
Abstract
Gain of chromosome 1q (+1q) is one of the most common recurrent cytogenetic abnormalities in multiple myeloma (MM), occurring in approximately 40% of newly diagnosed cases. Although it is often considered a poor prognostic marker in MM, +1q has not been uniformly adopted as a high-risk cytogenetic abnormality in guidelines. Controversy exists regarding the importance of copy number, as well as whether +1q is itself a driver of poor outcomes or merely a common passenger genetic abnormality in biologically unstable disease. Although the identification of a clear pathogenic mechanism from +1q remains elusive, many genes at the 1q21 locus have been proposed to cause early progression and resistance to anti-myeloma therapy. The plethora of potential drivers suggests that +1q is not only a causative factor or poor outcomes in MM but may be targetable and/or predictive of response to novel therapies. This review will summarize our current understanding of the pathogenesis of +1q in plasma cell neoplasms, the impact of 1q copy number, identify potential genetic drivers of poor outcomes within this subset, and attempt to clarify its clinical significance and implications for the management of patients with multiple myeloma.
Similar content being viewed by others


Introduction
Multiple myeloma (MM) is the second most common hematological malignancy1 and fortunately has seen substantial improvement in patient survival over the past two decades2. Although MM is characterized by marked biological and genetic heterogeneity, the improvement in outcomes has been driven primarily by treatment regimens that target key components of plasma cell biology rather than the underlying genomic aberrancies3. While almost all patients seem to benefit somewhat from novel therapies, many patients still experience an aggressive disease course with early relapse, clinical morbidity, and early mortality. Screening for recurrent cytogenetic abnormalities (CA) remains one of the most powerful means to identify patients who are at higher risk for early progression and death and who may be candidates for investigational approaches to improve survival.
MM is a biologically heterogeneous disease that has been historically been characterized by either having either having extra chromosome copies (or hyperdiploidy) or translocations associated with chromosome 14 where the immunoglobulin heavy chain (IgH) gene resides. Originally described as such using conventional karyotyping in the 1970s, fluorescent in situ hybridization was utilized in the late 1990s to better characterize the disease using this nomenclature. There is a growing recognition that disease biology dictates outcomes in uniformly treated MM patients and trials enriching for specific subgroups that have short survival outcomes with standard of care approaches need to be explored instead of the one-size-fits-all approach.
After discovery that large chromosomal structural changes were common among many cancer types, additional copies of chromosome 1q (+1q) was identified as one of the most common CA on metaphase cytogenetics in MM4,5 and other cancers6,7,8. Although abnormalities on karyotype were noted to impact prognosis in MM9, the sensitivity and accuracy of karyotyping to identify large chromosomal aberrations remains suboptimal due to the low proliferative rate of plasma cells10,11,12. Fluorescent in situ hybridization (FISH) overcame many of these barriers in MM13 and found that copy number alterations were not only common in MM11,13,14, but that abnormalities detected by FISH were also highly prognostic of outcomes15. Because of its high sensitivity and reproducibility, FISH has become the primary means to identify recurrent CA in MM and has been incorporated into the Revised International Staging System (R-ISS)16. By FISH, +1q is identified in approximately 40% of newly diagnosed MM cases17.
Despite its frequency, considerable debate remains regarding the prognostic impact of +1q in MM. One major barrier to understanding the impact of 1q abnormalities in MM is the lack of uniformity in reporting and annotation of cytogenetics18, and this is even more profound when reporting chromosome 1 abnormalities. This is particularly important because copy number of 1q has emerged as a very important feature of this CA. In general, +1q should be understood as denoting any patient who has at least one extra copy of some portion of chromosome 1q. Probes detecting DNA on chromosome 1 in region 2, band 1 (1q21) are used to detect this abnormality. Among patients with +1q, gain(1q) describes patients with only one additional copy of 1q (3 total copies, commonly reported in a clinical FISH report as “nuc ish 1q21x3”), whereas amp(1q) denotes patients who have amplification of 1q, defined as two or more extra copies (4 or more total copies). Although some labs report “duplication” of 1q or ratios of 1q21:1p32 (CKS1B:CDKN2C) of >1.1, these annotations do not specify copy number and should be avoided. We propose that these common definitions be uniformly applied in reporting chromosome 1q copy number in MM, and this has been highlighted for emphasis in Table 1. The present review summarizes the pathophysiology of 1q abnormalities in MM, compiles key data regarding the prognostic impact of chromosome 1q21 abnormalities in MM, and attempts to clarify clinical trial outcomes and management implications for patients with +1q myeloma.
Pathogenesis
In addition to the prognostic impact of cytogenetics, characterization of recurrent genetic abnormalities has helped aid in our understanding of myelomagenesis and clonal progression. In ~60% of patients, myeloma is characterized by having extra copies of odd numbered chromosomes, known as hyperdiploidy or trisomic MM, and the remaining cases typically involve translocations involving the IgH locus at chromosome 14q32. These events are almost always mutually exclusive19,20,21, but additional genetic changes including chromosome 1 abnormalities often occur as secondary events and can be seen among patients in both groups. The frequency and copy number of +1q seem to increase as the disease progresses from smoldering myeloma to newly diagnosed MM to relapsed/refractory disease17,22,23. In nearly all series, +1q is identified at a higher frequency among patients with high-risk IgH translocations, −13, del(1p), and other high-risk clinical features such as hypercalcemia, but it can still be found among patients with standard risk MM including trisomies24,25,26.
Genomic instability appears to be a hallmark of +1q MM. In what has been described as “jumping 1q syndrome” by the University of Arkansas for Medical Sciences Myeloma group (UAMS-MIRT), patients with +1q were noted to have frequent and common breakpoints in the pericentromeric heterochromatin27,28, with subsequent translocation of a 1q segment or whole arm to other chromosomes. This unstable chromatin persists after the original event, leading to a repetitive pattern of translocations or segmental duplications that causes the same segment of DNA from 1q to “jump” around the genome, with increasing copy number as genomic instability becomes worse, frequently upon disease progression29,30. Trisomy of chromosome 1 is also commonly seen, although it is not clear whether this is associated with a different clinical course compared to “jumping 1q”. Newer genomic techniques have helped to reveal that multiple myeloma is often characterized by complex structural variants. Within CoMMpass, a prospective study examining the impact of genomics on outcomes of >1000 MM patients, it is notable that +1q21 was most commonly found within the context of whole arm gains rather than chromothripsis31, consistent with the pathogenesis previously identified as large copy number gains. Chromosome 1 copy number variations (CNV), including both +1q and del(1p) are also frequently seen within the context of hypodiploidy in MM samples, and clustering analysis of individual CNV has shown that +1q does not cluster with gains or loss of 1p, and that these likely occur as separate events within a complex genetic landscape32. Ultimately, regardless of the mechanisms underlying the development of +1q, the main question is whether this occurs as a byproduct of high-risk biology and genomic instability, or whether increased expression of genes on extra copies of 1q causes high-risk disease, resulting in resistance to therapy and early progression/death.
Potential genetic drivers on chromosome 1q
As chromosome 1q is the largest chromosome arm in the human genome, the search for a particular driver of more aggressive disease has identified numerous candidates, but without clear identification of a gene or pathway that is consistently dysregulated among patients with +1q myeloma. As mentioned above, a key reason for this is that +1q occurs as a secondary event, often in conjunction with other high-risk cytogenetic abnormalities and genomic complexity that complicates the search for a main driver of outcomes and response to therapy. Furthermore, there are almost always multiple pathways involved, and although gene expression profiling (GEP) has helped to characterize patterns of gene dysregulation, the search for a precise, targetable gene has been difficult. As will be explored in this section, several key biological pathways, including cell cycle regulators, apoptosis, interactions with the bone marrow microenvironment, and others have been implicated in genetic aberrations found among MM patients with +1q and are potential candidates as key regulators of pathogenesis in +1q myeloma. Interestingly, despite the heterogeneity of genes involved, many of the cellular functions encoded by these genes seem to be inter-related and result in activation the JAK/STAT3 pathway, either directly or as a downstream effect.
The UAMS-MIRT group, through their pioneering of GEP in MM, found that one subset of MM with high risk features was defined by overexpression of genes localizing at 1q2133. In particular, they found that overexpression of CKS1B, a cell cycle regulator, was highly associated with an aggressive clinical course34,35 and was found to promote myeloma cell growth by activating cyclin-dependent kinases and through SKP2-mediated ubiquitination of the tumor suppressor p27Kip1 34,36. Subsequently, CKS1B was later shown to activate myeloma cell growth through upregulation of the STAT3 and MEK/ERK pathways37.
MCL-1 is a member of the BCL2 family of anti-apoptosis proteins with a genetic locus at 1q2138. It has been well described that, with the exception of the t(11;14) MM subset, myeloma cells and normal plasma cells are highly dependent on MCL1 for survival39,40. Within CoMMpass, it was found that by RNA sequencing, higher expression of MCL1 was found as copy number of 1q increases41, and conversely, very high MCL1 gene expression highly correlates with presence of +1q among patients with newly diagnosed MM42. MCL-1 dependency is also enhanced by signaling from IL-6 within the bone marrow microenvironment43, and notably, the IL-6 receptor (IL-6R) is also encoded at 1q21. Soluble IL-6R levels rise with increasing copy number of 1q, but the impact on outcomes has been unclear44,45,46.
Recently, ADAR1 has been identified as a potentially important gene located at 1q21. ADAR1 is an RNA editing protein involved in A to I post-transcriptional modification, and overexpression leads to aberrant hyperediting and MM cell proliferation47. In CoMMpass, higher editing levels as mediated by ADAR1 were associated with inferior survival, independent of 1q copy number48. The P150 isoform of ADAR1 was subsequently found to promote oncogenesis through the STAT3 pathway, and this was further enhanced when combined with increased activity of IL-6R49.
PDZK1 is overexpressed in MM cell lines and drives resistance to chemotherapy in vitro50. Although this has been largely underexplored, a recent study showed that PDZ binding kinase is induced by IL-6 and promotes growth through STAT3 pathway51, yet another example of a protein at the 1q21 locus that is involved in the IL6-STAT3 continuum. PSMD4 and ILF2 have been implicated in resistance to specific treatments and are summarized later in the review.
Outcomes
While +1q has been most extensively studied within the context of multiple myeloma, it has also been found in approximately 30% of patients with smoldering myeloma22. When detected by FISH or GEP-70 classification, +1q appears to be an independent risk factor for progression from SMM to MM22,52,53. The increased risk of progression to symptomatic myeloma is likely related to the association between +1q and genomic instability that has been described above, leading to additional mutational burden and progression to overt malignancy.
Prior to the widespread adoption of immunomodulatory drugs (IMiDs) and proteasome inhibitors (PIs) in the management of myeloma, the impact of +1q was evaluated in several clinical trials. In the IFM99-02 and IFM99-04 trials, induction with vincristine, doxorubicin, and dexamethasone (VAD) was evaluated prior to tandem autologous stem cell transplantation (ASCT)54. In these studies, gain(1q23) by comparative genome hybridization (CGH) and single nucleotide polymorphism (SNP) array was highly prognostic of early death on multivariate analysis55,56. Similarly, in the CMG2002 trial, patients with +1q21 by FISH who were treated with VAD induction had inferior outcomes than those without the abnormality57.
The Total Therapy 2 trial (TT2) performed by the UAMS group evaluated the implementation of thalidomide (in TT2) and other novel agents into the initial management strategy for patients along with chemotherapy induction, tandem transplantation, and consolidation/maintenance. In TT2, it was noted that patients with +1q at diagnosis had similar responses, but worse PFS and OS compared to those without +1q and ultimately derived no benefit from the addition of thalidomide17. In the Myeloma IX study, which also incorporated thalidomide into induction and maintenance for both transplant eligible and ineligible patients, +1q was also a poor prognostic factor58,59. When combined with adverse IgH translocations or del(17p), the impact of +1q was additive, and the effect was just as pronounced when found with hyperdiploid MM as with high-risk cytogenetics60,61.
Despite this early evidence suggesting inferior outcomes, a key meta-analysis raised questions about whether +1q was, in fact, an independent prognostic factor25. This study included three cohorts of patients—two from the Mayo clinic who were treated with high-dose therapy and ASCT, as well as the TT2 trial. In the analysis, 1q gain as determined by CKS1B copy number or the ratio of CKS1B to total chromosome 1 copies was again shown to be associated with high-risk clinical features, other high risk CAs, and poor survival. However, regardless of methodology to define +1q, when adjusted for other high-risk cytogenetic abnormalities, +1q was not found to be a significant prognostic factor on multivariate analysis. Subsequently, in an analysis of the GMMG-3 and GMMG-4 trials, that evaluated induction with doxorubicin/dexamethasone in combination with vincristine (VAD), thalidomide (TAD), or bortezomib (PAD) followed by transplantation, +1q21 was again found to be associated with high-risk disease but insignificant on multivariate analysis62. As such, although the IMWG has stated that +1q must be absent for a patient to qualify as standard risk63, +1q was not routinely evaluated at many centers for years and most Phase 3 clinical trials in newly diagnosed MM have not uniformly collected or reported data for +1q.
Several retrospective series have evaluated outcomes of patients with +1q21 myeloma after the implementation of PI/IMiD induction regimens. The Emory group found that among MM patients treated with lenalidomide, bortezomib, and dexamethasone (RVd) and risk-adapted maintenance among transplant-eligible patients, those with +1q by FISH had worse PFS and OS compared to those without +1q. Patients with amp(1q) had poor survival regardless of other cytogenetic abnormalities, however, there was a discrepancy among patients with gain.(1q) Patients with standard risk and gain(1q) had no significant impact on PFS compared to standard risk alone, whereas patients with gain(1q) plus t(4;14), t(14;16) or del(17p) had dismal PFS. Interestingly, patients with high risk cytogenetics but no +1q had outcomes similar to those with standard risk disease, suggesting a “double hit” effect26. In contrast, the Mayo group showed that OS was decreased among patients with +1q regardless of copy number, other cytogenetic abnormalities, and whether patients received a PI, IMiD, or both as a part of induction64. Two independent studies found that patients with chromosome 1 abnormalities, (C1A), including either +1q or del(1p), had inferior outcomes compared to those without C1A65,66. Outside of the United States, a study of outcomes within the Austrian myeloma registry, investigators found that the impact of +1q is dependent on copy number, but even gain(1q) was sufficient to abrogate the good prognosis of the hyperdiploid group defined by gains of 11q67.
Observational genomic analyses have shown similar results. In the Myeloma Genome Project, which collected comprehensive genomic data among 1273 patients with newly diagnosed myeloma, amp(1q) in combination with ISS stage 3 comprised a group of patients who had dismal outcomes, similar to those with the combination of del(17p) and mutated TP5368. In CoMMpass, PFS, and OS both decreased with each respective copy of 1q as determined by DNA sequencing26. Notably, although these retrospective and observational studies differ somewhat regarding methodology and the relative importance of copy number and other cytogenetic abnormalities, they were all conducted in the era of highly-active PI and IMiD induction therapy and the presence of +1q (or C1A) was found to be an independent prognostic factor for inferior outcomes, even when accounting for other CAs and clinical stage. As such, it appears that although outcomes for MM patients have improved with widespread implementation of PI/IMiD induction, patients with +1q have not benefitted as much, and clearly need to be evaluated in prospective clinical trials.
Two recent studies have reported data for +1q in newly diagnosed MM. The Myeloma XI trial, conducted in the UK, evaluated lenalidomide or thalidomide in combination with cyclophosphamide and dexamethasone (CRd and CTd, respectively), and later added an arm including carfilzomib with CRd (KCRd), followed by ASCT for eligible patients and maintenance with lenalidomide vs observation. In this study, patients with +1q had worse OS, and the prognostic effect was more prominent among those with amp(1q) or co-occurrence of additional high-risk cytogenetic abnormalities69. However, although +1q was an independent prognostic factor when evaluating cytogenetics alone, with the addition of the SKY92 GEP signature, even amp(1q) was no longer a significant factor, as nearly all of these patients were captured by the high-risk SKY92 signature. The FORTE study randomized transplant-eligible patients to receive 4 cycles of carfilzomib with cyclophosphamide and dexamethasone (KCd) followed by ASCT and 4 cycles of KCd consolidation, carfilzomib with lenalidomide/dexamethasone (KRd) for 4 cycles, followed by ASCT and 4 cycles of KRd consolidation, or 12 cycles of KRd without transplantation. In this study, patients with gain(1q) had worse survival in the KCd_ASCT and KRd12 arms, but the risk was abrogated in the KRd_ASCT arm. However, it was noted that patients with amp(1q) had dismal outcomes regardless of treatment arm, unless they were able to achieve negative minimal residual disease (MRD)70. More data regarding the outcomes of patients with and without +1q in Phase 3 clinical trials are compiled in Table 2.
In summary, amp(1q) is uniformly associated with poor survival and should be considered a high risk cytogenetic abnormality. Although most studies suggest that gain(1q) is also high risk, its impact does not appear to be as detrimental as amp(1q), and the presence of other high risk cytogenetic abnormalities or gene expression profiles may be important factors in whether this abnormality imparts additional risk. In order to truly determine whether gain(1q) is, itself, a key prognostic marker or predictive of treatment outcomes, it will be essential for clinicians to evaluate for and document the presence or absence of an abnormality and copy number of 1q in all patients, and to report this data in a uniform matter, alongside other frequently reported cytogenetics such as IgH translocations, del(17p), and trisomies.
Management implications
Ideally, standard management recommendations should be determined by prospective data from randomized controlled clinical trials. However, aside from the recent trials mentioned above, the standard of care in MM has evolved without much evidence regarding the relative benefit of novel combinations on patients with +1q. As such, clinicians are faced with the challenge of making treatment recommendations for a large population of patients with MM who are suggested to have inferior outcomes with the current standards of care, but no strong evidence to suggest benefit from alternative strategies.
It is generally believed that patients with high-risk disease derive benefit from proteasome inhibitors, particularly the t(4;14) and del(17p) subsets based on clinical data showing that bortezomib could abrogate the poor prognosis of these patients71,72. However, data among patients with +1q are conflicting, and as detailed below, several studies have suggested that +1q may actually confer resistance to bortezomib. In the Total Therapy 3 (TT3) trial, patients with +1q were noted to have early progression when treated with bortezomib compared to those without +1q, and GEP suggested that this may have been related to overexpression of PSMD4, a proteasome subunit encoded at 1q21 that was highly correlated with 1q copy number in TT373. PSMD4 was identified as a poor prognostic factor in both the GEP-70 and GEP-80 models developed by UAMS-MIRT group, and PSMD4 overexpression appears to promote resistance to bortezomib, with increased resistance seen with additional copies of 1q2173. On the contrary, a recent study suggested that a high MCL1 gene signature, typically seen in patients with +1q, was predictive of longer PFS and OS when treated with bortezomib42. An, et al. found that patients with +1q had inferior outcomes with bortezomib-based induction, whereas it did not affect outcomes when thalidomide was used74. In the HOVON65/GMMG-4 trial, although patients treated with bortezomib had superior OS at 96 months compared to VAD, the outcome of patients with +1q was far inferior to those who did not have +1q. In the relapsed/refractory setting, several studies showed that patients with +1q had inferior outcomes with bortezomib-based regimens compared to patients without +1q75,76,77, although this was not a universal finding78. Preclinical data suggest that a NEDD8 inhibitor overcomes resistance to bortezomib among CKS1B overexpressing cells in vitro79, though this has not yet been translated to patient care.
It has been postulated that carfilzomib, a more potent proteasome inhibitor, may be able to overcome resistance to bortezomib among patients with high-risk myeloma. Pharmacogenomic analysis of patients treated with carfilzomib after prior bortezomib exposure in the TT3 trials and in Total Therapy 6 showed that expression of PSMD4, as well as other proteasome subunits, was increased in response to carfilzomib but not bortezomib, perhaps indicating that carfilzomib may be able to more effectively inhibit this and other subunits of the proteasome in MM, with increased gene expression reflecting the cell’s attempt to restore functionality of the inhibited proteasome subunit80. Early phase clinical trials have shown impressive response rates and MRD negativity when treated with carfilzomib-based induction, including high-risk patients81,82. However, the only Phase 3 data comparing RVd to KRd showed no difference in response rates or PFS between the two regimens among patients with standard risk myeloma or t(4;14)83. Outcomes for patients with +1q in this trial have not yet been reported. Although outcomes of patients treated with KRd induction and transplant in FORTE are encouraging and could be used to justify the use of KRd induction and consolidation with ASCT in transplant-eligible patients, it must be noted that no prospective, randomized trial has demonstrated superiority of KRd to VRd in newly diagnosed MM.
Regarding the utility of ASCT for patients with +1q MM, data are conflicting. A retrospective series from MD Anderson showed that patients with high CKS1B had inferior PFS following ASCT compared to those without this abnormality84. ILF2, a gene located at 1q21, is involved in homologous recombination in response to high-dose chemotherapy, and promotes myeloma cell resistance to DNA damaging agents such as melphalan85. However, in a CIBMTR analysis, +1q patients were not found to have inferior outcomes after ASCT86, and data from the FORTE trial suggest that patients with +1q who are treated with KRd induction have longer PFS when ASCT is employed compared to KRd alone. Based upon these data, we recommend upfront ASCT for patients with +1q who are transplant eligible.
The use of monoclonal antibodies (mAb) in the treatment of patients with MM has become ubiquitous, and based upon impressive data in the frontline setting by adding daratumumab to standard therapies87,88,89,90, many experts have recommended that a CD38 mAb be added to standard induction regimens. However, although daratumumab has clearly demonstrated efficacy among high-risk patients in the relapsed/refractory setting, the benefit of adding daratumumab in newly diagnosed high-risk patients has been more difficult to establish91. A recent meta-analysis showed that, with pooled data, there is a benefit to adding daratumumab to induction therapy for high-risk patients, though not as substantial as among standard risk92. Additionally, the majority of this benefit was driven by the MAIA study, when daratumumab was added to a doublet, lenalidomide/dexamethasone, as opposed to PI/IMiD triplets in the other studies. Notably, data regarding the impact of CD38 mAbs in patients with +1q are extremely limited. In one retrospective series, +1q21 or GEP-70 high risk score portended poor prognosis among relapsed/refractory patients treated with daratumumab93. In the ICARIA-MM study, patients with +1q21 by FISH who were treated with isatuximab plus pomalidomide/dexamethasone (Pd) had superior PFS compared to those treated with Pd alone94, although patients with +1q21 were noted to have inferior ORR and PFS in both treatment groups compared to those without 1q. Recently, the JAK-STAT pathway has been implicated in downregulation of CD38 on the surface of MM cells95. If STAT3 overactivation is driven by +1q21, this could potentially explain a mechanism for resistance to daratumumab, though this has not been directly assessed.
There are no data reporting the specific impact of consolidation or maintenance therapy for patients with +1q MM. Lenalidomide maintenance until progression is recommended for most patients in the United States based upon the PFS benefit seen in the FIRST trial for transplant-ineligible patients96, and an overall survival benefit seen in the CALGB 100104 study97 and a pivotal meta-analysis of post-transplant maintenance98. However, patients with high risk disease do not derive as much benefit from this approach99, and alternative strategies for these patients have been explored. Bortezomib maintenance has been suggested to be beneficial among high-risk patients71,99, but the impact of bortezomib on outcomes of patients with +1q was not directly assessed in these studies. The Emory group has demonstrated PFS and OS rates far superior to historical controls among patients with high risk MM with the use of RVd maintenance after transplant, but +1q was not assessed in that study100. In a prospective study, the SWOG S1211 study, which exclusively enrolled patients with high-risk MM and employed VRd with or without elotuzumab (Elo) as induction and maintenance, showed no benefit for the addition of Elo, but demonstrated a median PFS for the RVd arm of 33.5 months, which outperformed the expected PFS for this group and is postulated to be due to prolonged PI/IMiD maintenance101. Recent data presented from the FORTE study suggests that carfilzomib plus lenalidomide maintenance improves PFS compared to lenalidomide monotherapy as post-transplant maintenance among all patients, including those with high-risk disease, further supporting the use of PI/IMiD maintenance in the post-transplant setting in high risk MM.
Given the plethora of potential drivers at 1q21, it is enticing to investigate the potential to target some of these genes and pathways. Particularly given the success of the BCL2 inhibitor, venetoclax, in other lymphoid malignancies and in generating impressive responses in the t(11;14) subset of MM102, many attempts have been made to target MCL1 in MM. Preclinical data suggest that patients with +1q may be less likely to be resistant to MCL1 inhibitors41. However, because MCL1 is a very labile protein and since the intrinsic apoptosis pathway has substantial complexity in its regulation, this approach has been largely unsuccessful. Recently, several small molecule MCL-1 inhibitors have been developed and are in early phase clinical trials103,104,105.
FcHR5 is a surface protein of unknown function that is highly expressed on the surface of MM cells and has a genetic locus in the chromosomal breakpoint at 1q21. Preclinical studies have demonstrated that FcHR5 is expressed at higher levels among patients with MM harboring +1q21 compared to those without +1q21106. Cevostamab is a bispecific antibody targeting FcHR5 and has shown promising activity in Phase 1 studies107. Although data among patients with +1q21 are unknown in that study, it will be interesting to see whether these patients have increased sensitivity to targeting by cevostamab. Targeting of CKS1B has also been investigated and is in early stages of development108. Based upon the common pathway of STAT3 activation of candidate genes at 1q21, this may also be an intriguing approach for patients with +1q MM.
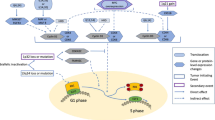
Improving outcomes and defining the optimal management strategy for patients with high-risk myeloma is one of the most important questions faced by myeloma researchers today. Risk-adapted treatment strategies have been reported109 and are becoming more frequently utilized, as the “one-size-fits-all” approach to the management of MM is becoming outdated. Furthermore, the implementation of advanced genomics and dynamic risk assessment based on MRD testing is likely to improve our ability to identify patients at risk of early relapse who are in need of alternate treatment strategies110. However, at this time cytogenetics remain the most widely utilized genomic factor in risk stratification of MM, and the importance of obtaining accurate cytogenetic information at diagnosis cannot be understated in order to identify patients who may need risk-adapted approaches. Ultimately, despite the frequency with which +1q is identified and its association with inferior outcomes, the optimal management of patients with +1q MM is poorly defined, largely due to the wide variability in assessment and reporting of 1q abnormalities. Based upon the data covered in this review, our general approach to management of newly diagnosed MM patients with +1q is summarized in Fig. 1.

V = bortezomib; R or Len = lenalidomide; d = dexamethasone; Dara or D = daratumumab; ASCT = autologous stem cell transplantation with high-dose melphalan conditioning; HR CA = high risk cytogenetic abnormalities. aCarfilzomib may be substituted for bortezomib if neuropathy is present.
Conclusion
Outcomes for patients with high-risk MM, including those with +1q, remain suboptimal and investigational approaches are recommended. As more effective treatment options including adaptive cellular therapy, bispecific antibodies, and targeted therapy continue to be developed and are implemented in the management of MM, these will likely lead to improved outcomes for patients with high-risk MM including those with +1q. Because these novel therapies have almost always shown to be effective for all subsets, but to a lesser degree in high risk patients, determining the optimal time point to utilize these treatment strategies will be of critical importance to improve survival and potentially cure patients. Clinical trials such as S1211101 and MASTER111 that selectively enroll high-risk patients are critically needed, and patients with +1q, particularly those with amp(1q), should be considered for enrollment in these trials. On the contrary, due to the profound efficacy of highly-active treatment options, some patients may not require such an aggressive approach, and could be considered for de-escalation of therapy. It will be critically important to determine whether +1q is a determining factor in predicting outcomes with these risk-adapted strategies, and it is essential that these abnormalities, identified in 30–40% of newly diagnosed MM patients, be documented and studied uniformly. Based upon the exciting progress in therapeutics for MM, optimism is clearly warranted for the exciting possibilities of long-term survival outcomes of patients with MM, including those with +1q. Hope abounds that with proper risk stratification and potential targeted approaches, these patients can experience prolonged survival with excellent quality of life for many years.
References
American Cancer Society. Cancer Facts & Figures 2019 (2019).
Kumar, S. K. et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 28, 1122–1128 (2014).
Boise, L. H., Kaufman, J. L., Bahlis, N. J., Lonial, S. & Lee, K. P. The Tao of myeloma. Blood 124, 1873–1879 (2014).
Dewald, G. W., Kyle, R. A., Hicks, G. A. & Greipp, P. R. The clinical significance of cytogenetic studies in 100 patients with multiple myeloma, plasma cell leukemia, or amyloidosis. Blood. 66, 380–390 (1985).
Sawyer, J. R., Waldron, J. A., Jagannath, S. & Barlogie, B. Cytogenetic findings in 200 patients with multiple myeloma. Cancer Genet. Cytogenet. 82, 41–49 (1995).
Gahrton, G., Friberg, K., Zech, L. & Lindsten, J. Duplication of part of chromosome no. 1 in myeloproliferative diseases. Lancet 1, 96–97 (1978).
Rowley, J. D. Mapping of human chromosomal regions related to neoplasia: evidence from chromosomes 1 and 17. Proc. Natl Acad. Sci. USA 74, 5729–5733 (1977).
Atkin, N. B. Chromosome 1 aberrations in cancer. Cancer Genet. Cytogenet. 21, 279–285 (1986).
Tricot, G. et al. Poor prognosis in multiple myeloma is associated only with partial or complete deletions of chromosome 13 or abnormalities involving 11q and not with other karyotype abnormalities. Blood 86, 4250–4256 (1995).
Weh, H. J., Fiedler, W. & Hossfeld, D. K. Cytogenetics in multiple myeloma: are we studying the ‘right’ cells? Eur. J. Haematol. 45, 236–237 (1990).
Zandecki, M., Lai, J. L. & Facon, T. Multiple myeloma: almost all patients are cytogenetically abnormal. B.r J. Haematol. 94, 217–227 (1996).
Poddighe, P. J. et al. Interphase cytogenetics of hematological cancer: comparison of classical karyotyping and in situ hybridization using a panel of eleven chromosome specific DNA probes. Cancer Res. 51, 1959–1967 (1991).
Lee, W., Han, K., Drut, R. M., Harris, C. P. & Meisner, L. F. Use of fluorescence in situ hybridization for retrospective detection of aneuploidy in multiple myeloma. Genes Chromosomes Cancer. 7, 137–143 (1993).
Drach, J. et al. Multiple myeloma: high incidence of chromosomal aneuploidy as detected by interphase fluorescence in situ hybridization. Cancer Res. 55, 3854–3859 (1995).
Konigsberg, R. et al. Predictive role of interphase cytogenetics for survival of patients with multiple myeloma. J. Clin. Oncol. 18, 804–812 (2000).
Palumbo, A. et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J. Clin. Oncol. 33, 2863–2869 (2015).
Hanamura, I. et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood 108, 1724–1732 (2006).
Yu, Y. et al. Variability in cytogenetic testing for multiple myeloma: a comprehensive analysis from across the United States. JCO Oncol. Pract. 16, e1169–e1180 (2020).
Kuehl, W. M. & Bergsagel, P. L. Multiple myeloma: evolving genetic events and host interactions. Nat. Rev. Cancer. 2, 175–187 (2002).
Smadja, N. V. et al. Further cytogenetic characterization of multiple myeloma confirms that 14q32 translocations are a very rare event in hyperdiploid cases. Genes Chromosomes Cancer. 38, 234–239 (2003).
Fonseca, R. et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood 102, 2562–2567 (2003).
Neben, K. et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol. 31, 4325–4332 (2013).
Bustoros, M. et al. Genomic profiling of smoldering multiple myeloma identifies patients at a high risk of disease progression. J Clin Oncol. 38, 2380–2389 (2020).
Chang, H. et al. 1p21 deletions are strongly associated with 1q21 gains and are an independent adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma. Bone Marrow Transplant. 45, 117–121 (2010).
Fonseca, R. et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 20, 2034–2040 (2006).
Schmidt, T. M. et al. Gain of Chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J. 9, 94 (2019).
Brito-Babapulle, V. & Atkin, N. B. Break points in chromosome #1 abnormalities of 218 human neoplasms. Cancer Genet. Cytogenet. 4, 215–225 (1981).
Le Baccon, P. et al. Novel evidence of a role for chromosome 1 pericentric heterochromatin in the pathogenesis of B-cell lymphoma and multiple myeloma. Genes Chromosomes Cancer 32, 250–264 (2001).
Sawyer, J. R., Tricot, G., Mattox, S., Jagannath, S. & Barlogie, B. Jumping translocations of chromosome 1q in multiple myeloma: evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood 91, 1732–1741 (1998).
Sawyer, J. R. et al. An acquired high-risk chromosome instability phenotype in multiple myeloma: Jumping 1q Syndrome. Blood Cancer J. 9, 62 (2019).
Rustad, E. H. et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov. 1, 258–273 (2020).
Debes-Marun, C. S. et al. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma. Leukemia 17, 427–436 (2003).
Shaughnessy, J. D. Jr. et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 109, 2276–2284 (2007).
Shaughnessy, J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27Kip1 and an aggressive clinical course in multiple myeloma. Hematology. 10, 117–126 (2005).
Chang, H. et al. Multiple myeloma patients with CKS1B gene amplification have a shorter progression-free survival post-autologous stem cell transplantation. Br. J. Haematol. 135, 486–491 (2006).
Spruck, C. et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol. Cell. 7, 639–650 (2001).
Shi, L. et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget. 1, 22–33 (2010).
Craig, R. W. MCL-1 maps to human chromosome 1q21. Genomics 23, 457–463 (1994).
Zhang, B., Gojo, I. & Fenton, R. G. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 99, 1885–1893 (2002).
Peperzak, V. et al. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 14, 290–297 (2013).
Matulis, S. M. et al. Preclinical activity of novel MCL1 inhibitor AZD5991 in multiple myeloma. Blood 132, 952 (2018).
Samo, A. A. et al. MCL1 gene co-expression module stratifies multiple myeloma and predicts response to proteasome inhibitor-based therapy. Genes Chromosomes Cancer 57, 420–429 (2018).
Gupta, V. A. et al. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood 129, 1969–1979 (2017).
Pulkki, K. et al. Soluble interleukin-6 receptor as a prognostic factor in multiple myeloma. Finnish Leukaemia Group. Br. J. Haematol. 92, 370–374 (1996).
Stephens, O. W. et al. An intermediate-risk multiple myeloma subgroup is defined by sIL-6r: levels synergistically increase with incidence of SNP rs2228145 and 1q21 amplification. Blood 119, 503–512 (2012).
Stasi, R. et al. The prognostic value of soluble interleukin-6 receptor in patients with multiple myeloma. Cancer 82, 1860–1866 (1998).
Lazzari, E. et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 8, 1922 (2017).
Teoh, P. J. et al. Aberrant hyperediting of the myeloma transcriptome by ADAR1 confers oncogenicity and is a marker of poor prognosis. Blood 132, 1304–1317 (2018).
Phaik, Ju. T., Tae-Hoon, C., Pamela, Y. Z. C., Sabrina, H. M. T. & Wee Joo, C. IL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematologica 105, 1391–1404 (2020).
Inoue, J. et al. Overexpression of PDZK1 within the 1q12-q22 amplicon is likely to be associated with drug-resistance phenotype in multiple myeloma. Am. J. Pathol. 165, 71–81 (2004).
Ota, A. et al. Novel interleukin-6 inducible gene PDZ-binding kinase promotes tumor growth of multiple myeloma cells. J. Interferon Cytokine Res. 40, 389–405 (2020).
Merz, M. et al. Cytogenetic subclone formation and evolution in progressive smoldering multiple myeloma. Leukemia 34, 1192–1196 (2020).
Dhodapkar, M. V. et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120). Blood 123, 78–85 (2014).
Moreau, P. et al. Tandem autologous stem cell transplantation in high-risk de novo multiple myeloma: final results of the prospective and randomized IFM 99-04 protocol. Blood 107, 397–403 (2006).
Avet-Loiseau, H. et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J. Clin. Oncol. 30, 1949–1952 (2012).
Avet-Loiseau, H. et al. Prognostic significance of copy-number alterations in multiple myeloma. J. Clin. Oncol. 27, 4585–4590 (2009).
Nemec, P. et al. Gain of 1q21 is an unfavorable genetic prognostic factor for multiple myeloma patients treated with high-dose chemotherapy. Biol. Blood Marrow Transplant. 16, 548–554 (2010).
Morgan, G. J. et al. Cyclophosphamide, thalidomide, and dexamethasone (CTD) as initial therapy for patients with multiple myeloma unsuitable for autologous transplantation. Blood 118, 1231–1238 (2011).
Morgan, G. J. et al. Cyclophosphamide, thalidomide, and dexamethasone as induction therapy for newly diagnosed multiple myeloma patients destined for autologous stem-cell transplantation: MRC Myeloma IX randomized trial results. Haematologica 97, 442–450 (2012).
Boyd, K. D. et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC Myeloma IX trial. Leukemia 26, 349–355 (2012).
Pawlyn, C. et al. Coexistent hyperdiploidy does not abrogate poor prognosis in myeloma with adverse cytogenetics and may precede IGH translocations. Blood. 125, 831–840 (2015).
Neben, K. et al. Combining information regarding chromosomal aberrations t(4;14) and del(17p13) with the International Staging System classification allows stratification of myeloma patients undergoing autologous stem cell transplantation. Haematologica 95, 1150–1157 (2010).
Chng, W. J. et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 28, 269–277 (2014).
Abdallah, N. et al. Clinical characteristics and treatment outcomes of newly diagnosed multiple myeloma with chromosome 1q abnormalities. Blood Adv. 4, 3509–3519 (2020).
Giri, S. et al. Chromosome 1 abnormalities and survival of patients with multiple myeloma in the era of novel agents. Blood Adv. 4, 2245–2253 (2020).
Varma, A. et al. Outcome of multiple myeloma with Chromosome 1q gain and 1p deletion after autologous hematopoietic stem cell transplantation: propensity score matched analysis. Biol. Blood Marrow Transplant. 26, 665–671 (2020).
Locher, M. et al. The prognostic value of additional copies of 1q21 in multiple myeloma depends on the primary genetic event. Am. J. Hematol. 95, 1562–1571 (2020).
Walker, B. A. et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 33, 159–170 (2018).
Shah, V. et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular profiles of 1905 trial patients. Leukemia 32, 102–110 (2018).
D’Agostino, M. et al. Impact of gain and amplification of 1q in newly diagnosed multiple myeloma patients receiving carfilzomib-based treatment in the forte trial. Blood 136, 38–40 (2020).
Neben, K. et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood 119, 940–948 (2012).
Cavo, M. et al. Bortezomib-thalidomide-dexamethasone is superior to thalidomide-dexamethasone as consolidation therapy after autologous hematopoietic stem cell transplantation in patients with newly diagnosed multiple myeloma. Blood 120, 9–19 (2012).
Shaughnessy, J. D. Jr. et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with Total Therapy 3. Blood 118, 3512–3524 (2011).
An, G. et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica 99, 353–359 (2014).
Chen, M. H., Qi, C., Reece, D. & Chang, H. Cyclin kinase subunit 1B nuclear expression predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with bortezomib. Hum. Pathol. 43, 858–864 (2012).
Chang, H. et al. Impact of genomic aberrations including chromosome 1 abnormalities on the outcome of patients with relapsed or refractory multiple myeloma treated with lenalidomide and dexamethasone. Leuk. Lymphoma 51, 2084–2091 (2010).
Nahi, H. et al. Proteasome inhibitors and IMiDs can overcome some high-risk cytogenetics in multiple myeloma but not gain 1q21. Eur. J. Haematol. 96, 46–54 (2016).
Smetana, J. et al. Gain(1)(q21) is an unfavorable genetic prognostic factor for patients with relapsed multiple myeloma treated with thalidomide but not for those treated with bortezomib. Clin. Lymphoma Myeloma Leuk. 13, 123–130 (2013).
Huang, J. et al. NEDD8 inhibition overcomes CKS1B-induced drug resistance by upregulation of p21 in multiple myeloma. Clin. Cancer Res. 21, 5532–5542 (2015).
Zhang, Q. et al. Pharmacogenomic study of carfilzomib in relapsed/refractory multiple myeloma suggests that carfilzomib is a more potent proteasome inhibitor than bortezomib. Blood 120, 5015 (2012). -.
Korde, N. et al. Treatment with carfilzomib-lenalidomide-dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. 1, 746–754 (2015).
Jasielec, J. et al. Carfilzomib, lenalidomide, and dexamethasone plus transplant in newly diagnosed multiple myeloma. Blood 136, 2513–2523 (2020).
Kumar, S. K. et al. Carfilzomib or bortezomib in combination with lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma without intention for immediate autologous stem-cell transplantation (ENDURANCE): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 21, 1317–1330 (2020).
Bock, F. et al. Outcome of patients with multiple myeloma and CKS1B gene amplification after autologous hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 22, 2159–2164 (2016).
Marchesini, M. et al. ILF2 is a regulator of RNA splicing and DNA damage response in 1q21-amplified multiple myeloma. Cancer Cell. 32, 88–100.e6 (2017).
Scott, E. C. et al. Post-transplant outcomes in high-risk compared with non-high-risk multiple myeloma: a CIBMTR analysis. Biol. Blood Marrow Transplant. 22, 1893–1899 (2016).
Facon, T. et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N. Engl. J. Med. 380, 2104–2115 (2019).
Mateos, M. V. et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N. Engl. J. Med. 378, 518–528 (2018).
Moreau, P. et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet 394, 29–38 (2019).
Voorhees, P. M. et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood 136, 936–945 (2020).
Premkumar, V., Pan, S., Lentzsch, S. & Bhutani, D. Use of daratumumab in high risk multiple myeloma: a meta-analysis. eJHaem. 1, 267–271 (2020).
Giri, S. et al. Evaluation of daratumumab for the treatment of multiple myeloma in patients with high-risk cytogenetic factors: a systematic review and meta-analysis. JAMA Oncol. 6, 1–8 (2020).
Mohan, M. et al. Daratumumab in high-risk relapsed/refractory multiple myeloma patients: adverse effect of chromosome 1q21 gain/amplification and GEP70 status on outcome. Br. J. Haematol. 189, 67–71 (2020).
Richardson P. et al. Isatuximab plus pomalidomide and dexamethasone in relapsed/refractory multiple myeloma patients with 1q21 gain: insights from phase 1 and phase 3 studies. EHA Library EP1017 (2020).
Ogiya, D. et al. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: therapeutic implications. Blood 136, 2334–2345 (2020).
Benboubker, L. et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N. Engl. J. Med. 371, 906–917 (2014).
Holstein, S. A. et al. Updated analysis of CALGB (Alliance) 100104 assessing lenalidomide versus placebo maintenance after single autologous stem-cell transplantation for multiple myeloma: a randomised, double-blind, phase 3 trial. Lancet Haematol. 4, e431–e442 (2017).
McCarthy, P. L. et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J. Clin. Oncol. 35, 3279–3289 (2017).
Chakraborty, R. et al. Outcomes of maintenance therapy with lenalidomide or bortezomib in multiple myeloma in the setting of early autologous stem cell transplantation. Leukemia 32, 712–718 (2018).
Nooka, A. K. et al. Consolidation and maintenance therapy with lenalidomide, bortezomib and dexamethasone (RVD) in high-risk myeloma patients. Leukemia 28, 690–693 (2014).
Usmani, S. Z. et al. Bortezomib, lenalidomide, and dexamethasone with or without elotuzumab in patients with untreated, high-risk multiple myeloma (SWOG-1211): primary analysis of a randomised, phase 2 trial. Lancet Haematol. 8, e45–e54 (2021).
Kumar, S. et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 130, 2401–2409 (2017).
Tron, A. E. et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 9, 5341 (2018).
Kotschy, A. et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482 (2016).
Nguyen, M. et al. Obatoclax is a direct and potent antagonist of membrane-restricted Mcl-1 and is synthetic lethal with treatment that induces Bim. BMC Cancer 15, 568 (2015).
Li, J. et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by Anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell 31, 383–395 (2017).
Cohen, A. D. et al. Initial clinical activity and safety of BFCR4350A, a FcRH5/CD3 T-cell-engaging bispecific antibody, in relapsed/refractory multiple myeloma. Blood. 136, 42–43 (2020).
Shi, W. et al. CKS1B as drug resistance-inducing gene-A potential target to improve cancer therapy. Front. Oncol. 10, 582451 (2020).
Joseph, N. S. et al. Long-term follow-up results of lenalidomide, bortezomib, and dexamethasone induction therapy and risk-adapted maintenance approach in newly diagnosed multiple myeloma. J. Clin. Oncol. 38, 1928–1937 (2020).
Costa, L. J. & Usmani, S. Z. Defining and managing high-risk multiple myeloma: current concepts. J. Natl Compr. Canc. Netw. 18, 1730 (2020).
Costa, L. J. et al. Daratumumab, carfilzomib, lenalidomide and dexamethasone (Dara-KRd) induction, autologous transplantation and post-transplant, response-adapted, measurable residual disease (MRD)-based Dara-Krd consolidation in patients with newly diagnosed multiple myeloma (NDMM). Blood 134, 860 (2019).
Goldschmidt, H. et al. Bortezomib before and after high-dose therapy in myeloma: long-term results from the phase III HOVON-65/GMMG-HD4 trial. Leukemia 32, 383–390 (2018).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
T.M.S. declares no conflict of interest. R.F. served as a consultant for AbbVie, Aduro, Amgen, Bayer, BMS/Celgene, GlaxoSmithKline, Janssen, Juno, Kite, Merck, Novartis, OncoTracker, Pharmacyclics, Sanofi, and Takeda; and participated in an advisory board for Adaptive Biotechnologies, Caris Live Sciences, and OncoTracker. S.Z.U. served as a consultant/advisor for Amgen, Bristol-Myers Squibb, Celgene, Janssen, Merck, SkylineDx, and Takeda; participated in a Speakers Bureau for Amgen, Celgene, Janssen, Sanofi, and Takeda; and received research grants from Amgen, Array Biopharma, Bristol-Myers Squibb, Celgene, Janssen, Merck, Pharmacyclics, Sanofi, and Takeda.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schmidt, T.M., Fonseca, R. & Usmani, S.Z. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 11, 83 (2021). https://doi.org/10.1038/s41408-021-00474-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-021-00474-8
This article is cited by
-
Real-world advantage and challenge of post-autologous stem cell transplantation MRD negativity in high-risk patients with double-hit multiple myeloma
BMC Cancer (2024)
-
Fluorescence in situ hybridization reveals the evolutionary biology of minor clone of gain/amp(1q) in multiple myeloma
Leukemia (2024)
-
1q amplification and PHF19 expressing high-risk cells are associated with relapsed/refractory multiple myeloma
Nature Communications (2024)
-
Fc receptor-like 5 (FCRL5)-directed CAR-T cells exhibit antitumor activity against multiple myeloma
Signal Transduction and Targeted Therapy (2024)
-
Alterations in chromosome 1q in multiple myeloma randomized clinical trials: a systematic review
Blood Cancer Journal (2024)
