
Abstract
Background
A phase Ib study of binimetinib and capecitabine for gemcitabine-pretreated biliary tract cancer (BTC) patients was conducted.
Methods
Binimetinib and capecitabine were dosed twice daily on days 1–14, in 3-week cycles. In the dose-escalation (DE) part, three dose levels (DL) were tested (DL1: binimetinib/capecitabine, 15 mg/1000 mg/m2; DL2: 30 mg/1000 mg/m2; DL3: 30 mg/1250 mg/m2).
Results
In the DE part, nine patients were recruited and no dose-limiting toxicity was noted. Therefore, the recommended phase 2 dose was determined as DL3. In the expansion part, 25 patients were enrolled. In total, 34 patients, 25 (73.5%) and 9 patients (26.5%) were second-line and third-line settings, respectively. The 3-month progression-free survival (PFS) rate was 64.0%, and the median PFS and overall survival (OS) were 4.1 and 7.8 months. The objective response rate and disease control rate were 20.6% and 76.5%. In total, 68.4% of stable diseases were durable (> 12 weeks). Furthermore, patients with RAS/RAF/MEK/ERK pathway mutations (38.5%) showed significantly better tumour response (p = 0.028), PFS (5.4 vs. 3.5 months, p = 0.010) and OS (10.8 vs. 5.9 months, p = 0.160) than wild type. Most of the adverse events were grade 1/2 and manageable.
Conclusions
A combination of binimetinib and capecitabine shows acceptable tolerability and promising antitumor efficacy for gemcitabine-pretreated BTC, especially in patients with RAS/RAF/MEK/ERK pathway mutations.
Clinical trial registration
ClinicalTrials.gov (Identifier: NCT02773459).
Similar content being viewed by others
Background
Biliary tract cancer (BTC) arises from bile duct epithelial cells, and is a heterogeneous family of malignant diseases that includes intrahepatic cholangiocarcinoma, extrahepatic cholangiocarcinoma, gallbladder cancer and ampulla of Vater cancer. The incidence of BTC is higher in Korea, China and Thailand than the rest of the world.1 Currently, gemcitabine-based chemotherapy is globally considered the first-line treatment for recurrent or metastatic BTC.2 However, even with chemotherapy, the median overall survival (OS) of recurrent or metastatic BTC is ~1 year.2 There is no solid evidence for the role of second-line chemotherapy, although fluoropyrimidine-based chemotherapy is widely used in practice.3 ABC-06 trial has been evaluating the clinical efficacy of second-line mFOLFOX with active symptom control compared with active symptom control alone in BTC. Furthermore, no targeted therapy has been approved for BTC, although many interesting genetic alterations in BTC have been identified.4 There is, therefore, a significant unmet need for new and effective BTC treatment strategies.
In BTC, the RAS/RAF/MEK/ERK pathway is known to be activated in up to 20–40% of cases, which is mediated by mutations in KRAS, NRAS, BRAF and so on.4,5 The RAS/RAF/MEK/ERK pathway is a series of proteins that mediates communication between the cell surface and nucleus, which has been linked to several vital cellular functions, such as proliferation, apoptosis and metabolism.6,7 Therefore, this pathway could have a potential to be a good therapeutic target in BTC. Furthermore, mutations in RAS or BRAF have been suggested as predictive biomarkers for MEK inhibition.8,9 In previous clinical trials, selumetinib, an inhibitor of MEK1/2, demonstrated interesting activity and acceptable tolerability in patients with metastatic BTC.10,11 Similarly, binimetinib is a potent, adenosine triphosphate-uncompetitive, highly selective allosteric inhibitor of MEK1/2 with on-target activity that has been demonstrated both in vitro and in vivo.12 Moreover, phase I study of binimetinib showed a manageable safety profile, target inhibition and dose-proportional exposure.13
In this study, we evaluated the preclinical synergistic activity of binimetinib and fluoropyrimidine against BTC cell lines. Supported by the preclinical results, we then conducted a phase Ib study of binimetinib and capecitabine in gemcitabine-pretreated BTC patients to assess the safety and early antitumor activity. Furthermore, we identified genetic alterations to the RAS/RAF/MEK/ERK pathway and determined plasma biomarker concentrations.
Methods
Preclinical study
The effect of binimetinib, 5-fluorouracil (5-FU) or a combination of these drugs on cell viability was evaluated using eight BTC cell lines. In brief, confluent monolayers, which had been grown in 96-well plates, were exposed to drugs for 72 h. Subsequently, cell viability was measured at 540 nm with a Multiskan GO microplate reader, using a commercially available MTT assay, according to the manufacturer’s directions. The Chou–Talalay method was used to assess a combination effect.14,15 Cells were also treated with drugs for 48 h, and the expression of thymidylate synthase (TS), programmed death-ligand 1 (PD-L1) and β-actin was determined using western blot analysis according to a published protocol.15 The experimental material sources are provided in Supplementary Table S1.
Clinical study design
The clinical study was a phase Ib of dose-escalation and expansion part. The dose-escalation part was conducted as a standard 3 + 3 design to determine the maximum tolerated dose (MTD). The recommended phase 2 dose (RP2D) based on the results of the dose-escalation part was used in the expansion part. The primary endpoint of the dose-escalation part was determination of the MTD, and the secondary endpoints included identification of dose-limiting toxicity (DLT), the RP2D and safety. In the expansion part, the primary endpoint was determination of the 3-month progression-free survival (PFS) rate, and the secondary endpoints were the objective response rate (ORR), response duration, disease control rate (DCR), PFS, OS, safety, quality of life (QOL) and biomarker quantitation.
Patients
The target population of this study was BTC patients in their second- or third-line treatment setting, who had failed a gemcitabine-based first-line chemotherapy. The major inclusion criteria were age ≥ 20 years; histologically confirmed BTC; unresectable or recurrent disease; prior gemcitabine-based chemotherapy; Eastern Cooperative Oncology Group (ECOG) performance status 0–1; evaluable or measurable lesions by Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1); adequate bone marrow and organ function; corrected QT interval ≤ 480 ms. The major exclusion criteria were active central nervous system disease; brain metastasis; risk or history of retinal vein occlusion; transplantation history; Gilbert syndrome; major heart disease within 6 months; neuromuscular disease related with elevation of creatine kinase. Biliary drainage was allowed.
Dosing and dose modification
Binimetinib and capecitabine were orally administered twice daily, on days 1–14, in 3-week cycles. In the dose-escalation part, four predefined dose levels (DLs) were applied (DL-1: binimetinib/capecitabine, 15 mg/800 mg/m2; DL1: 15 mg/1000 mg/m2; DL2: 30 mg/1000 mg/m2; DL3: 30 mg/1250 mg/m2). The starting dose was DL1. If no patient experienced DLT in DL3, then DL3 was to be declared as the RP2D. DLT was predefined as grade 4 neutropenia with fever and/or infection; grade 4 neutropenia for ≥ 7 days; grade 3/4 thrombocytopenia with haemorrhage or transfusion; grade 4 thrombocytopenia for ≥ 7 days; grade 3 or 4 non-haematologic adverse events, except alopecia, anorexia, nausea or vomiting; grade 3 or 4 nausea, diarrhoea or vomiting, despite maximum supportive care. Dose reductions of binimetinib to 15 mg twice daily and capecitabine to 75 or 50% of the dose were permitted based on the protocol-defined treatment modifications.
Assessment of response, adverse events and QOL
Radiologic assessment was completed by computed tomography (CT) every 6 weeks, and tumour response was evaluated based on RECIST v1.1. Routine evaluation, including physical examinations and vital signs, and assessment of adverse events was completed weekly during the first cycle, and then at the end of every cycle. Adverse events were recorded using the NCI—Common Terminology Criteria of Adverse Events version 4.03. QOL was evaluated through the use of the EORTC-QLQ-C30 and EQ5D questionnaires,16,17 which were collected at baseline, after cycles 1, 2, 4, 6 and 8, and then after every third cycle until the end of the study.
Biomarker analysis
All patients were required to provide tumour tissues at screening, and blood samples at screening, after the first cycle, after the second cycle and at disease progression. Genetic alteration was assessed using targeted sequencing by next-generation sequencing (NGS) to determine mutations of the RAS/RAF/MEK/ERK pathway. To predict treatment efficacy, interleukin-6 (IL-6) plasma concentrations were evaluated. The plasma concentrations of IL-6 were measured using an enzyme-linked immunosorbent assay (Human IL-6, Quantikine ELISA Kit, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Each sample was analysed in duplicate.
Statistical analysis
The safety and efficacy analysis were completed in an intent-to-treat population who received at least one dose of binimetinib. Cut-point values of IL-6 at baseline and changes of IL-6 between baseline and the second cycle for OS and PFS predictions were determined by finding the optimal cut point for continuous covariates with time-to-event outcomes.18 The results of the EORTC-QLQ-C30 were interpreted in line with the method of Osoba et al.19 The PFS was calculated from the date of the first cycle to the development of progressive disease (PD) or death, regardless of the cause. The OS was calculated from the date of the first cycle to death, regardless of the cause. All analyses were performed using PASW Statistics 18 (SPSS Inc., Chicago, IL) and R Statistical Software (R version 3.4.4).
Results
Preclinical study
Exposure of the BTC cell lines to binimetinib was associated with significant decreases in cell viability (Fig. 1a). The combination of binimetinib and 5-FU in SNU245, SNU1196, SNU869 and HuCCT1 demonstrated synergistic effects (combination index < 1 at the fraction affected = 0.5; Fig. 1b). Of these cell lines, SNU869 and HuCCT1 have the KRAS mutation (p.G12D).20 To evaluate the underlying synergistic mechanism, protein expression was evaluated by western blot. Accordingly, TS, a marker of 5-FU resistance, was upregulated by 5-FU treatment. However, TS was downregulated by binimetinib monotherapy (Fig. 1c). When the BTC cells were treated with both binimetinib and 5-FU, the TS levels induced by 5-FU were partially downregulated by addition of binimetinib, which may increase the sensitivity of 5-FU. Interestingly, 5-FU induced PD-L1 expression, and co-administration with binimetinib and 5-FU decreased this expression (Fig. 1c).

The preclinical efficacy of binimetinib and 5-fluorouracil. a Binimetinib demonstrates antitumour activity in biliary tract cancer cell lines. b Synergistic properties of the combination of binimetinib and 5-FU are identified in SNU245, SNU1196, SNU869 and HuCCT1 (CI < 1 at Fa = 0.5). c TS, a 5-FU resistance marker, is upregulated by 5-FU treatment. TS levels induced by 5-FU are downregulated by the addition of binimetinib. Similarly, 5-FU-induced expression of PD-L1 is abrogated by the addition of binimetinib. Actin was included as a loading control. The data are representative of three independent experiments. CI combination index, Fa fraction affected, 5-FU 5-fluorouracil, TS thymidylate synthase, PD-L1 programmed death-ligand 1
Patients
Nine patients were recruited for the dose-escalation part. None of the patients experienced DLT up to DL3; therefore, the RP2D was determined as DL3 (binimetinib 30 mg, capecitabine 1250 mg/m2, twice daily on days 1–14, in 3-week cycles). For the expansion part, 25 patients were enrolled (Supplementary Fig. S1). At the data cut-off time, 22 patients had died, and treatment was ongoing in three patients. The median follow-up duration was 6.8 months (range, 2.0–17.8) and patients received a median of five cycles (range, 1–16).
The median age of enrolled patients was 63 years old (range, 48–73) (Table 1). The primary tumour origins were the gallbladder (n = 10, 29.4%), intrahepatic bile duct (n = 10, 29.4%), extrahepatic bile duct (n = 9, 26.5%) and ampulla of Vater (n = 5, 14.7%). Twenty-five (73.5%) patients were in their second-line treatment setting, and nine patients (26.5%) were in their third-line setting. Twelve patients (35.3%) had previously been exposed to fluoropyrimidine as an adjuvant or during a palliative chemotherapy period, and among them, four patients (33.3%) had experienced fluoropyrimidine failure as first- or second-line treatment.
Adverse events
The majority of the adverse events were both manageable and reversible. Of note, during the first cycle in the dose-escalation part, one G3 neutropenia and one G3 thrombocytopenia occurred, but there was no DLT (Supplementary Table S2). The most common adverse events in the whole population (n = 34, dose-escalation and expansion part) were stomatitis (61.7%), oedema (50.0%), nausea (41.2%), papulopustular rash (41.2%), palmar–plantar erythrodysesthesia syndrome (41.2%) and fatigue (41.2%) (Table 2; Supplementary Table S3). There were no ocular adverse events and only one patient experienced grade 4 toxicity (hypokalaemia). There were no treatment-related deaths.
Treatment efficacy
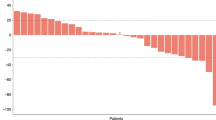
All patients had at least one measurable lesion. Of 34 patients, 7 patients (20.6%) and 19 patients (55.9%) demonstrated partial response (PR) and stable disease (SD), respectively (Table 3). The ORR and DCR were 20.6% (95% confidence interval (CI), 7.0–34.2) and 76.5% (95% CI, 62.2–90.8), respectively. Twenty-five patients (73.5%) experienced tumour shrinkage with any grade (Fig. 2a), and the median response duration was 4.7 months (95% CI, 2.4–7.0). Of the 19 patients with SD, 13 (68.4%) demonstrated durable disease control with SD duration for >12 weeks. The tumour response was similar between second- and third-line settings (Table 3). Tumour origin also did not alter tumour response. Furthermore, of the four patients who had failed fluoropyrimidine at the first- or second-line setting, one patient (25%) showed PR and the other three patients (75%) showed SD.

The efficacy of the combination of binimetinib and capecitabine. a Waterfall plot of tumour shrinkage, b progression-free survival and overall survival are present. Patients with mutant type on RAS/RAF/MEK/ERK exhibit better results in c waterfall plot, d progression-free survival, e swimmer plot of treatment duration and f overall survival compared with those with wild type. PD progressive disease, SD stable disease, PR partial response, PFS progression-free survival, OS overall survival, CI confidence interval, NR not reached
In all patients, the 3-month PFS rate was 64.0% and the median PFS was 4.1 months (95% CI, 2.8–5.7; Fig. 2b). The median OS was 7.8 months (95% CI, 5.9–12.2). Neither the PFS nor OS were significantly different between second- and third-line settings (p = 0.064, p = 0.796, respectively). There was also no significant difference in the PFS or OS when patients were grouped according to tumour origin (p = 0.158, p = 0.091, respectively).
Biomarker analysis
In all patients, tumour tissues were obtained during screening. However, genomic sequence information using NGS techniques was obtained for 26 of the 34 participants (76.5%). Genetic alterations in the RAS/RAF/MEK/ERK pathway were identified in 10 (38.5%) of 26 patients (Supplementary Table S4). Seven of these mutations were identified in KRAS, one in NRAS and two in MEK. Furthermore, patients with mutations in the RAS/RAF/MEK/ERK pathway responded significantly better to therapy than those with wild type (40.0% vs. 12.5%; Fig. 2c; Supplementary Table S5). Patients with mutant type also showed longer PFS (5.4 vs. 3.5 months; Fig. 2d, e) and OS (10.8 vs. 5.9 months; Fig. 2f) than those with wild type.
In terms of IL-6 plasma concentrations, the mean value (± standard deviation) of baseline plasma IL-6 was 11.5 pg/ml (± 12.6). Patients with higher baseline IL-6 showed significantly shorter PFS and OS (p = 0.025, p = 0.033, respectively; Fig. 3a, b). Similarly, the baseline value of IL-6 was associated with tumour response, that is, IL-6 was higher in PD than PR patients. The mean concentrations were 19.9, 9.2 and 7.7 pg/ml for PD, SD and PR, respectively (p = 0.085). With regard to changes between baseline and after the second cycle, a greater increase in the IL-6 concentration (Δ > 14.8 pg/ml) was associated with shorter PFS and OS (Fig. 3c, d). Furthermore, the plasma concentrations of IL-6 when PD was confirmed were also significantly increased relative to baseline (mean ± standard deviation: 32.0 ± 29.8 vs. 9.0 ± 5.9 pg/ml, respectively; paired t test, p = 0.008).

Progression-free survival and overall survival according to IL-6 concentration. a, b The baseline plasma concentrations of IL-6 are significantly associated with survival. c, d Alterations in the IL-6 concentration from baseline to after the second cycle are associated with survival. IL-6 interleukin-6, CI confidence interval, NR not reached
QOL
Based on the EORTC-QLQ-C30 questionnaire, most QOLs regarding global health status and functioning were altered between the degrees of ‘a little’ to ‘very much’ as the cycles proceeded (Supplementary Table S6). QOLs related to symptoms demonstrably improved at some time points. Compared with best status to baseline, role functioning and financial difficulties improved with ‘a little’ degree (p = 0.028 and p = 0.032, respectively), and QOL related to pain improved with ‘moderate’ degree (p = 0.039). The EQ5D questionnaire also demonstrated alterations of the QOL throughout the treatment period (Supplementary Table S7).
Discussion
This study is the first proof-of-concept trial to evaluate the safety and efficacy of binimetinib in combination with capecitabine for gemcitabine-pretreated BTC, and was supported by preclinical data suggestive of the synergism between binimetinib with fluoropyrimidine. This combination demonstrated promising antitumour efficacy, especially in BTC patients with mutations in the RAS/RAF/MEK/ERK pathway.
A previous phase Ib study of binimetinib monotherapy in advanced or metastatic BTC patients reported an ORR of 8% and a DCR of 51%, as well as PFS of 2.1 months and OS of 4.8 months, respectively.21 Similarly, in a phase II study of selumetinib for metastatic BTC, the ORR, DCR, PFS and OS were 12%, 80%, 3.7 months and 9.8 months, respectively.10 In contrast, the ORR, DCR, PFS and OS were 20.6%, 76.5%, 4.1 months and 7.8 months, in this study. Considering that our study consisted of patients in their second- (73.5%) and third-line settings (26.5%), the efficacy of this study may be better than the aforementioned studies, in which more than half of the patient populations were chemotherapy-naive in a metastatic setting (57 and 75%, respectively). Furthermore, a systematic review, of patients in the second- and third-line setting for BTC, described ORR and DCR of <10% and <50%; the PFS and OS were around 3 months and 6 months.3 Moreover, our previous study, which included both second- and third-line setting BTC patients treated with a 5-FU-based combination treatment (infusional FAM regimen), had a similar population to this study, and reported PFS and OS values of 2.4 months and 6.1 months, respectively.22 Considering these results, the efficacy of binimetinib and capecitabine in this study is very encouraging and is likely attributed to the synergism between binimetinib and capecitabine. Indeed, preclinical experiments demonstrated the downregulation of TS and PD-L1 induced by 5-FU in response to binimetinib. Synergistic properties between a MEK inhibition and fluoropyrimidine have also been reported for other cancer types.23,24,25 Interestingly, in this study, all patients who had previously failed fluoropyrimidine-based chemotherapy achieved either a PR or SD with this combination, which is likely attributed to synergism.
Of note, activation of the MEK pathway, such as through RAS or BRAF mutations, has been reported as a predictive marker for the success of MEK inhibitors.8,9 In this study, genetic mutations within the RAS/RAF/MEK/ERK pathway were identified in 10 (38.5%) out of 26 patients, whose NGS data were obtained. This incidence was in accordance with previous studies.4,5 Importantly, this study demonstrates that a tissue-based biomarker selection strategy for BTC patient management or enrolling BTC patients into clinical trials is doable and feasible. Interestingly, patients with mutations in the RAS/RAF/MEK/ERK pathway were associated with a higher ORR and longer survival than those with wild type. Considering that our patients were in the second- or third-line setting of BTC, these results of ORR (40%), PFS (5.4 months) and OS (10.8 months) were very promising in patients with mutant type of RAS/RAF/MEK/ERK pathways. Therefore, mutations in the RAS/RAF/MEK/ERK pathway could be a predictive biomarker for effective binimetinib treatment of BTC.
Immune modulation is another antitumour mechanism of MEK inhibitors,26 as MEK inhibition was reported to reduce the secretion of IL-6, which is associated with BTC tumour growth.26,27,28,29 In this study, higher baseline concentrations of IL-6 were associated with worse prognosis, which is similar to the results from other studies regarding the prognostic value of IL-6.30,31 Furthermore, after the second cycle of treatment, patients with a larger increase were associated with worse prognosis. Therefore, early comparative determination of IL-6 between baseline and after treatment may predict disease outcomes in the binimetinib treatment.
In the previous phase I study of binimetinib monotherapy, 60 mg twice daily was identified as the MTD.13,21 However, due to the frequent treatment-related ocular toxicity at this MTD, 45 mg twice daily was used as the RP2D.13,32 In this study, predefined four DLs were tested, and no DLT was observed. The highest DL (binimetinib 30 mg, capecitabine 1250 mg/m2, twice daily on days 1–14, in 3-week cycles) was determined as the RP2D. This dosage of binimetinib was relatively low in comparison with the monotherapy study, and we introduced a 1-week drug holiday in 3-week cycles. This combination dosing schedule, which allowed full-dose capecitabine, was tolerable and adverse events were manageable. There was also no ocular toxicity in our study. Furthermore, this combination resulted in promising efficacy outcomes.
In conclusion, the preclinical synergistic activity of binimetinib with 5-FU against BTC was demonstrated to support the clinical development of this combination. We next completed a clinical phase Ib trial, assessing the combination of binimetinib and capecitabine in patients with gemcitabine-pretreated advanced BTC. The RP2D was determined during the dose-escalation part of the trial and was identified as 30 mg binimetinib with 1250 mg/m2 capecitabine, twice daily on days 1–14 in 3-week cycles. The drug combination was well tolerated, associated with manageable adverse events, and demonstrated promising antitumour efficacy, especially in patients with RAS/RAF/MEK/ERK pathway mutations. These findings support future clinical development of MEK inhibition strategies for BTC management.
References
Banales, J. M., Cardinale, V., Carpino, G., Marzioni, M., Andersen, J. B., Invernizzi, P. et al. Expert consensus document: cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 13, 261–280 (2016).
Valle, J., Wasan, H., Palmer, D. H., Cunningham, D., Anthoney, A., Maraveyas, A. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281 (2010).
Lamarca, A., Hubner, R. A., David Ryder, W. & Valle, J. W. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann. Oncol. 25, 2328–2338 (2014).
Nakamura, H., Arai, Y., Totoki, Y., Shirota, T., Elzawahry, A., Kato, M. et al. Genomic spectra of biliary tract cancer. Nat. Genet. 47, 1003–1010 (2015).
Tannapfel, A., Sommerer, F., Benicke, M., Katalinic, A., Uhlmann, D., Witzigmann, H. et al. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut. 52, 706–712 (2003).
O’Neill, E. & Kolch, W. Conferring specificity on the ubiquitous Raf/MEK signalling pathway. Br. J. Cancer 90, 283–288 (2004).
Khokhlatchev, A. V., Canagarajah, B., Wilsbacher, J., Robinson, M., Atkinson, M., Goldsmith, E. et al. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93, 605–615 (1998).
Horiuchi, H., Kawamata, H., Fujimori, T. & Kuroda, Y. A. MEK inhibitor (U0126) prolongs survival in nude mice bearing human gallbladder cancer cells with K-ras mutation: analysis in a novel orthotopic inoculation model. Int. J. Oncol. 23, 957–963 (2003).
Solit, D. B., Garraway, L. A., Pratilas, C. A., Sawai, A., Getz, G., Basso, A. et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 439, 358–362 (2006).
Bekaii-Saab, T., Phelps, M. A., Li, X., Saji, M., Goff, L., Kauh, J. S. et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J. Clin. Oncol. 29, 2357–2363 (2011).
Adjei, A. A., Cohen, R. B., Franklin, W., Morris, C., Wilson, D., Molina, J. R. et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J. Clin. Oncol. 26, 2139–2146 (2008).
Winski, S., Anderson, D., Bouhana, K., Impastato, R., Woessner, R., Zuzack, J. et al. MEK162 (ARRY-162), a novel MEK 1/2 inhibitor, inhibits tumor growth regardless of KRas/Raf pathway mutations. EJC Suppl. 8, 56 (2010).
Bendell, J. C., Javle, M., Bekaii-Saab, T. S., Finn, R. S., Wainberg, Z. A., Laheru, D. A. et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br. J. Cancer 116, 575–583 (2017).
Chou, T. C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 (2010).
Kim, J. W., Kim, H. P., Im, S. A., Kang, S., Hur, H. S., Yoon, Y. K. et al. The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines. Cancer Lett. 272, 296–306 (2008).
Aaronson, N. K., Ahmedzai, S., Bergman, B., Bullinger, M., Cull, A., Duez, N. J. et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J. Natl Cancer Inst. 85, 365–376 (1993).
EuroQol, G. EuroQol–a new facility for the measurement of health-related quality of life. Health Policy 16, 199–208 (1990).
Williams, B., Mandrekar, J. N., Mandrekar, S. J., Cha, S. S., Furth, A. F. Finding optimal cutpoints for continuous covariates with binary and time-to-event outcomes. Tech. Rep. Ser. 79, 2006.
Osoba, D., Rodrigues, G., Myles, J., Zee, B. & Pater, J. Interpreting the significance of changes in health-related quality-of-life scores. J. Clin. Oncol. 16, 139–144 (1998).
Jin, L., Jin, M. H., Nam, A. R., Park, J. E., Bang, J. H., Oh, D. Y. et al. Anti-tumor effects of NVP-BKM120 alone or in combination with MEK162 in biliary tract cancer. Cancer Lett. 411, 162–170 (2017).
Finn, R. S., Ahn, D. H., Javle, M. M., Tan, B. R. Jr., Weekes, C. D., Bendell, J. C. et al. Phase 1b investigation of the MEK inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Invest. New Drugs 36, 1037–1043 (2018).
Lim, K. H., Kim, T. Y., Lee, K. H., Han, S. W., Oh, D. Y., Im, S. A. et al. Efficacy of infusional 5-fluorouracil, doxorubicin, and mitomycin-C (iFAM) in the treatment of patients with gemcitabine-pretreated pancreatic cancer and analysis of prognostic factors in a salvage setting. Cancer Chemother. Pharmacol. 68, 1017–1026 (2011).
Gong, J., Chen, Y., Yang, L. X., Pillai, R., Shirasawa, S. & Fakih, M. MEK162 enhances antitumor activity of 5-fluorouracil and trifluridine in KRAS-mutated human colorectal cancer cell lines. Anticancer Res. 37, 2831–2838 (2017).
Urick, M. E., Chung, E. J., Shield, W. P. 3rd, Gerber, N., White, A., Sowers, A. et al. Enhancement of 5-fluorouracil-induced in vitro and in vivo radiosensitization with MEK inhibition. Clin. Cancer Res. 17, 5038–5047 (2011).
Pereira, D. M., Simoes, A. E., Gomes, S. E., Castro, R. E., Carvalho, T., Rodrigues, C. M. et al. MEK5/ERK5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget. 7, 34322–34340 (2016).
Tai, Y. T., Fulciniti, M., Hideshima, T., Song, W., Leiba, M., Li, X. F. et al. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood 110, 1656–1663 (2007).
Park, J., Tadlock, L., Gores, G. J. & Patel, T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology 30, 1128–1133 (1999).
Meng, F., Yamagiwa, Y., Ueno, Y. & Patel, T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J. Hepatol. 44, 1055–1065 (2006).
Wehbe, H., Henson, R., Meng, F., Mize-Berge, J. & Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 66, 10517–10524 (2006).
Gastl, G. & Plante, M. Bioactive interleukin-6 levels in serum and ascites as a prognostic factor in patients with epithelial ovarian cancer. Methods Mol. Med. 39, 121–123 (2001).
Bachelot, T., Ray-Coquard, I., Menetrier-Caux, C., Rastkha, M., Duc, A. & Blay, J. Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 88, 1721–1726 (2003).
Watanabe, K., Otsu, S., Hirashima, Y., Morinaga, R., Nishikawa, K., Hisamatsu, Y. et al. A phase I study of binimetinib (MEK162) in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 77, 1157–1164 (2016).
Acknowledgements
This study was partly presented at the 2018 American Society of Clinical Oncology (ASCO) annual meeting, Chicago, IL, USA.
Author information
Authors and Affiliations
Contributions
Conception and design: D.Y.O. Provision of study materials or patients: J.W.K., K.H.L., J.W.K., K.J.S., Y.J.B. and D.Y.O. Collection and assembly of data: J.W.K., A.R.N. and J.H.B. Data analysis and interpretation: J.W.K., A.R.N. and D.Y.O. Paper writing: J.W.K. and D.Y.O. All authors have approved the final paper. All authors are accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All participating patients provided a written informed consent. This study was approved by the institutional review boards of each hospital (Seoul National University Hospital: H-1407-152-597, Seoul National University Bundang Hospital: B-1610/365-405). The study was performed in accordance with the Declaration of Helsinki.
Funding
This study was partly funded by Array BioPharma, and by grant No. 03-2017-0100 from the Seoul National University Hospital Research Fund. Capecitabine was kindly provided by Ildong Pharmaceutical Co., Seoul, Korea.
Consent to publish
Not applicable.
Data availability
All data and materials are available by inquiring to the corresponding author.
Note
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, J.W., Lee, KH., Kim, JW. et al. Enhanced antitumor effect of binimetinib in combination with capecitabine for biliary tract cancer patients with mutations in the RAS/RAF/MEK/ERK pathway: phase Ib study. Br J Cancer 121, 332–339 (2019). https://doi.org/10.1038/s41416-019-0523-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0523-5
This article is cited by
-
Mucocutaneous toxicities from MEK inhibitors: a scoping review of the literature
Supportive Care in Cancer (2024)
