
Abstract
Background
Human epidermal growth factor 2 (HER2) is an effective therapeutic target in breast cancer; however, resistance to anti-HER2 agents such as trastuzumab and lapatinib develops. In a preclinical model, an HDAC inhibitor epigenetically reversed the resistance of cancer cells to trastuzumab and showed synergistic efficacy with lapatinib in inhibiting growth of trastuzumab-resistant HER2-positive (HER2+) breast cancer.
Methods
A phase 1b, dose escalation study was performed to assess maximum tolerated dose, safety/toxicity, clinical efficacy and explored pharmacodynamic biomarkers of response to entinostat combined with lapatinib with or without trastuzumab.
Results
The combination was safe. The MTD was lapatinib, 1000 mg daily; entinostat, 12 mg every other week; trastuzumab, 8 mg/kg followed by 6 mg/kg every 3 weeks. Adverse events included diarrhoea (89%), neutropenia (31%), and thrombocytopenia (23%). Neutropenia, thrombocytopenia and hypokalaemia were noted. Pharmacodynamic assessment did not yield conclusive results. Among 35 patients with evaluable response, PR was observed in 3 patients and CR in 3 patients, 1 maintained SD for over 6 months.
Discussion
This study identified the MTD of the entinostat, lapatinib, and trastuzumab combination that provided acceptable tolerability and anti-tumour activity in heavily pre-treated patients with HER2+ metastatic breast cancer, supporting a confirmatory trial.
Similar content being viewed by others

Background
Human epidermal growth factor receptor 2 (HER2) is overexpressed in 20–25% of all breast cancers,1 thus accounting for near 60,000 patients each year in the U.S.2 Use of trastuzumab to suppress HER2 activity in HER2-positive (HER2+) breast cancer opened up a new era of therapeutics in HER2+ breast cancers using monoclonal antibodies.3 However, almost all patients with HER2+ metastatic breast cancer eventually develop resistance to trastuzumab after first-line treatment; therefore, novel therapy is needed. Lapatinib co-targets both epidermal growth factor receptor (EGFR; also called HER1) and HER2, via competitive binding to the intracellular ATP binding domain of the HER2. Lapatinib (Tykerb; Novartis Pharmaceuticals Co., East Hanover, New Jersey) has shown clinical benefit in combination with capecitabine or trastuzumab, with activity noted in patients with brain metastases and multiple prior lines of treatment.4,5,6,7 Notably, Overall survival (OS) benefit of lapatinib combined with trastuzumab has been shown in heavily pre-treated patients with HER2+ metastatic breast cancer.8 Formerly, these lapatinib-based therapy combinations were implemented in the second-line setting after progression on trastuzumab plus chemotherapy.9,10
Histone deacetylase (HDAC) inhibitors, e.g., entinostat (SNDX-275), have been recognised as cell “switches” that can reverse the therapeutic response of cancer cells from insensitive to sensitive when combined with cancer therapeutics.11 Our group studied this activity of entinostat in a preclinical trastuzumab-resistant HER2+ breast cancer model, in combination with lapatinib.12 The drug combination had anti-tumour efficacy against HER2+ cell lines that were resistant to single-agent trastuzumab or lapatinib. In another preclinical study, entinostat enhanced the efficacy of trastuzumab in HER2-overexpressing breast cancer cells and exhibited potential to overcome trastuzumab resistance.13
With our preclinical study showing synergy between entinostat and anti-HER2 therapeutics as a scientific rationale, we conducted a phase Ib study investigating the novel combinations of entinostat and lapatinib plus/minus trastuzumab in patients with metastatic HER2+ breast cancer after progression during treatment with a trastuzumab combination regimen. The primary objective was to determine the maximum tolerated dose (MTD) of entinostat and lapatinib, when combined with a fixed dose of trastuzumab. The secondary objectives were determining the safety and tolerability of the combination regimen and its clinical efficacy, including the rates of complete response (CR), partial response (PR), and stable disease (SD) at 6 months from enrolment in the study. As exploratory endpoints, pharmacodynamic markers, including HER2 receptor family members EGFR, HER2 and their downstream molecule AKT, were analysed in patients’ tissue and blood samples using gene/protein detection assays.
Methods
The trial was a single-centre, open-label, single-arm study (ClinicalTrials.gov identifier NCT01434303; NCI identifier: #8871) approved by the Investigational Drug Branch of the Cancer Therapy Evaluation Program, National Cancer Institute. The study was conducted in accordance with the Declaration of Helsinki and was approved by institutional review boards at The University of Texas MD Anderson Cancer Center. All enroled patients provided written informed consent.
Patient eligibility
Both male and female adult patients with metastatic HER2+ breast cancer were eligible. HER2 overexpression was defined either by immunohistochemical staining intensity (3+) or fluorescence in situ hybridisation (FISH) testing (HER2/CEP17 ratio >2.0).14 An ECOG performance status of 0–1, normal organ function, and prior exposure to trastuzumab were required, but it was not mandated that trastuzumab to be the immediately preceding regimen. If the patient had had progression to metastatic disease within 6 months of a previous trastuzumab regimen given during treatment of local disease (e.g., adjuvant therapy), the patient was eligible. Patients had to be able to swallow and retain oral medication. Measurable disease by Response Evaluation Criteria in Solid Tumours (RECIST) v 1.1,15 left ventricular ejection fraction equal to or greater than 50% were required. Lactating or pregnant patients were excluded. Information about the patients’ demographic characteristics, previous lines of therapy, biomarker measurements (including oestrogen receptor and progesterone receptor) from archived tissue obtained at diagnosis or recurrence, and HER2 measurements (immunohistochemistry and FISH assay) were collected for this analysis.
Study design
Patients were enroled in the study between [January 2012] and [November 2015]. Initially patients only received entinostat and lapatinib without trastuzumab, since the study was designed prior to the confirmed safety and efficacy of dual anti-HER2 therapy with lapatinib and trastuzumab, which were published in 2012.8 For the initial dose-escalation phase, patients received various doses of oral entinostat (5–15 mg every other week) and oral lapatinib (1000 or 1250 mg daily) with 28 days as one cycle and, once trastuzumab was added, the standard dose of trastuzumab: an 8 mg/m2 initial loading dose with subsequent doses of 6 mg/m2 every 3 weeks via intravenous infusion. Following the schedule of oral regimens, 28 days were considered as one cycle. A 3 + 3 design, in which each new dose level was tested in 3 consecutive patients, was used to determine the MTD (dose level summarised in Table 2). After the MTD was determined, an additional 10 evaluable patients were enroled as an expansion cohort to confirm the treatment safety and tolerability. No diarrhoeal prophylaxis was introduced.
Dose modification and toxicity assessment
Adverse events (AEs) and laboratory results were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v 4.03.16 Dose-limiting toxicity (DLT) was defined as one of the following AEs with an attribution of possibly, probably, or definitely related to the study agents and occurring within 28 days after the first dose: grade 4 neutropenia lasting >7 days or any febrile neutropenia; grade 4 thrombocytopenia; non-haematologic toxicity ≥grade 3; or >14 days of treatment delay due to any therapy-related toxicity of any grade. Nausea/vomiting, diarrhoea, and electrolyte imbalances were considered DLT if they persisted for 48 h despite adequate supportive care. Toxicity was evaluated on days 15 and 28 for first 2 cycles, and at the end of each cycle thereafter.
Efficacy evaluation
Tumour assessments were conducted based on RECIST v1.1.15 Clinical efficacy assessment measured the patient’s best response: complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD) after the first 2 cycles and every 2 cycles subsequently unless there was a clear progression on skin in patients with inflammatory breast cancer (IBC). The clinical benefit rate was defined as the percentage of patients combined who had SD lasting at least 6 months, PR, or CR. For survival analysis, OS and PFS were measured from the day the patients started trial drugs to the times the patients died or had disease progression, respectively. OS was assessed based on death reports and last available follow-up in the clinic as of April 6, 2017 when the final analysis was performed. For patients who had obvious clinical progression prior to the first scan, the date of clinical progression was annotated as the date of progression.
Pharmacodynamic markers
For exploratory biomarker analysis, archived tumour samples obtained from biopsies and prospectively collected blood samples were analysed using at Apocell, Inc. (Houston, TX). Tissue samples were analysed for protein expression of EGFR, HER2, and AKT and their phosphorylated forms, and for gene levels of EGFR and HER2. The expression of each gene was measured by FISH. Circulating tumour cells (CTCs) from peripheral blood were collected at baseline and after cycle 1. The Wilcoxon signed-rank test was used to examine the change in target molecule expression measures from baseline to after cycle 1. While blood-based markers including CTC were collected before and after the therapeutic intervention, tissues were collected retrospectively, thus mostly baseline biopsy of surgical samples were utilised for PD tissue biomarker analysis.
Statistical analysis
Data were summarised using standard descriptive statistics such as mean, standard deviation, median, and range for continuous variables and frequency and proportion for categorical variables. Association between categorical variables was examined by the chi-square test or Fisher exact test when appropriate. The Wilcoxon rank-sum test was used to examine differences in continuous variables between patient characteristic groups. OS time and PFS time were estimated using the Kaplan-Meier method, and the comparison between or among patient characteristic groups was evaluated by the log-rank test. Univariate and multivariate Cox regression models were applied to assess the effect of covariates of interest on OS and PFS. Computations were carried out in SAS 9.3 (SAS Institute Inc., Cary, NC, USA) and R 3.2.4.
Results
Patient characteristics
Among 37 patients who signed the consent form and enroled in the study, 1 patient was found prior to starting therapy to have negative HER2 status on institutional pathology review, and another patient was admitted to the hospital for a non-trial-related severe illness and did not pursue further treatment after the first several doses. The remaining 35 patients were evaluable for study endpoints. Demographic characteristics are summarised in Table 1. Only 1 patient was male. Twenty-four were white, 6 were Hispanic, and 5 were black. The median age was 52 years (range: 26–72).
The median number of previous lines of therapy for metastatic disease was 3 (range: 0–15). The median best progression-free survival (PFS) on a prior trastuzumab-containing regimen for all patients was 4 months (range: 0–33). The mean duration of prior trastuzumab exposure was 14.5 months, and the maximum duration of response to trastuzumab had been 77 months prior to study.
Dose escalation and MTD
The first 14 patients received entinostat and lapatinib in combination. Eleven patients received entinostat, lapatinib, and trastuzumab in combination until the MTD was reached. Once the MTD was defined, 10 additional patients were enroled and treated. The median number of cycles of treatment was 3 (counting 28 days as 1 cycle). The median follow-up time was 2.53 years (95% CI: 1.77, 4.30).
No patients in the entinostat/lapatinib group experienced DLT. Among patients who received all three agents in combination, 2 of 5 patients experienced DLT at 15 mg of entinostat, 1000 mg of lapatinib, and standard-dose trastuzumab; 1 patient had grade 3 thrombocytopenia, and 1 patient had grade 3 diarrhoea that required dose reduction. Thus, the MTD was designated as one dose level down (Table 2, cohort 6): 12 mg of entinostat, the same 1000-mg dose of lapatinib, and standard-dose trastuzumab, and the cohort was expanded to 6. One of 6 patients treated at the MTD had grade 4 hypokalaemia that required hospitalisation. Among the 10 out of 11 evaluable patients who were enroled in the expansion cohort (excluding 1 patient who was not evaluable because the patient withdrew the consent before reaching 28 days), 1 patient experienced grade 3 diarrhoea consistent with DLT during the first 28 days.
Safety and tolerability of combination
All patients experienced at least grade 1 AEs during the study and experienced a total of 244 treatment-related AEs (Table 3; Supplemental Table S1). Of the 21 patients who received entinostat, lapatinib, and trastuzumab in combination, grade 3 neutropenia, leukopenia, thrombocytopenia were observed throughout the treatment. One had grade 4 thrombocytopenia. Three patients developed infections. Several electrolyte abnormalities were observed. Of the 14 patients who received entinostat and lapatinib without trastuzumab, grade 3 neutropenia and thrombocytopenia were observed.
Diarrhoea was the most frequently reported AE, seen in 31 patients (89%). Not surprisingly given that the starting dose of lapatinib was higher in the first 14 patients who received entinostat and lapatinib alone without trastuzumab, the frequency of diarrhoea was higher in that group: 13 of 14 patients (92.9%) compared to 18 of 21 (85.7%) in the triple combination group (Table 3). Among the patients with reported diarrhoea, 27 patients had grade 1–2 diarrhoea, while 4 patients had grade 3. One patient withdrew consent after 4 months on the study due to intolerance from diarrhoea.
Efficacy
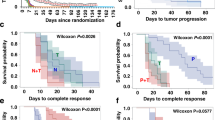
The overall clinical benefit rate was 20%: 3 patients with CR, 3 patients with PR, and 1 patient who maintained SD for over 6 months (Table 4). A total of 17 patients (48.6%) had PD as the best response. The clinical benefit rate in IBC patients was not inferior to that in non-IBC patients (23.1 vs 18.2%, respectively; P = 1). There was no significant difference in clinical benefit rate between patients who had received previous lapatinib use vs not, pertuzumab vs not, and T-DM1 vs not. The best response for each patient is summarised in a swimmer plot in Fig. 1.

Swimmer plot showing clinical response for each patient. Each bar represents an individual patient. Different clinical responses are indicated by different patterns, as shown in the legend on the right
PFS and OS curves for the full cohort are shown in Fig. 2. The median PFS time was 3.48 months (95% CI: 1.81, 3.78). Analysis by oestrogen receptor and progesterone receptor positivity, IBC vs non-IBC, and prior therapies did not show association of these factors with PFS.

Overall survival and progression-free survival of all patients from cycle 1, day 1 (C1D1) of study treatment. E/N, Number of patients with the event (death for OS and disease progression for PFS) / Total number of evaluable patients. Among the 35 patients, 19 (54.2%) died. The median OS time was 2.63 years (95% CI: 1.36-NA), and the median follow-up time was 2.53 years (95% CI: 1.77, 4.30)
As of final data analysis (April 6, 2017), over half of the patients had died (54.2%, 19/35). Two patients died within 30 days of study completion. Both patients were confirmed to have PD at day 42 of the study enrolment prior to the first staging scan by other exams. The median OS was 2.63 years (95% CI: 1.36, NA). Age was found to be significantly associated with OS (HR = 1.07; 95% CI: 1.02, 1.12; P = 0.0071). There was a favourable but not statistically significant trend for PFS in patients who had previously received pertuzumab. Other variables (race, receptor status, IBC vs non-IBC, and prior therapies) were not associated with OS. OS rates at 1, 2, and 5 years were 77%, 52%, and 35%, respectively.
Pharmacodynamics
The median FISH HER2/CEP17 ratios were 7.84 in the entinostat/lapatinib treatment group vs 5.5 in the entinostat/lapatinib/trastuzumab group; however, the difference was not statistically significant (Supplementary Table S2). In 20 of 35 patients (57.1%), more than 10% of cells were positive for oestrogen receptor and/or progesterone receptor expression.
A total of 23 baseline tissue samples were analysed for HER2, EGFR, AKT, and the phosphorylated forms of these proteins, as well as the HER2 and EGFR genes by FISH. No statistical significance was found for these markers with regard to clinical efficacy.
A total of 52 CTC samples were obtained and analysed for correlative biomarkers in 20 patients from the entinostat/lapatinib/trastuzumab treatment group, among whom 17 had paired samples collected at baseline and after the first cycle of treatment. For these 17 patients, the median time from cycle 1, day 1, to the date of the second CTC measurement was 28 days (Supplemental Table S3). Neither baseline CTC counts or changes in CTCs and CTC-based biomarkers showed significant correlation with clinical efficacy.
Discussion
We report results of a phase Ib trial evaluating the combination of entinostat, lapatinib, and trastuzumab at a single institution. The MTD was defined, and the treatment was safe and well tolerated, with mainly grade 1 and 2 toxicities. There was also encouraging early clinical benefit noted in this heavily pre-treated patient population.
The side effect profile of HDAC inhibitors includes pancytopenia, and the potential for enhancing off-target effects (i.e., cardiac toxicity or diarrhoea) of lapatinib and/or trastuzumab was anticipated. In our study, diarrhoea was the most common side effect. Major DLTs included grade 3 and 4 haematologic toxicities and grade 3 diarrhoea. Despite the known high frequency of high-grade diarrhoea induced by lapatinib, the combination with entinostat did not result in high-grade diarrhoea; the rate of grade 3 or above diarrhoea was lower compared to previous lapatinib-related studies,17,18,19 which was positive aspect to this combination. However, the rate of high grade diarrhoea remains to be significantly high at 19% grade 3 or above, that further needs to be confirmed in larger size trials. Whether the epigenetic modulation of entinostat contributes to this lower rate of diarrhoea is unclear and should be studied further. We did not observe any cases of cardiac toxicity during the trial period. One case of grade 4 hypokalaemia was observed without treatment-related diarrhoea, and the correlation between the therapy and the occurrence of hypokalaemia was unclear.
HDAC inhibitors have been recognised to have a role in reversing resistance to anti-HER2 therapies.13 Indeed, entinostat has shown the ability to reverse resistance to endocrine therapy in hormone receptor-positive breast cancer,20 and currently being studied in hormone receptor-positive metastatic breast cancer in combination with an aromatase inhibitor in a phase III trial (NCT02115282). In our preclinical models, addition of HDAC inhibitor induced the apoptotic process in reversing the resistance to trastuzumab,12 and this is the first study to report entinostat and anti-HER2 therapy combination in the clinical setting, to our knowledge.
The encouraging clinical benefit rate of 20% in a heavily pre-treated population is of interest for further study. Patients with multiple previous lines of treatment still showed clinical benefit. There was no significant difference based on previous use of T-DM1. On the other hand, previous pertuzumab use had a trend of association with longer OS, although the difference was not statistically significant. We can hypothesise that when HER2+ breast cancer has already been treated by co-inhibition of heterodimerization partner HER3 by the use of pertuzumab and developed resistance, then another partner of the HER2 receptor, EGFR, may be overexpressed as a compensatory mechanism to the therapeutics, in which case, an anti-EGFR inhibitor maybe more effective compare to lapatinib. The efficacy was also shown in patients who had previous lapatinib containing regimen, which suggests a role of entinostat in reversal of resistance. Although lapatinib has shown activity against HER2 positive breast cancer that has metastasised to brain, we designed to exclude patients given the concern over rapid progression of patients with brain metastasis and the heterogeneous patient population that could potentially affect our clinical outcome analysis. To test this, a separate clinical trial dedicated to study an efficacy for patients with brain metastasis using our combination can be considered.
We analysed CTCs and tissue-based biomarkers to correlate changes in expression among key EGFR/HER2-PI3K-AKT pathway molecules with clinical efficacy, however we could not show meaningful association with clinical outcome. There were several limitations with regard to the CTC results. First, the collection of CTCs was mandated only during the triple-therapy phase of the trial; therefore, only 17 had CTCs collected from both baseline and after the first cycle of treatment, and 5 among those did not have detectable CTCs. Another limitation was the method of CTC detection. In 2010, EpCAM-negative (mesenchymal) CTCs were not included among the detected cells from the blood, which may have lowered the sensitivity of detection. Moreover, while the number of CTCs in patients with metastatic breast cancer was shown to correlate with long term survival,21,22 changes made by treatment, including our proposed entinostat-containing combination therapy has never been investigated before. In addition, the BEACON trial demonstrated significant correlations in mechanistic biomarkers in CTCs and overall survival.23 Circulating tumour DNA has shown its value as a real-time predictor of resistance to ongoing therapy in metastatic breast cancer before,24 however our study mainly tested CTC instead of ctDNA for two reasons. First, at the time of our original study development, ctDNA assay was not readily available due to sensitivity issue. Second and more importantly, we used CTC to detect protein changes, which is limited when ctDNA is used as liquid biopsy tools given detection of mutation-based data. In terms of the changes in the number of CTC, we hypothesised that effective therapy in metastatic breast cancer can induce the reduction of peripheral CTCs, thus can be utilised as a biomarker of response as itself while it enables the functional changes of circulating markers based on protein changes. This hypothesis needs further investigation in future studies with larger number of samples. Lastly, given limited available cells for testing, we had to omit two important biomarkers of HER2 resistance, HER3 and IGF-1R as critical biomarkers on CTC, and this is another limitation of our biomarker analysis. We plan to study these markers, as well as other biomarkers that have shown to confer resistance, e.g., Bim in our future studies. Taken together, although the analysis did not reveal any correlations with many limitations as we summarised here, we did demonstrate the feasibility of using CTC-based testing as a complement to tissue-based testing in future trials.
The primary objective of assessing the safety and MTD of the novel drug combination was fulfilled. Completing this assessment of clinical efficacy requires a larger phase II study of entinostat in combination with a HER2-targeted agent as a therapeutic option for trastuzumab-resistant HER2+ metastatic breast cancer, and while there were limitations in clinical efficacy assessment and biomarker, we believe our phase Ib data supports the rationale to conduct such trial. To strengthen the data on potential relevant biomarkers, we plan to analyse the mechanism of action of entinostat and serum-based biomarker expression changes in future trials.
References
Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A., McGuire, W. L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 (1987).
Breast cancer by the numbers. 39, 213–214 (2014). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4005124/
Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792 (2001).
Blackwell, K. L., Burstein, H. J., Storniolo, A. M., Rugo, H., Sledge, G., Koehler, M. et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J. Clin. Oncol. 28, 1124–1130 (2010).
Geyer, C. E., Forster, J., Lindquist, D., Chan, S., Romieu, C. G., Pienkowski, T. et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743 (2006).
Bachelot, T., Romieu, G., Campone, M., Dieras, V., Cropet, C., Dalenc, F. et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol. 14, 64–71 (2013).
Lin, N. U., Dieras, V., Paul, D., Lossignol, D., Christodoulou, C., Stemmler, H. J. et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin. Cancer Res. 15, 1452–1459 (2009).
Blackwell, K. L., Burstein, H. J., Storniolo, A. M., Rugo, H. S., Sledge, G., Aktan, G. et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J. Clin. Oncol. 30, 2585–2592 (2012).
Esteva, F. J., Valero, V., Booser, D., Guerra, L. T., Murray, J. L., Pusztai, L. et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 20, 1800–1808 (2002).
Burstein, H. J., Keshaviah, A., Baron, A. D., Hart, R. D., Lambert-Falls, R., Marcom, P. K. et al. Trastuzumab plus vinorelbine or taxane chemotherapy for HER2-overexpressing metastatic breast cancer: the trastuzumab and vinorelbine or taxane study. Cancer 110, 965–972 (2007).
Johnstone, R. W. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287–299 (2002).
Lee, J., Bartholomeusz, C., Mansour, O., Humphries, J., Hortobagyi, G. N., Ordentlich, P. et al. A class I histone deacetylase inhibitor, entinostat, enhances lapatinib efficacy in HER2-overexpressing breast cancer cells through FOXO3-mediated Bim1 expression. Breast Cancer Res Treat. 146, 259–272 (2014).
Huang, X., Wang, S., Lee, C. K., Yang, X., Liu, B. HDAC inhibitor SNDX-275 enhances efficacy of trastuzumab in erbB2-overexpressing breast cancer cells and exhibits potential to overcome trastuzumab resistance. Cancer Lett. 307, 72–79 (2011).
Wolff, A. C., Hammond, M. E., Schwartz, J. N., Hagerty, K. L., Allred, D. C., Cote, R. J. et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 25, 118–145 (2007).
Eisenhauer, E. A., Therasse, P., Bogaerts, J., Schwartz, L. H., Sargent, D., Ford, R. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
US Department of Health and Human Services NIoH, National Cancer Institute. Common terminology criteria for adverse events (CTCAE). https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed on 30 Jan 2018.
Robidoux, A., Tang, G., Rastogi, P., Geyer, C. E. Jr., Azar, C. A., Atkins, J. N. et al. Lapatinib as a component of neoadjuvant therapy for HER2-positive operable breast cancer (NSABP protocol B-41): an open-label, randomised phase 3 trial. Lancet Oncol. 14, 1183–1192 (2013).
Guan, Z., Xu, B., DeSilvio, M. L., Shen, Z., Arpornwirat, W., Tong, Z. et al. Randomized trial of lapatinib versus placebo added to paclitaxel in the treatment of human epidermal growth factor receptor 2-overexpressing metastatic breast cancer. J. Clin. Oncol. 31, 1947–1953 (2013).
Goss, P., Smith, I., O’Shaughnessy, J., Ejlertsen, B., Kaufmann, M., Boyle, F. et al. Adjuvant lapatinib for women with early-stage HER2-positive breast cancer: a randomised, controlled, phase 3 trial. Lancet Oncol. 14, 88–96 (2013).
Yardley, D. A., Ismail-Khan, R. R., Melichar, B., Lichinitser, M., Munster, P. N., Klein, P. M. et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 31, 2128–2135 (2013).
Kallergi, G., Konstantinidis, G., Markomanolaki, H., Papadaki, M. A., Mavroudis, D., Stournaras, C. et al. Apoptotic circulating tumor cells in early and metastatic breast cancer patients. Mol. Cancer Ther. 12, 1886–1895 (2013).
Giuliano, M., Giordano, A., Jackson, S., Hess, K. R., De Giorgi, U., Mego, M. et al. Circulating tumor cells as prognostic and predictive markers in metastatic breast cancer patients receiving first-line systemic treatment. Breast Cancer Res. 13, R67 (2011).
Rugo, H. S., Cortes, J., Awada, A., O’Shaughnessy, J., Twelves, C., Im, S. A. et al. Change in topoisomerase 1-positive circulating tumor cells affects overall survival in patients with advanced breast cancer after treatment with etirinotecan pegol. Clin. Cancer Res. 24, 3348–3357 (2018).
Stover, D. G., Parsons, H. A., Ha, G., Freeman, S. S., Barry, W. T., Guo, H. et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. J. Clin. Oncol. 36, 543–553 (2018).
Acknowledgements
We acknowledge other research staff: Angela Marx, Charla Parker at the Morgan Welch Inflammatory Breast Cancer program who are not listed here as authors for their help in managing the study. Tissue and CTC biomarker assays and initial basic statistical analysis were performed by ApoCell. Lapatinib was provided by Novartis, and entinostat was provided by Syndax Pharmaceuticals. Sunita Patterson of the Department of Scientific Publications provided editorial assistance.
Author contributions
First author B.L. and Corresponding author N.T.U. collected, analysed the data along with other members of the team and formulated the whole manuscript. Second author R.K.M. provided her perspective on Her2 therapy in metastatic Her2 cancer in discussion and background session. J.L. helped initial design of clinical trial based on his pre-clinical data, S.A.J., J.S.W. had provided significant clinical trial regulatory efforts. T.I. helped collecting the updated clinical information of the patients. D.W.D. helped with circulating biomarker analysis. J.W. and Y.S. had the primary statistical analysis of the study. D.T., R.A., N.K.I., A.M.B., C.H.B., P.H.B., S.H.G., S.L.M., D.J.B., V.V. enrolled their patients to the study and review the result of the study, validate the data quality of the manuscript. J.A.M. and R.P. provided the guidance of the trial development and conduct from CTEP perspective.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and was approved by institutional review boards at The University of Texas MD Anderson Cancer Center. All enroled patients provided written informed consent.
Consent to publish
All authors consent the material to publish.
Competing interests
The authors declare no competing interests.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Note
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Funding
This study was supported by the National Cancer Institute at the National Institutes of Health through a Cancer Therapy Evaluation Program UM1 grant [NCI #8871] and MD Anderson’s Cancer Center Support Grant [P30CA016672; used the Clinical Trials Support Resource and Biostatistics Resource Group]; MD Anderson’s Center for Genetics and Genomics (used the Molecular Cytogenetics Facility); and the Morgan Welch Inflammatory Breast Cancer Research Program and Clinic.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lim, B., Murthy, R.K., Lee, J. et al. A phase Ib study of entinostat plus lapatinib with or without trastuzumab in patients with HER2-positive metastatic breast cancer that progressed during trastuzumab treatment. Br J Cancer 120, 1105–1112 (2019). https://doi.org/10.1038/s41416-019-0473-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0473-y
This article is cited by
-
FOXA1 and adaptive response determinants to HER2 targeted therapy in TBCRC 036
npj Breast Cancer (2021)
