Sullivan, A., Rael, L. T., Thomas, G. W., Craun, M. et al. Plasma albumin cysteinylation is regulated by cystathionine beta-synthase. Biochem. Biophys. Res. Commun. 325, 1449–1453 (2004).
Schulman, J. D., Bradley, K. H. & Seegmiller, J. E. Cystine: compartmentalization within lysosomes in cystinotic leukocytes. Science 166, 1152–1154 (1969).
Yamazaki, S., Su, Y., Maruyama, A., Makinoshima, H., Suzuki, J., Tsuboi, M. et al. Uptake of collagen type I via macropinocytosis cause mTOR activation and anti-cancer drug resistance. Biochem. Biophys. Res. Commun. 526, 191–198 (2020).
Wyant, G. A., Abu-Remaileh, M., Wolfson, R. L., Chen, W. W., Freinkman, E., Danai, L. V. et al. mTORC1 Activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–54 e12 (2017).
Pisoni, R. L., Acker, T. L., Lisowski, K. M., Lemons, R. M. & Thoene, J. G. A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: possible role in supporting lysosomal proteolysis. J. Cell Biol. 110, 327–335 (1990).
Sumayao, R., Jr., Newsholme, P. & McMorrow, T. The role of cystinosin in the intermediary thiol metabolism and redox homeostasis in kidney proximal tubular cells. Antioxidants 7, 179 (2018).
Simizu, S., Suzuki, T., Muroi, M., Lai, N. S., Takagi, S., Dohmae, N. et al. Involvement of disulfide bond formation in the activation of heparanase. Cancer Res. 67, 7841–7849 (2007).
Auclair, J. R., Brodkin, H. R., D’Aquino, J. A., Petsko, G. A., Ringe, D. & Agar, J. N. Structural consequences of cysteinylation of Cu/Zn-superoxide dismutase. Biochemistry 52, 6145–6150 (2013).
Auclair, J. R., Johnson, J. L., Liu, Q., Salisbury, J. P., Rotunno, M. S., Petsko, G. A. et al. Post-translational modification by cysteine protects Cu/Zn-superoxide dismutase from oxidative damage. Biochemistry 52, 6137–6144 (2013).
Cortese-Krott, M. M., Koning, A., Kuhnle, G. G. C., Nagy, P., Bianco, C. L., Pasch, A. et al. The reactive species interactome: evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxid. Redox Signal. 27, 684–712 (2017).
Haque, M. A., Hawes, J. W. & Blum, J. S. Cysteinylation of MHC class II ligands: peptide endocytosis and reduction within APC influences T cell recognition. J. Immunol. 166, 4543–4551 (2001).
Finkelstein, J. D. Pathways and regulation of homocysteine metabolism in mammals. Semin. Thromb. Hemost. 26, 219–225 (2000).
Konno, M., Asai, A., Kawamoto, K., Nishida, N., Satoh, T., Doki, Y. et al. The one-carbon metabolism pathway highlights therapeutic targets for gastrointestinal cancer. Int. J. Oncol. 50, 1057–1063 (2017).
Bailey, H. H., Ripple, G., Tutsch, K. D., Arzoomanian, R. Z., Alberti, D., Feierabend, C. et al. Phase I study of continuous-infusion l-S,R-buthionine sulfoximine with intravenous melphalan. J. Natl Cancer Inst. 89, 1789–1796 (1997).
Cao, X., Ding, L., Xie, Z. Z., Yang, Y., Whiteman, M., Moore, P. K. et al. A review of hydrogen sulfide synthesis, metabolism, and measurement: is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid. Redox Signal. 31, 1–38 (2019).
Cramer, S. L., Saha, A., Liu, J., Tadi, S., Tiziani, S., Yan, W. et al. Systemic depletion of l-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med 23, 120–127 (2017).
Kshattry, S., Saha, A., Gries, P., Tiziani, S., Stone, E., Georgiou, G. et al. Enzyme-mediated depletion of l-cyst(e)ine synergizes with thioredoxin reductase inhibition for suppression of pancreatic tumor growth. NPJ Precis. Oncol. 3, 16 (2019).
Nunes, S. C. & Serpa, J. Glutathione in ovarian cancer: a double-edged sword. Int. J. Mol. Sci. 19 1882 (2018).
Zhou, B., Liu, J., Kang, R., Klionsky, D. J., Kroemer, G. & Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 14, 30006–30009 (2019).
Conrad, M. & Pratt, D. A. The chemical basis of ferroptosis. Nat. Chem. Biol. 15, 1137–1147 (2019).
Szczesny, B., Marcatti, M., Zatarain, J. R., Druzhyna, N., Wiktorowicz, J. E., Nagy, P. et al. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 6, 36125 (2016).
Stokes, E., Shuang, T., Zhang, Y., Pei, Y., Fu, M., Guo, B. et al. Efflux inhibition by H2S confers sensitivity to doxorubicin-induced cell death in liver cancer cells. Life Sci. 213, 116–125 (2018).
Chen, S., Yue, T., Huang, Z., Zhu, J., Bu, D., Wang, X. et al. Inhibition of hydrogen sulfide synthesis reverses acquired resistance to 5-FU through miR-215-5p-EREG/TYMS axis in colon cancer cells. Cancer Lett. 466, 49–60 (2019).
Hanaoka, K., Sasakura, K., Suwanai, Y., Toma-Fukai, S., Shimamoto, K., Takano, Y. et al. Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteine persulfide. Sci. Rep. 7, 40227 (2017).
Thorson, M. K., Majtan, T., Kraus, J. P. & Barrios, A. M. Identification of cystathionine beta-synthase inhibitors using a hydrogen sulfide selective probe. Angew. Chem. 52, 4641–4644 (2013).
Druzhyna, N., Szczesny, B., Olah, G., Modis, K., Asimakopoulou, A., Pavlidou, A. et al. Screening of a composite library of clinically used drugs and well-characterized pharmacological compounds for cystathionine beta-synthase inhibition identifies benserazide as a drug potentially suitable for repurposing for the experimental therapy of colon cancer. Pharm. Res. 113, 18–37 (2016).
Chao, C., Zatarain, J. R., Ding, Y., Coletta, C., Mrazek, A. A., Druzhyna, N. et al. Cystathionine-beta-synthase inhibition for colon cancer: enhancement of the efficacy of aminooxyacetic acid via the prodrug approach. Mol. Med. 22, 361–379 (2016).
Asimakopoulou, A., Panopoulos, P., Chasapis, C. T., Coletta, C., Zhou, Z., Cirino, G. et al. Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br. J. Pharmacol. 169, 922–932 (2013).
Dong, Q., Yang, B., Han, J. G., Zhang, M. M., Liu, W., Zhang, X. et al. A novel hydrogen sulfide-releasing donor, HA-ADT, suppresses the growth of human breast cancer cells through inhibiting the PI3K/AKT/mTOR and Ras/Raf/MEK/ERK signaling pathways. Cancer Lett. 455, 60–72 (2019).
Nin, D. S., Idres, S. B., Song, Z. J., Moore, P. K. & Deng, L. W. Biological effects of morpholin-4-Ium 4 methoxyphenyl (morpholino) phosphinodithioate and other phosphorothioate-based hydrogen sulfide donors. Antioxid. Redox Signal. 32, 145–158 (2020).
Xu, S., Pan, J., Cheng, X., Zheng, J., Wang, X., Guan, H. et al. Diallyl trisulfide, a H2 S donor, inhibits cell growth of human papillary thyroid carcinoma KTC-1 cells through a positive feedback loop between H2 S and cystathionine-gamma-lyase. Phytother. Res. 34, 1154–1165 (2020).
Wangpaichitr, M., Wu, C., Li, Y. Y., Nguyen, D. J. M., Kandemir, H., Shah, S. et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 8, 49275–49292 (2017).
Kim, E. H., Shin, D., Lee, J., Jung, A. R. & Roh, J. L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 432, 180–190 (2018).
Su, Y., Zhao, B., Zhou, L., Zhang, Z., Shen, Y., Lv, H. et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 483, 127–136 (2020).
Ogihara, K., Kikuchi, E., Okazaki, S., Hagiwara, M., Takeda, T., Matsumoto, K. et al. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci. 110, 1431–1441 (2019).
Gout, P. W., Buckley, A. R., Simms, C. R. & Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640 (2001).
Ishimoto, T., Nagano, O., Yae, T., Tamada, M., Motohara, T., Oshima, H. et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 19, 387–400 (2011).
Timmerman, L. A., Holton, T., Yuneva, M., Louie, R. J., Padro, M., Daemen, A. et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465 (2013).
Okazaki, S., Umene, K., Yamasaki, J., Suina, K., Otsuki, Y., Yoshikawa, M. et al. Glutaminolysis-related genes determine sensitivity to xCT-targeted therapy in head and neck squamous cell carcinoma. Cancer Sci. 110, 3453–3463 (2019).
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Viswanathan, V. S., Ryan, M. J., Dhruv, H. D., Gill, S., Eichhoff, O. M., Seashore-Ludlow, B. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Ye, J., Jiang, X., Dong, Z., Hu, S., Xiao, M. & Low-Concentration, P. T. X. And RSL3 inhibits tumor cell growth synergistically by inducing ferroptosis in mutant p53 hypopharyngeal squamous carcinoma. Cancer Manag. Res. 11, 9783–9792 (2019).
Rajani, C., Borisa, P., Karanwad, T., Borade, Y., Patel, V., Rajpoot, K. et al. in Pharmaceutical Applications of Dendrimers (eds Chauhan, A. & Kulhari, H.) (Elsevier, 2020).
Mota, P., Pires, R. F., Serpa, J. & Bonifacio, V. D. B. l-Buthionine sulfoximine detection and quantification in polyurea dendrimer nanoformulations. Molecules 24, 3111 (2019).
Santos, I., Ramos, C., Mendes, C., Sequeira, C. O., Tome, C. S., Fernandes, D. G. H. et al. Targeting glutathione and cystathionine beta-synthase in ovarian cancer treatment by selenium-chrysin polyurea dendrimer nanoformulation. Nutrients 11, 2523 (2019).
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–22 e21 (2018).
Author information
Authors and Affiliations
Contributions
V.D.B.B. wrote the first draft, discussed and revised the final version of the manuscript. S.A.P. wrote the first draft, discussed and revised the final version of the manuscript. J.S. wrote the first draft, discussed and revised the final version of the manuscript. J.B.V. wrote the first draft, discussed and revised the final version of the manuscript. Since the authors contributed equally for the final version of the paper, the authors list is presented in alphabetic order according to the authors’ surname.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
All data presented in the review paper is published and referenced.
Competing interests
The authors declare no competing interests.
Funding information
iNOVA4Health—UID/Multi/04462/, a programme financially supported by Fundação para a Ciência e a Tecnologia-Ministério da Educação e Ciência (FCT-MCTES) through national funds.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bonifácio, V.D.B., Pereira, S.A., Serpa, J. et al. Cysteine metabolic circuitries: druggable targets in cancer. Br J Cancer 124, 862–879 (2021). https://doi.org/10.1038/s41416-020-01156-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-01156-1
This article is cited by
-
Epigenetic repression of de novo cysteine synthetases induces intra-cellular accumulation of cysteine in hepatocarcinoma by up-regulating the cystine uptake transporter xCT
Cancer & Metabolism (2024)
-
Disulfidptosis decoded: a journey through cell death mysteries, regulatory networks, disease paradigms and future directions
Biomarker Research (2024)
-
Upregulation of BRCA1 and 2 protein expression is associated with dysregulation in amino acids profiles in breast cancer
Molecular Biology Reports (2024)
-
Dietary cysteine and methionine promote peroxisome elevation and fat loss by induction of CG33474 expression in Drosophila adipose tissue
Cellular and Molecular Life Sciences (2024)
-
Dissecting hair breakage in alopecia areata: the central role of dysregulated cysteine homeostasis
Amino Acids (2024)
