
Abstract
CD19-targeted chimeric antigen receptor (CAR) T cell therapy is an effective treatment for diffuse large B cell lymphoma (DLBCL). In addition to cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity (ICANS), B cell aplasia and hypogammaglobulinemia are common toxicities predisposing these patients to infections. We analyzed 60 patients with DLBCL treated with FDA-approved CD19 CAR T cells and report the incidence, risk factors, and management of infections during the first year after treatment. A total of 101 infectious events were observed, including 25 mild, 51 moderate, 23 severe, 1 life-threatening, and 1 fatal infection. Bacteria were the most common causative pathogens. The cumulative incidence of overall, bacterial, severe bacterial, viral, and fungal infection at 1 year were 63.3%, 57.2%, 29.6%, 44.7%, and 4%, respectively. In multivariate analyses, the use of systemic corticosteroids for the management of CRS or ICANS was associated with an increased risk of infections and prolonged admission. Impaired performance status and history of infections within 30 days before CAR T cell therapy was a risk factor for severe bacterial infection. In conclusion, infections were common within the first 60 days after CAR T cell therapy, however, they were not associated with an increased risk of death.
Similar content being viewed by others


Introduction
CD19-directed chimeric antigen receptor (CAR) T cell is a major breakthrough that has revolutionized the treatment paradigm of relapsed/refractory (RR) diffuse large B cell lymphoma (DLBCL) over the past recent years1,2. Despite the significant anti-lymphoma activity, CD19 CAR T cells possess unique toxicities. Besides immune-mediated toxicities, B cell aplasia, and resultant hypogammaglobulinemia are common consequences of CD19 CAR T cell therapy, which put patients at risk for infectious complications3,4. Although there have been some initial reports on the infectious complications of CAR T cell therapy, most studies included patients treated in clinical trials or with multiple underlying B cell malignancies5,6,7,8,9,10. Currently, there are limited real-world data on infectious risks in patients treated with CD19 CAR T cell therapy for DLBCL. Moreover, little is known about proper prophylaxis and management strategies for these patients. Herein, we describe the pattern, incidence, impact of infections, including infection prophylactic strategies, in patients with DLBCL who received FDA-approved CAR T cell therapy at Memorial Sloan Kettering Cancer Center (MSKCC).
Sample and methods
The study cohort included 60 consecutive patients with RR DLBCL who received FDA-approved CAR T cell therapy (axicabtagene ciloleucel—Yescarta; Kite Pharma, Santa Monica, CA or tisagenlecleucel—Kymriah; Novartis, Basel, Switzerland) at MSKCC between January 2018 and June 2019. Baseline clinical characteristics, patterns of antimicrobial prophylaxis, treatment of infection, and laboratory data, including blood count, CD4 lymphocyte, and immunoglobulin (Ig) level before lymphodepletion (LD) chemotherapy were abstracted from the electronic health records (EHR). Systemic bridging therapy were classified to intensive or non-intensive regimens. Intensive regimens included multi-agent immunochemotherapy e.g. CHOP-like, bendamustine-based, gemcitabine-based, high dose cytarabine-based, and ICE regimens. Non-intensive regimens included single-agent rituximab, immunomodulatory agent or small molecule inhibitor. The LD chemotherapy before CAR T cell infusion was selected based on recommended regimens in the package insert of each approved CAR T product. In patients who received axicabtagene ciloleucel, LD chemotherapy consisted of fludarabine 30 mg/m2, and cyclophosphamide 500 mg/m2 daily for 3 days. For patients receiving tisagenlecleucel, fludarabine 25 mg/m2 and cyclophosphamide 250 mg/m2 daily for 3 days or bendamustine 90 mg/m2 daily for 2 days was given for LD.
Prior to March 2019, there was no formal institutional guideline for antimicrobial prophylaxis or infection surveillance in patients treated with CAR T cell therapy, and treatment regimen was based on the autologous stem cell transplant (SCT) protocol or left to the primary physician’s preference. Standardized guidelines for CAR T cell patients were implemented in March 2019 and included acyclovir for herpes simplex virus (HSV) prophylaxis, fluconazole for antifungal prophylaxis, and trimethoprim/sulfamethoxazole or aerosolized pentamidine for Pneumocystis jiroveci prophylaxis (Table 1).
All infections were documented from the day of CAR T cell infusion through 1-year post-CAR T cell therapy, last follow-up, or relapse/progression, whichever came first. Infection events included both confirmed infections in which causative pathogens were identified, and probable infections diagnosed by the presence of fever plus localized physical exam and/or radiological findings. Any episode of culture-negative neutropenic fever in the absence of localized infection within the first 30 days after CAR T cell infusion was excluded from the analysis owing to the high probability of overlap with cytokine release syndromes (CRS). We classified types of infection according to causal pathogens, including bacterial, viral, fungal, and protozoal infection. Bacterial infections were further classified into organ-specific infection or bacteremia without localizing organ involvement. Infection severity was graded as mild, moderate, severe, life-threatening, or fatal, according to published criteria5,11. Mild infection was defined as not requiring antimicrobial therapy. Moderate infection entailed therapy with an oral antimicrobial medication. Severe infection was defined as infection requiring receipt of intravenous antimicrobial treatment. Life-threatening infection was defined as the presence of end-organ or cardiovascular compromise. Cumulative incidence of any infection, bacterial infection, severe bacterial infection, and viral infection were reported and separated by time after CAR T cell infusion (0–30 days, 31–100 days, 101–180 days, and 181–365 days). The data cutoff for statistical analysis was December 31, 2019.
Statistical analysis
We reported continuous variables using median and range. Categorical data were presented as a percentage. Overall survival was analyzed by Kaplan–Meier methodology and infections were treated as time-dependent covariates. Cumulative incidence of time to the first infection was evaluated with progression of disease, relapse, and death from non-infection causes as competing events. Factors associated with infection were identified by univariate analysis using cause-specific hazard ratios and 95% confidence intervals. CRS, immune effector cell-associated neurotoxicity syndrome (ICANS), corticosteroid, tocilizumab, and intravenous immunoglobulin (IVIG) were treated as time-dependent covariates. P-values less than 0.10 were considered for multivariate analysis using cause-specific hazard ratios. All statistical analyses were performed by R program version 3.6.0. The cmprsk package was used for the cumulative incidence of infection. The institutional review board and the ethic committee of MSKCC granted approval for conducting the study.
Results
Baseline clinical characteristics
Table 2 summarizes the baseline clinical characteristics of the 60 patients in this cohort. The median age at the time of CAR T treatment was 63 years (19.5–85.9 years). Thirty-five patients (58%) had de novo DLBCL. Patients had a median of 42,3,4,5,6,7,8,9 prior lines of treatment before CAR T cells, and 16 (26.7%) underwent prior hematopoietic cell transplantation. Thirty-eight patients (63.3%) received bridging therapy before CAR T cells (4 radiation therapy, 33 immunochemotherapy and 1 combined modality). Forty-three patients (71.7%) were treated with axicabtagene ciloleucel, and 17 (28.3%) received tisagenlecleucel. The median length of hospital stay for CAR T cell admission was 17 days (0–72 days). Thirteen patients (21.7%) received CAR T cell therapy after the institutional antimicrobial prophylactic protocol was implemented.
Baseline infection and antimicrobial prophylaxis
Nineteen patients (31.7%) received systemic antibacterial treatment for an infection within 30 days before the CAR T cell infusion. Three of these patients continued antibiotics through the admission of CAR T cell therapy. Thirty-one patients (51.7%) received antibacterial prophylaxis (Supplementary Table S1). All patients received antiviral prophylaxis for HSV, and 6 received entecavir due to the positive hepatitis B core antibody. Fifty-five patients (91.7%) were given prophylaxis for Pneumocytis jiroveci. Forty-eight patients (80%) received antifungal prophylaxis, 26 were initiated before CAR T cell infusion whereas the other 22 had antifungal prophylaxis started once absolute neutrophil count (ANC) was less than 500 µ/mL (median time from CAR T infusion to antifungal prophylaxis initiation of 7 days).
Recovery of leukocytes and immunoglobulin levels (Table 3)
Baseline median ANC and absolute lymphocyte counts (ALC) before LD chemotherapy were 3850 (200–10,600) and 600 (100–2700) cells/µL, respectively. Two patients had grade 4 neutropenia (ANC < 500 cells/µL) before LD therapy, as defined by CTCAE version 5.012. Forty-seven of the remaining 58 patients (81.0%) developed neutropenia grade 4 after LD therapy. The median duration of neutropenia grade 4 was 12 days (3–66 days). Ten patients had grade 4 neutropenia after day 30 (5 of which had persistent grade 4 neutropenia from the first 30 days). Thirty patients required at least one dose of growth factor support after CAR T cell therapy. Grade 3–5 lymphopenia beyond day 30 after CAR T cell was observed in 35 patients (58.3%). Of 19 patients who had lymphocyte subset analysis at day 30, all had B cell aplasia and the median CD4+ lymphocyte count was 116 cells/µL (41–630 cells/µL). Supplementary Table S2 summarizes the status of leukocyte subset reconstitution after CAR T cell infusion.
In 59 patients with available baseline IgG level before LD chemotherapy, the median IgG level was 487 mg/dL (163–1399 mg/dL), and 15 patients (25%) had baseline hypogammaglobulinemia (IgG ≤ 400 mg/dL). Immunoglobulin levels were checked at day 30 after CAR T cell therapy in 32 patients; 14 (43.8%) had hypogammaglobulinemia. An additional 12 patients (37.5%) developed hypogammaglobulinemia at later follow-up timepoints. Nineteen patients (31.7%) received at least 1 dose of IVIG replacement, including 10 (52.6%) who had IVIG after history of recurrent infections. Figure 1 illustrates patterns of leukocyte and IgG level during the CD19 CAR T cell treatment course.

Immune status at baseline before lymphodepletion and recovery by time post chimeric antigen receptor T cell therapy (number in the boxplot indicates median value of each parameter at each timepoint). a Immunoglobulin G (IgG) Level. b Absolute neutrophil count. c Absolute lymphocyte count. d CD4 lymphocyte count. D Day, mo Month.
CRS and ICANS
CRS was observed in 48 patients (80%) (grade ≥ 3 in 7 patients; 11.7%) at a median onset of 2 days after CAR T cell infusion (0–11 days). ICANS was observed in 24 patients (40%) and was grade ≥3 in 13 patients. The median onset of ICANS was 5 days after infusion. Of patients who developed CRS or ICANS, 25 (53.2%) received systemic corticosteroid with a median duration of 4 days (1–58 days). The median prednisone equivalent dose intensity of corticosteroid was 1.12 mg/kg/day (0.33–4.23 mg/kg/day) with the corresponding median cumulative dose of 380 mg (66.7–4586 mg) or 8.1 mg/kg (0.33–69.6 mg/kg) prednisone equivalent. Patients who received systemic corticosteroid had longer median hospital stay than patients who did not (27 days vs. 14 days, P < 0.001).
Baseline c-reactive protein, procalcitonin, and IL-6 before CAR T cell infusion were elevated in 52 (86.7%), 4 (7.4%), and 38 (63.3%) patients, respectively. The median peak procalcitonin level was 0.46 (0.05–27.65) µg/L. The trends of procalcitonin and IL-6 are shown in Supplementary Figs. S1 and S2, respectively.
Incidence, characteristics, and patterns of infection after CAR T cell therapy
Fifty-two of 60 patients (86.7%) developed neutropenic fever within the first 30 days after CAR T cell infusion. With the median follow-up of 6 months (0.8–12 months), after excluding neutropenic fever without identified pathogen or localizing organ, there was a total of 101 infection events (60 bacterial, 38 viral, 2 fungal, and 1 protozoal) in 40 patients during the entire study period. Pathogenic organism were identified in 73 infection events (72%) (60% of bacterial, 92.1% of viral, 50% of fungal, and 100% of protozoal infection). Thirty-seven infection episodes (34.6%) occurred within the first 30 days after CAR T cell therapy. Of 101 events, 23 (22.8)% infections were classified as severe, 1 (1.0%) as life-threatening (Escherichia coli biliary sepsis), and 1 (1.0%) fatal (influenza A pneumonia). Of all 101 infection events, 32 occurred during the initial CAR T cell admission. Among the other 69 infection episodes (bacterial; n = 42, viral; n = 26, fungal; n = 1), which occurred following hospital discharge from CAR T cell therapy, 21 (bacterial; n = 14, viral; n = 6, fungal; n = 1) required hospital readmission with the median hospital stay of 5 (2–37) days.
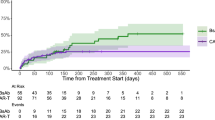
Figure 2 shows the distribution of bacterial or viral infection at each period post-CAR T cell infusion. The distribution of organ involvement and causative organism of bacterial infection are shown in Fig. 3 and Supplementary Table S3. The 1-year cumulative incidence of all infections, bacterial, viral, and fungal infections were 63.3, 57.2, 44.7%, and 4.0%, respectively (Fig. 4, Table 4).

Distribution of bacterial and viral infection by time post chimeric antigen receptor T cell and severity.

a By localization—primary bacteremia vs. Localized infection. b Identified organism in localized bacterial infection. c Localized bacterial infection by involved organs.

a Any infection. b Bacterial Infection. c Severe Bacterial Infection. d Viral Infection.
Infection within the first 30 days of CAR T cell infusion
Of 37 infection episodes that occurred within the first 30 days after CAR T cell infusion (Supplementary Tables S3–S6), bacterial infections were the most frequent with 25 events (15 moderate, 10 severe) in 20 patients. The median onset of the first bacterial infection was day 12 (0–30). A total of 15 events were organ-specific infections, whereas 10 were primary bacteremias. Organisms were identified in 19 definite infectious episodes with Clostridium difficile (colitis) being the most common causative bacterial pathogen (n = 7). The other 6 events were probable infections (including 3 lobar pneumonia, 3 soft tissue infection). Piperacillin/tazobactam was the most common empirical anti-bacterial agent for neutropenic fever during the first 30 days in 38 patients (63.3%), followed by cefepime in 7 patients.
Ten viral infections occurred during the first 30 days (8 mild, 2 moderate) with the median onset at day 8 (0–26 days). Viral pathogens included respiratory syncytial virus (n = 5), cytomegalovirus (n = 2), polyoma BK virus (n = 2), and norovirus (n = 1). Both cytomegalovirus infection were viremia without end organ involvement. There was 1 probable invasive aspergillosis pulmonary infection (elevated serum galactomannan antigen and consistent radiographic imaging), and 1 protozoal infection (Cryptosporidium parvum).
Infection after day 30 post CAR T cell infusion
Thirty-five bacterial infections were observed in 16 patients after day 30, including 12 events during day 31–100, 7 during day 101–180, and 16 beyond day 180 (Supplementary Tables S3–S6). Six of these 16 patients had previous bacterial infection within the first 30 days. Among 35 bacterial infections, there were 26 moderate, 8 severe, and 1 fatal infection. Ninety-seven percent were organ-specific infections with urinary tract infection being the most common presentation (n = 11). Of 28 viral infections, 10 occurred during day 31–100, 8 occurred during day 101–180, and 10 occurred after day 180. Approximately 50% of viral infections were of mild severity (n = 17). Respiratory tract infections (n = 21) were the most common with rhinovirus being the most frequently recovered. There were 1 cytomegalovirus reactivation (viremia without organ dysfunction), 2 BK virus cystitis, and 2 herpes zoster reactivation (both of whom were on acyclovir prophylaxis). Pneumocystis jiroveci infection was identified in 1 patient at 9 months after CAR T cell infusion (4 months after pentamidine prophylaxis discontinuation).
Risk factors associated with infection
In the univariate analysis, impaired baseline performance status, ICANS grade ≥2, and systemic corticosteroid exposure after CAR T cell infusion were associated with a higher incidence of overall infection (Table 5). In multivariate analysis, systemic corticosteroid was the only risk factor of infectious complications. Impaired performance status and previous infection 30 days before LD chemotherapy was independent predictors for severe bacterial infection (HR 3.98, 95% CI 1.3–12.2). Patients with low IgG before LD chemotherapy had higher risk of viral infection after CAR T cells (HR 5.7, 95% CI 2.3–14.3; Supplementary Table S7), however, IVIG replacement did not decrease the incidence of infection. CRS, tocilizumab administration, and procalcitonin were not associated with infection or severe bacterial infection. The incidence of infection was comparable between 47 patients who received CAR T cell therapy before March 2019 and 13 patients who were treated with CAR T cells after March 2019, when the standardized antimicrobial prophylaxis guideline was implemented (HR 1.22, 95% CI 0.6–2.5).
Impact of infection on patients’ survival outcomes
Of all infectious complications, one resulted in death attributed to influenza pneumonia despite a 10-day course of oseltamivir treatment. There was no association between infectious complications and mortality risk in CAR T cell-treated patients when analyzed by univariate cox regression.
Discussion
Our study reported comprehensive real-world data on infectious complications in DLBCL patients treated with commercially available CD19 CAR T cell products. Data from the pivotal studies of CD19 CAR T cell therapy in DLBCL demonstrated an incidence of 15–30% for severe infection13,14,15. The incidence of overall infection in our study was comparable to these landmark trials. Moreover, the patterns of infection in our cohort were similar to the findings from previous reports5,7,10. Bacterial and viral infections were commonly observed, with bacteria being the most common pathogen, especially during the first 30 days5,7,10. Recently, Cordeiro and colleagues reported the incidence of adverse events beyond day 90 from CAR T cell infusion16 and described a relatively low incidence of late infections (2.08 per patient-year), with most being of mild to moderate severity. Our study observed similar results with 71% of all infections considered mild to moderate. Upper respiratory tract infections were the most common infectious events. Serious infections occurred in 23.4% of DLBCL patients treated with commercial CD19 CAR T cell products; nonetheless, most infections were manageable and infection-related mortality was low similar to the results of previous reports5,7,17. In our study, one patient died from influenza A pneumonia at day +159 despite treatment with oseltamivir. Fungal infection was uncommon in patients treated with CAR T cells likely due to short duration of neutropenia5,18. One patient had Pneumocystis jirovecii pneumonia 4 months after pentamidine prophylaxis was stopped. Retrospectively, the patient’s CD4 count was 44 cells/µL at the time of infection, thus emphasizing the importance of implementing immune monitoring protocols to guide the duration of antimicrobial prophylaxis in these patients.
General practice for infection prophylaxis in patients treated with CAR T cell are heterogeneous and vary by institutions19. The recent European guidelines for antimicrobial prophylaxis and IVIG replacement in patients treated with CAR T cells20 was primarily based on the evidence from SCT patients21. Currently, the appropriate prophylactic approach in these patients remains unknown and requires further understanding of the immune reconstitution pattern and longer follow-up data. B cell aplasia is a well-known “off tumor and on target” phenomenon after CD19 CAR T cell therapy contributing to hypogammaglobulinemia in these patients. A quarter of patients in our cohort had IgG < 400 mg/dL, and 60% had IgG < 600 mg/dL. This finding was comparable to data from the JULIET trial14, which highlighted the baseline humoral immune defect in these patients. However, there are data showing persistent long-lived plasma cells after CAR T cell therapy22. In addition, Hill and colleagues recently demonstrated preserved anti-viral humoral immune response in patients treated with CD19 CAR T cells23. The authors reported sustained anti-measle IgG level independent of the total IgG level in 95% of patients. Moreover, overall anti-virome was preserved in most patients. Moreover, there was a low incidence of viral infection after day 90. In our study, there were 18 viral infectious episodes in 15 patients after day 100 with mild respiratory tract infection as the most common presentation similar to previous reports. Interestingly, we observed an increased risk of viral infection in patients with hypogammaglobulinemia, but no such correlation was seen with other types of infection (supplementary data). We hypothesize that IgG deficiency at baseline might indicate pre-existing depleted plasma cell and antibody repertoire, which may have a more critical impact on the ability to mount viral-specific neutralizing IgG and predispose patients to infection after CAR T cell therapy. The significance of hypogammaglobulinemia on the risk of infection in patients treated with CD19 CAR T cells warrants further study. In the ELIANA trial, all pediatric patients with precursor B acute lymphoblastic leukemia (ALL) received IVIG replacement. In contrast, the proportion of IVIG replacement among DLBCL patients treated in pivotal studies was lower and ranged around 20–60%14,15,24. Only 30% of our patients received IVIG replacement, of which half had a history of preceding recurrent infection after CAR T cell therapy. The primary malignancy probably has a critical contribution to the risk of infection attributed to underlying immune function and kinetics of immune recovery. Data from patients with B-ALL indicated that CD8+ lymphocyte recovered early whereas CD4+ lymphocyte had delayed recovery after CAR T cell therapy25. Further studies on infection prophylaxis, immunization, and immune reconstitution in CAR T cell-treated patients are warranted.
In our study, we identified systemic corticosteroid as a predictor for infection after CAR T cell therapy whereas history of infection within 30 days before CAR T cell infusion was associated with severe bacterial infection, which may contribute to longer hospital stays in these patients. This observation is similar to another previous retrospective study8. We did not see an association between CRS and infectious complications in our lymphoma cohort. Park and colleagues previously demonstrated severe CRS (grade ≥ 3) as a risk factor for bacterial infection in adult B-ALL treated with CD19 CAR T cells7,10. Other risk factors for infection after CAR T cell therapy shown by previous studies included higher number of prior treatments, higher doses of CAR T cells, older age, previous history of infection, and CD22-specific CAR T cells5,8. Finally, a recent study described the association between double peak IL-6 pattern (second surge of serum IL-6 after initial normalization) and life-threatening infection26. Along with the current interest of anti-IL6 therapy in severe acute respiratory syndrome coronavirus 2 patients with respiratory failure27,28,29,30,31,32,33,34, it is at least worth noting that we did not observe an association between the use of tocilizumab and infections in this cohort.
Our study has unique strengths. We comprehensively analyzed the real-world data on patterns of infection, detailed information on relevant immune status and prophylactic strategies during the first year after CAR T cell therapy in patients with DLBCL from a dedicated lymphoma patient cohort. We acknowledge several limitations of this study. Besides its retrospective nature, immune function monitoring and infection prophylaxis were not prospectively studied in a systematic manner. Lastly, the relatively small number of patients included in the study could limit its statistical power.
In summary, infection is common in DLBCL patients treated with CD19 CAR T cells. However, most events occurred early after CAR T cell therapy and were largely manageable. The mechanism of infection in these patients is complex and multifactorial. Appropriate infection prophylaxis in these patients remain to be determined, and prospective clinical trials are warranted.
References
Yanez, L., Sanchez-Escamilla, M. & Perales, M. A. CAR T cell toxicity: current management and future directions. Hemasphere. 3, e186 (2019).
Jain, T. et al. Use of chimeric antigen receptor T cell therapy in clinical practice for relapsed/refractory aggressive B cell non-Hodgkin lymphoma: an expert panel opinion from the american society for transplantation and cellular therapy. Biol Blood Marrow Transplant. 25, 2305–2321 (2019).
Nahas, G. et al. Persistent cytopenias after chimeric antigen receptor T-cell immunotherapy for CD19+ aggressive lymphoma: a single institution experience. Biol. Blood Marrow Transplant. 25, S180 (2019).
Fried, S. et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 54, 1643–1650 (2019).
Hill, J. A. et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 131, 121–130 (2018).
Zhu, F., Hu, Y., Guoqing, W., Huang, H. & Wu, W. Incidence and risk factors associated with infection after chimeric antigen receptor T cell therapy for relapsed/refractory B-cell malignancies. Blood. 134(Supplement_1), 3220 (2019).
Park, J. H. et al. Cytokine release syndrome grade as a predictive marker for infections in patients with relapsed or refractory B-cell acute lymphoblastic leukemia treated with chimeric antigen receptor T cells. Clin. Infect. Dis. 67, 533–540 (2018).
Mikkilineni, L. et al. Infectious complications associated with CAR T-cell therapy. Blood. 134(Supplement_1), 4449 (2019).
Syed, T. I. S. et al. Infections associated with CAR T therapy for treatment of hematological malignancies. Blood. 134(Supplement_1), 4442 (2019).
Vora, S. B. et al. Infectious complications following CD19 chimeric antigen receptor t-cell therapy for children, adolescents, and young adults. Open Forum Infect. Dis. 7, ofaa121 (2020).
Young, J. H. et al. Infections after transplantation of bone marrow or peripheral blood stem cells from unrelated donors. Biol. Blood Marrow Transplant. 22, 359–370 (2016).
Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf (2020).
Neelapu, S. S. et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Schuster, S. J. et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380, 45–56 (2019).
Abramson, J. S. et al. Pivotal safety and efficacy results from transcend NHL 001, a multicenter phase 1 study of Lisocabtagene Maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas. Blood. 134(Supplement_1), 241 (2019).
Cordeiro, A. et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biol. Blood Marrow Transplant. 26, 26–33 (2020).
Riedell, P. A. et al. A multicenter retrospective analysis of clinical outcomes, toxicities, and patterns of use in institutions utilizing commercial axicabtagene ciloleucel and tisagenlecleucel for relapsed/refractory aggressive B-cell lymphomas. Blood. 134(Supplement_1), 1599 (2019).
Haidar, G. et al. Invasive mold infections after chimeric antigen receptor-modified T-cell Therapy: a case series, review of the literature, and implications for prophylaxis. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciz1127 (2019).
Mahmoudjafari, Z. et al. American Society for Blood and Marrow Transplantation Pharmacy Special Interest Group Survey on chimeric antigen receptor T cell therapy administrative, logistic, and toxicity management practices in the United States. Biol. Blood Marrow Transplant. 25, 26–33 (2019).
Yakoub-Agha, I. et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica. 105, 297–316 (2020).
Bhella, S. et al. Choosing wisely BMT: American Society for Blood and Marrow Transplantation and Canadian Blood and Marrow Transplant Group’s list of 5 tests and treatments to question in blood and marrow transplantation. Biol. Blood Marrow Transplant. 24, 909–913 (2018).
Bhoj, V. G. et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. 128, 360–370 (2016).
Hill, J. A. et al. Durable preservation of antiviral antibodies after CD19-directed chimeric antigen receptor T-cell immunotherapy. Blood Adv. 3, 3590–3601 (2019).
Locke, F. L. et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 20, 31–42 (2019).
Wang, Y. et al. Kinetics of immune reconstitution after CD19 CAR-T cell therapy in ALL patients. Blood 134(Supplement_1), 1301 (2019).
Luo, H. et al. Inflammatory signatures for quick diagnosis of life-threatening infection during the CAR T-cell therapy. J. Immunother. Cancer. 7, 271 (2019).
Xu, X. et al. Effective treatment of severe COVID-19 patients with Tocilizumab. Proc Natl. Acad. Sci. USA 117, 10970–10975 (2020).
Tocilizumab in COVID-19 pneumonia (TOCIVID-19). https://ClinicalTrials.gov/show/NCT04317092 (2020).
A Study to evaluate the safety and efficacy of tocilizumab in patients with severe COVID-19 pneumonia. https://ClinicalTrials.gov/show/NCT04320615 (2020).
Favipiravir combined with Tocilizumab in the treatment of corona virus disease 2019. https://ClinicalTrials.gov/show/NCT04310228 (2020).
Tocilizumab vs CRRT in management of cytokine release syndrome (CRS) in COVID-19. https://ClinicalTrials.gov/show/NCT04306705 (2020).
Tocilizumab for SARS-CoV2 severe pneumonitis. https://ClinicalTrials.gov/show/NCT04315480 (2020).
Anti-il6 treatment of serious COVID-19 disease with threatening respiratory failure. https://ClinicalTrials.gov/show/NCT04322773 (2020).
Zhang, X. et al. First case of COVID-19 in a patient with multiple myeloma successfully treated with tocilizumab. Blood Adv. 4, 1307–1310 (2020).
Acknowledgements
This research was supported in part by NIH award P01 CA23766 and NIH/NCI Cancer Center Support Grant P30 CA008748. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. K.W. receives salary support from Parker Institute for Cancer Immunotherapy at Memorial Sloan Kettering Cancer Center. M.P. received salary support from an American Italian Cancer Foundation Post-Doctoral Research Fellowship and by AIL Milano e Provincia ONLUS.
Author information
Authors and Affiliations
Contributions
K.W., M.L.P., S.K.S., and M.A.P. designed the study and wrote the paper. K.W., M.P., and M.G.R. collected the data and conducted the analysis. M.L.S., A.A., and M.A.M. participated in data collection. M.P. and M.G.R participated in the data analysis. J.R.F. and S.M.D. conducted the statistical analysis. C.L.B., G.L.S., M.S., P.B.D., C.S.S., B.D.S., and E.M. took care of the patients. All the authors reviewed and approved the paper.
Corresponding author
Ethics declarations
Conflict of interest
K.W., M.P., J.R.F., S.M.D., A.A., M.L.S., M.A.M., and S.K.S. have no relevant conflict of interest. M.G.R. reports receiving honoraria from Takeda and Janssen Pharmaceutical. M.S. has served as a paid consultant for McKinsey & Company, Angiocrine Bioscience, Inc., and Omeros Corporation. He has received research funding from Angiocrine Bioscience, Inc. He has served on an ad hoc advisory board for Kite—A Gilead Company. G.L.S. receives research funding from Amgen and Janssen Pharmaceutical. C.L.B. serves as a paid consultant for Life Sci, GLG, Juno/Celgene, Seattle Genetics and Kite/Gilead. She reports receiving research funding from Janssen Pharmaceutical, Novartis, Epizyme, Xynomics and Bayer. She receives honorarium from Dava Oncology. M.L.P. has served on ad hoc advisory board for Kite and Novartis. P.B.D. serves on the advisory board for Kite/Gilead. C.S.S. has served as a paid consultant on advisory boards for Juno Therapeutics, Sanofi-Genzyme, Spectrum Pharmaceuticals, Novartis, Genmab, Precision Biosciences, Kite, a Gilead Company, Celgene, Gamida Cell and GSK. He has received research funds for clinical trials from Juno Therapeutics, Celgene, Precision Biosciences and Sanofi-Genzyme. B.D.S. provides consultancy for Kite/Gilead, Juno/Celgene and Janssen. She also receives research support from ADC Therapeutics. M.A.P. reports honoraria from Kite/Gilead, Abbvie, Bellicum, Bristol-Myers Squibb, Incyte, Merck, Novartis, Nektar Therapeutics, Omeros, and Takeda. He serves on DSMBs for Servier and Medigene, and the scientific advisory boards of MolMed and NexImmune. He has received research support for clinical trials from Incyte, Kite/Gilead and Miltenyi Biotec. He serves in a volunteer capacity as a member of the Board of Directors of the American Society for Transplantation and Cellular Therapy (ASTCT) and Be the Match (National Marrow Donor Program, NMDP), as well as on the CIBMTR Cellular Immunotherapy Data Resource (CIDR) Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wudhikarn, K., Palomba, M.L., Pennisi, M. et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 10, 79 (2020). https://doi.org/10.1038/s41408-020-00346-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-020-00346-7
This article is cited by
-
Infectious complications in pediatric patients undergoing CD19+CD22+ chimeric antigen receptor T-cell therapy for relapsed/refractory B-lymphoblastic leukemia
Clinical and Experimental Medicine (2024)
-
Cause-specific mortality in a population-level cohort of diffuse large B-cell lymphoma following chemotherapy in the early 21st century
Annals of Hematology (2024)
-
Management of chimeric antigen receptor T (CAR-T) cell-associated toxicities
Intensive Care Medicine (2024)
-
Fungal Infections Associated with CD19-Targeted Chimeric Antigen Receptor T Cell Therapy
Current Fungal Infection Reports (2023)
-
CAR-T Cells and the Kidney: Insights from the WHO Safety Database
BioDrugs (2023)
