
Abstract
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare, male-predominant hematologic malignancy with poor outcomes and with just one recently approved agent (tagraxofusp). It is characterized by the abnormal proliferation of precursor plasmacytoid dendritic cells (pDCs) with morphologic and molecular similarities to acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS)/chronic myelomonocytic leukemia (CMML) in its presentation within the bone marrow and peripheral blood. To identify disease-specific molecular features of BPDCN, we profiled the bone marrow, peripheral blood, and serum samples from primary patient samples using an in-house hematologic malignancy panel (“T300” panel), transcriptome microarray, and serum multiplex immunoassays. TET2 mutations (5/8, 63%) were the most prevalent in our cohort. Using the transcriptome microarray, genes specific to pDCs (LAMP5, CCDC50) were more highly expressed in BPDCN than in AML specimens. Finally, the serum cytokine profile analysis showed significantly elevated levels of eosinophil chemoattractants eotaxin and RANTES in BPDCN as compared with AML. Along with the high levels of PTPRS and dendritic nature of the tumor cells, these findings suggest a possible pre-inflammatory context of this disease, in which BPDCN features nonactivated pDCs.
Similar content being viewed by others
Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare, highly clinically aggressive hematologic malignancy thought to arise from proliferation of precursor plasmacytoid dendritic cells (pDC). Disease presentation includes skin lesions and disseminated disease in the bone marrow, peripheral blood, and lymph nodes1,2,3. Normal pDCs generate interferons during chronic viral infection, can present antigens, and have potential roles in reducing autoimmune responses4. However, little is known about this autoimmune aspect in the context of BPDCN. BPDCN has predominance in males (incidence of 0.05 versus 0.02 in females per 100,000 population), generally occurs in those older than 60, and short overall survivals (8–14 months median overall survivals) that decline with age5. Patients are predisposed to acute leukemia transformation, despite multi-agent chemotherapy5. After multiple attempts to reclassify the disease due to its protean manifestations, BPDCN was initially labeled under the umbrella of acute myeloid leukemia (AML) and associated neoplasms by the World Health Organization (WHO) in 2008. Then in 2016, it was re-classified by WHO under its own separate category under myeloid malignancies, highlighting its unique biology and clinical features6,7. In the absence of standard treatment, current chemotherapy regimens borrowed from lymphoma or acute leukemia have been the preferred approach treating patients with BPDCN. However, long-term effectiveness of these chemotherapy regimens and response vary greatly. One potential breakthrough in the field was the observation of near-universal expression of IL3Rα (IL3RA/CD123) in BPDCN, which led to the first approved therapy that uses a recombinant human interleukin 3α (IL3A) protein conjugated to diphtheria toxin (SL-401)8. Therefore, further characterization of its pathobiology is warranted. BPDCN is currently distinguished from AML using positive markers, such as: CD4, CD56, CD123, and TCL1, TCF-4, and CD303; and negative markers: CD3, CD8, CD13, CD19, CD34, and myeloperoxidase (MPO)9.
Similar to myeloid hematologic disorders AML and myelodysplastic syndromes (MDS), BPDCN patients acquire somatic point mutations in TET2 and TP53, ASXL1, IDH2, NRAS, and NPM1, with TET2 truncating mutations being the most prevalent and recurrent genomic alteration reported9,10,11,12. Also consistent with AML, the somatic missense and truncating mutations in TET2 are mutually exclusive with co-occurring DNMT3A and IDH1/2 in BPDCN11. Yet their differential responses to similar therapeutic regimens in clinical trial testing suggests that there are key underlying etiologies that are yet to be determined. We sought to further understand the pathobiologic differences between AML and BPDCN, with emphasis on molecular and cytokine analyses.
Materials and methods
Specimens
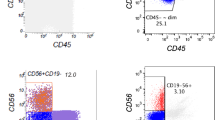
Collection of specimens was through a protocol approved by the UT MD Anderson Cancer Center Institutional Review Board that included informed consent for tissues used for research purposes. For DNA and RNA assays, we used specimens with >60% blasts, specimens with <60% blasts for which CD56+ flow sorting was successful. Several specimens had insufficient yields for the assays and could not be used. Two patients had mixed BPDCN/AML diagnoses at the time of specimen collection (BPDCN-1, BPDCN-4). We were able to sort for CD45 low blasts for BPDCN-1, but not the second patient BPDCN-4 due to specimen limitations. AML samples with TET2 mutations were identified by searching clinical records for physician-ordered gene-panel results. In total, we profiled bone marrow, peripheral blood and serum samples from primary patient samples of BPDCN (N = 16) and TET2-mutated AML (AMLTET2m) (N = 9) (Table 1).
Gene panel sequencing
Genomic DNA (gDNA) was extracted from eight peripheral blood and bone marrow samples of seven patients with BPDCN using the Frozen Tissue protocol 389 from the QIAamp DNA Mini kit (Qiagen, Inc., Valencia, CA). Two timepoints were sequenced for BPDCN-12. Sequencing was then performed on a new-generation version of our in-house gene panel composed of genes commonly associated with hematological malignancies13 using Illumina HiSeq 2000 (Illumina Inc., San Diego, CA) (Supplemental Table 1). An in-house virtual normal control was used to identify somatic point mutation and copy-number alterations as previously described13. Because our virtual common normal could not be gender-matched, we were unable to assess alterations in chrX. MutationMapper (cBioPortal)14 was used to compile and visualize TET2 mutations.
Transcriptome microarray
RNA extraction was performed using the Cell Suspension/Body Fluid protocol from the QIAamp RNeasy Mini kit (Qiagen Inc., Valencia, CA) with elution in 35 µL of RNase-free water. Six BPDCN samples had sufficient quantity and quality for use on the ThermoFisher ThermoFisher Scientific ClariomTM D Pico Assay, human. Thus, 100 ng of RNA from each BPDCN (N = 6) and AML (N = 7) bone marrow specimens were submitted for processing on this microarray at The University of Texas Southwestern Genomics and Microarray Core Facility. The results were subsequently analyzed using the Transcriptome Analysis Console (TAC) (ThermoFisher Scientific, Waltham, MA). Differentially expressed genes among diagnoses were defined as those genes having gene-level fold changes of <−2 or >2 with gene-level FDR diagnostic F-test < 0.01. Differenitally enriched pathways were identified using the TAC Wiki pathway tool on genes with fold changes of <−4 or >4. Shared pathways were determined by using Gene Set Variation Analysis15 on each sample using the C2 subset of the Molecular Signatures Database (MSigDB) that includes 4762 gene sets16. The enrichment scores were summed across all BPDCN and AML samples, respectively. Those pathways with the lowest and highest enrichment score sums (<−1 and >1) were compared between BPDCN and AML to determine overlapping pathways.
Serum multiplex immunoassay
Available serum from BPDCN (N = 13) and AMLTET2m (N = 8) were profiled in triplicate using the Cytokine/Chemokine/Growth Factor 45-Plex Human ProcartaPlex™ Panel 1 (EPX450-12171-901, ThermoFisher Scientific) with the addition of IL-3 Human ProcartaPlex™ Simplex Kit (Invitrogen™). The protocol was followed with the option of overnight incubation of antibody with beads. The samples were profiled in triplicate. Out of 45 factors profiled, we kept those factors (N = 21) for which >70% of samples had the data within the standard curves for further analysis. Wilcoxon Rank-Sum test on mean observed concentrations were used to determine differentially expressed proteins. The Benjamini & Hochberg method was applied to calculate the false discovery rate (FDR).
Results
Historically, therapies have not been developed specifically for BPDCN due to incomplete knowledge on the underpinnings of the disease. Therefore, we conducted molecular profiling of BPDCN using gene panel sequencing, transcriptome microarray, and serum multiplex immunoassays and cytokine analysis (Table 1). We first assessed somatic point mutations and gene-level copy-number alterations on BPDCN specimens (N = 8) using a 300-gene panel that was designed to profile frequently mutated genes in hematologic malignancies and disorders13. There, we observed TET2 mutations in 5/8 (63%) of BPDCN patients, with single or compound truncating and missense mutations scattered throughout the gene (Fig. 1; Supplementary Table 2). Additional mutations were seen in SRSF2 (recurrent position p.P95L (BPDCN-12) and p.P95R (BPDCN-15)), STAG2 p.R216X (BPDCN-12), ASXL1 p.P721fs (BPDCN-4), EZH2 p.22_22del (BPDCN-12), and ZRSR2 p.15_18del (BPDCN-10) (Supplementary Table 2). Copy-number alterations were mostly consistent with cytogenetic profiles (Supplementary Table 3). Losses were from three patients (BPDCN-4, BPDCN-10, and BPDCN-12) in chromosomes 3, 5, 7, 9, 12, 13, 17, and 20 (Supplementary Table 3b). Along with the cytogenetics reports, we concluded that our cohort was composed of patients with predominantly TET2 mutations.

Annotations are based on NM_001127208.2. The S1674fs and R1476fs mutations in BPDCN-12 were found only in the bone marrow sample that was taken 1 month after the specimen from the peripheral blood, which contained only the R1425X mutation for TET2.
Since TET2 mutations occur frequently in other myeloid malignancies, these were unlikely to be disease-specific alterations. Therefore, we sought to enhance our ability to observe disease-specific expression signals by comparing BPDCN to AML specimens that had TET2 mutations. We used available AMLTET2m specimens for use in the transcriptome (N = 7) and serum (N = 8) profiling (Supplementary Table 4). In the transcriptome assay, three of the seven (78%) AMLTET2m patients had normal cytogenetics, which is the subset that is reported to have poorer prognosis based on TET2-mutated status17,18. Healthy control dendritic cells were not readily available for this analysis.
Out of the 7957 genes with significantly greater than twofold difference in expression between the BPDCN and AMLTET2m samples, 4646 were upregulated and 3311 downregulated in BPDCN as compared with AMLTET2m (Fig. 2a–c). Among them, we validated previous findings of higher levels of TCL1A and lower levels of MPO in BPDCN as compared with AMLTET2m (FDR adjusted P < 0.05 and P < 0.1, respectively) (Fig. 2c; Supplementary Fig. 1a–b, Supplementary Table 5). The 272 significantly differentially expressed pathways included those that were lower in BPDCN than in AMLTET2m: Cytoplasmic Ribosomal Proteins (Rank: 1), Electron Transport Chain (Rank: 2), Generic Transcription Pathway (Rank: 11). These types of pathways may indicate that BPDCN is more indolent as compared with AML (Supplementary Table 5). None of the most highly enriched pathways (top 400 of 4762 interrogated) in both diseases were shared. Two pathways with the lowest enrichment scores were present in both BPDCN and AML: BIOCARTA_ASBCELL_PATHWAY, the antigen-dependent activation of B cells, and REACTOME_NA_CL_DEPENDENT_NEUROTRANSMITTER. Both of these gene sets had low expression in four of six BPDCN samples, and five of seven AML samples (Supplementary Table 6a, b).

Pie charts of the type of transcripts upregulated (a) or downregulated (b) in BPDCN as compared with AMLTET2. Coding: transcript results in a protein. Noncoding: transcript does not result in a protein. Pseudogene: transcript may not be expressed and if expressed, does not produce a functional protein. Precursor-microRNA: unprocessed, premature microRNA. Unassigned: uncharacterized transcript. Multiple_Complex: fits into multiple categories of the above. c Volcano plot showing the genes that are differentially expressed between BPDCN and AMLTET2. The genes in red (upregulated in BPDCN) and green (downregulated in BPDCN) are at fold changes −4 > x > 4, with FDR F-test adjusted P < 0.03.
pDC markers LAMP519 and CCDC5020 were higher in BPDCN than in AMLTET2m (P < 0.01, P < 0.001, respectively) (Fig. 2c; Supplementary Fig. 1c, d, Supplementary Table 5). LAMP5 is expressed in pDCs that are not activated19. Thus, the elevated levels here may indicate the state of dendritic nature in BPDCN cells21. Given the stimulatory role of NFkB hyperactivation in BPDCN22, the upregulated expression of CCDC50 may negatively regulate NFkB in these patients. From a therapeutic standpoint, this may validate recent efforts to suppress NFkB activation with the proteasome inhibitor bortezomib in order to inhibit cell proliferation, induce cell death, and prolong the survival of BPDCN patients23.
Our data indicated possible links between BPDCN and immune deficiency. PTPRS is higher in BPDCN as compared with AMLTET2m (FDR adjusted P < 0.00001; Supplementary Fig. 1e). Since PTPRS inhibits the activation of nonneoplastic pDCs so as to prevent immune-mediated inflammation22, this high expression may be imply that BPDCN cells are derived from nonactivated pDCs in an environment that is poised to become inflammatory. Interestingly, the serum cytokine profiles showed significant higher levels of eotaxin and RANTES in the BPDCN cohort than in the AMLTET2m group (Fig. 3a, b; Supplementary Table 7). Both of these are implicated in allergic and autoimmune reactions by behaving as eosinophil chemoattractants. There is little evidence of underlying inflammatory processes since white blood cell counts were low, while there may be an indication for eosinopenia based on absent or low absolute count and proportion of eosinophils (Supplementary Table 8). Therefore, BPDCN may exist in a context in which there is a defect in inflammatory response.

a Eotaxin, **P < 0.01; b RANTES, *P < 0.05.
Finally, we observed lower JAK-STAT signaling in BPDCN (Rank:14), which is related to the signaling events that occur downstream of IL-3Rα activation. Lower levels of downstream molecules (STAT3, STAT5B, CBL, and PTPN6) and related upstream molecule (CSF3R) were notable (Supplementary Figs. 2, 3). In addition, LAMP5 is protein that is relocated to the cell surface of pDCs 24 h after stimulation by IL-3. Its high levels in BPDCN may be inhibiting further IL-3/STAT3 signaling. In contrast, constitutive STAT3 signaling contributes to AML cell proliferation and survival24,25.
We also observed several other notable genes: CPA3, ATP8B4, SPINK2, TRGJP1, and CDK6, which were expressed at lower levels in BPDCN as compared with AMLTET2m as well as SULF2, miR297, and miR708 that were higher in BPDCN (Fig. 2c). These are potential markers in distinguishing BPDCN from AML, but currently have unclear implications in the etiology of disease.
Discussion
Previous single-cell sequencing of CD45 + HLA-DR + CD123+ blasts from four BPDCN patients revealed that BPDCN is most closely related to pDCs, with expression of B-cell markers, such as FCRLA, IGLL1, TCL1A, and IGLL526. At the same time, patients with BPDCN often share features with patients with AML, including anemia, blasts in the bone marrow, and somatic point mutations in genes such as: TET2, ASXL1, IDH2, and NPM1. BPDCN can also progress to AML5, has been associated with co-existence of/or prior MDS/CMML in ~20% of patients in one major cohort27, and has been detected in a patient with Felty’s syndrome and myelodysplastic syndrome5,28. Therefore, BPDCN harbor mixed dendritic and myeloid characteristics. However, the features that distinguish BPDCN from AML have not been explored to great depth. These features are not only potential biomarkers, but could also determine the molecular context for which a dendritic cell that acquires such similar mutations would succumb to a similar etiology as AML. We sought to uncover these types of disease-specific features by conducting transcriptome and serum analyses comparisons of BPDCN and AML samples. We chose to compare BPDCN with TET2-mutated AML in order to rule out epigenetic-driven expression patterns that are the result of TET2 mutations. This group of BPDCN represents the majority of patients, but may exclude other minor subgroups that are yet to be characterized.
In examining the serum, we found elevated levels of eotaxin and RANTES, which suggests that BPDCN patients may be more prone to autoimmunity. Previously, Zhan et al. proposed that susceptibility to autoimmunity could depend on pDC lifespans29. This idea was based on the fact that the NZB mouse strain has a longer pDC lifespan than C57BL/6 mice, and is more prone to developing lupus29. Transgenic mice that are overexpressing the anti-apoptotic gene BCL2 have longer-lived pDCs30. In BPDCN, BCL2 levels are higher as compared with normal pDCs31, which may indicate that BPDCN cells are comprised long-lived malignant pDCs in patients who are susceptible to autoimmunity. At the same time, BCL2 is advantageous for promoting tumor cell survival, which is consistent with the effectiveness of BCL2 inhibition in BPDCN32,33. Moreover, autoimmune pathologies have been hypothesized to damage the bone marrow and induce destruction of myeloid precursor cells34. Thus, these links may give insights into the progression of BPDCN to AML. The role of pDC lifespan and state of autoimmunity in these patients should be examined further.
Among the top differentially expressed genes between BPDCN and AMLTET2m were specific dendritic markers that we believe are insightful into the dendritic nature of the disease: (LAMP519, CCDC5020). LAMP5 is within the list of genes that have overlapping expression between healthy pDCs and cDCs by single-cell RNASeq26. In addition, the high levels of LAMP5 is consistent with the immunohistochemical predominance (78%) that was noted in 33 other BPDCN patients21.
In its natural context, pDCs serve to recognize foreign particles such as viruses and synthetic oligonucleotides by becoming activated35. The canonical activation pathway is through toll-like receptors TLR7/9 that leads to the production of pro-inflammatory cytokines by pDCs in order to regulate activity and to stimulate T lymphocytes35. Using both normal pDC and BPDCN cells (CAL-1), Combes et al. demonstrated that initial high levels of LAMP5 (BAD-LAMP) in both of these cell types actively traffic TLR9 to late endosomes as a possible mechanism for preventing cytokine release36. BAD-LAMP subsequently decreases upon receiving a CpG activation signal. In our data, no differences in the levels of TLR pathways were seen between BPDCN and AMLTET2m at the expression level36. This, along with the high levels of LAMP5, suggest that the TLR pathways are not differentially activated in BPDCN. Thus, inhibiting this particular pDC activation pathway may be contributing to a state of immune tolerance. Additional support for this are the high levels of PTPRS that prevent activation as well as the elevated serum levels of eotaxin and RANTES in BPDCN that would be predicted to elicit an activation response37.
By integrating our findings with those of others, we surmise that BPDCN consists of pDCs in a nonactivated survival state within a pre-inflammatory context. BPDCN and AML are distinct, yet related diseases. Understanding the initiation and formation of BPDCN could serve to prevent further progression into AML and also link progenitor cells from the bone marrow to both malignancies and pathogeneses of other autoimmune disorders such as systemic lupus erythematosus (SLE) that have hallmark cutaneous lesions38.
Data availability
Gene panel sequencing and transcriptome microarray data are deposited in the European Genome-phenome Archive under EGAS00001003453.
References
Riaz, W., Zhang, L., Horna, P. & Sokol, L. Blastic plasmacytoid dendritic cell neoplasm: update on molecular biology, diagnosis, and therapy. Cancer Control 21, 279–289 (2014).
Herling, M., Teitell, M. A., Shen, R. R., Medeiros, L. J. & Jones, D. TCL1 expression in plasmacytoid dendritic cells (DC2s) and the related CD4+ CD56+ blastic tumors of skin. Blood 101, 5007–5009 (2003).
Shi, Y. & Wang, E. Blastic plasmacytoid dendritic cell neoplasm: a clinicopathologic review. Arch. Pathol. Lab. Med. 138, 564–569 (2014).
Swiecki, M. & Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 15, 471–485 (2015).
Pagano, L. et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian multicenter study. Haematologica 98, 239–246 (2013).
Feuillard, J. et al. Clinical and biologic features of CD4(+)CD56(+) malignancies. Blood 99, 1556–1563 (2002).
Vardiman, J. W. et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114, 937–951 (2009).
Pemmaraju, N. et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. N. Engl. J. Med. 380, 1628–1637 (2019).
Alayed, K. et al. TET2 mutations, myelodysplastic features, and a distinct immunoprofile characterize blastic plasmacytoid dendritic cell neoplasm in the bone marrow. Am. J. Hematol. 88, 1055–1061 (2013).
Taylor, J. et al. Loss-of-function mutations in the splicing factor ZRSR2 are common in blastic plasmacytoid dendritic cell neoplasm and have male predominance. Blood 122, 741 (2013).
Menezes, J. et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia 28, 823–829 (2014).
Jardin, F. et al. TET2 and TP53 mutations are frequently observed in blastic plasmacytoid dendritic cell neoplasm. Br. J. Haematol. 153, 413–416 (2011).
Takahashi, K. et al. Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 131, 1820–1832 (2018).
Vohra, S. & Biggin, P. C. Mutationmapper: a tool to aid the mapping of protein mutation data. PLoS ONE 8, e71711 (2013).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Chou, W. C. et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 118, 3803–3810 (2011).
Metzeler, K. H. et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J. Clin. Oncol. 29, 1373–1381 (2011).
Zhang, H. et al. LAMP5 distribution in and on plasmacytoid dendritic cells (P5019). J. Immunol. 190, 110.110 (2013).
Worah, K. et al. Proteomics of human dendritic cell subsets reveals subset-specific surface markers and differential inflammasome function. Cell Rep. 16, 2953–2966 (2016).
Defays, A. et al. BAD-LAMP is a novel biomarker of nonactivated human plasmacytoid dendritic cells. Blood 118, 609–617 (2011).
Bunin, A. et al. Protein tyrosine phosphatase PTPRS is an inhibitory receptor on human and murine plasmacytoid dendritic cells. Immunity 43, 277–288 (2015).
Dalod, M., Chelbi, R., Malissen, B. & Lawrence, T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J. 33, 1104–1116 (2014).
Redell, M. S., Ruiz, M. J., Alonzo, T. A., Gerbing, R. B. & Tweardy, D. J. Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 117, 5701–5709 (2011).
Hossain, D. M. et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood 123, 15–25 (2014).
Villani, A. C. et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017).
Pemmaraju, N. et al. Long-term outcomes in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood 130, 3855 (2017).
Chamoun, K. et al. Early detection of transformation to BPDCN in a patient with MDS. Exp. Hematol. Oncol. 7, 26 (2018).
Zhan, Y. et al. Plasmacytoid dendritic cells are short-lived: reappraising the influence of migration, genetic factors and activation on estimation of lifespan. Sci Rep 6, 25060 (2016).
Carrington, E. M. et al. Prosurvival Bcl-2 family members reveal a distinct apoptotic identity between conventional and plasmacytoid dendritic cells. Proc. Natl Acad. Sci. USA 112, 4044–4049 (2015).
Sapienza, M. R. et al. Molecular profiling of blastic plasmacytoid dendritic cell neoplasm reveals a unique pattern and suggests selective sensitivity to NF-kB pathway inhibition. Leukemia 28, 1606–1616 (2014).
Montero, J. et al. Blastic plasmacytoid dendritic cell neoplasm is dependent on BCL2 and sensitive to venetoclax. Cancer Discov. 7, 156–164 (2017).
Pemmaraju, N., Konopleva, M. & Lane, A. A. More on blastic plasmacytoid dendritic-cell neoplasms. N. Engl. J. Med. 380, 695–697 (2019).
Anderson, L. A. et al. Risks of myeloid malignancies in patients with autoimmune conditions. Br. J. Cancer 100, 822–828 (2009).
Mellman, I. & Steinman, R. M. Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258 (2001).
Combes, A. et al. BAD-LAMP controls TLR9 trafficking and signalling in human plasmacytoid dendritic cells. Nat. Commun. 8, 913 (2017).
Garcia-Zepeda, E. A. et al. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat. Med. 2, 449–456 (1996).
Ronnblom, L. & Pascual, V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus 17, 394–399 (2008).
Acknowledgements
We thank Jairo Matthews, Evan Cohen, James Reuben, and the Department of Genomic Medicine Cancer Genomics Laboratory for research support, and Kiyomi Morita for discussions. This work was supported in part by the MD Anderson Cancer Support Grant CA016672 and Award number P01 CA049639. Dr Pemmaraju was supported by the 2016 Affymetrix Tumor Profiling Grant Award (now part of ThermoFisher) as well as the Sager Strong Foundation.
Author information
Authors and Affiliations
Contributions
H.C.B., M. Khan, P.A.F., M. Konopleva, and N.P. conceived the study design. J.K. reviewed specimens. H.C.B. and T.C. conducted experiments. M.A. collected the clinical data. H.C.B., M. Khan, and F.W. analyzed the data. H.C.B., P.A.F., M. Khan, L.Z. and N.P. interpreted the data. H.C.B., M. Khan, M. Konopleva, and N.P. wrote the paper. All authors have reviewed and approve of the paper.
Corresponding author
Ethics declarations
Conflict of interest
M. Konopleva receives consulting/honorarium from: AbbVie, Genentech, F. Hoffman La-Roche, Stemline Therapeutics, Amgen, Forty-Seven. M. Konopleva receives research funding/clinical trials support from AbbVie, Genentech, F. Hoffman La-Roche, Eli Lilly, Cellectis, Calithera, Ablynx, Stemline Therapeutics, Agios, Ascentage. M. Konopleva holds stock options/royalties in Reata Pharmaceutical. For N.P.: research funding and honorarium were given by ThermoFisher, Stemline Therapeutics, and Cellectis. N.P. receives consulting/honorarium from:AbbVie; Celgene; Stemline; Incyte; Novartis; MustangBio; Roche Diagnostics, LFB; research funding/clinical trials support from:Stemline; Novartis; AbbVie; Samus; Cellectis; Plexxikon; Daiichi-Sankyo; Affymetrix; grants/funding from:Affymetrix, SagerStrong Foundation.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These final results are based, in part, on preliminary ASH 2018 poster presentation Beird et al. 2018.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beird, H.C., Khan, M., Wang, F. et al. Features of non-activation dendritic state and immune deficiency in blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood Cancer J. 9, 99 (2019). https://doi.org/10.1038/s41408-019-0262-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-019-0262-0
This article is cited by
-
Allogeneic hematopoietic cell transplantation for patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN)
Bone Marrow Transplantation (2022)
-
Divergent clonal evolution of blastic plasmacytoid dendritic cell neoplasm and chronic myelomonocytic leukemia from a shared TET2-mutated origin
Leukemia (2021)
-
Blastic Plasmacytoid Dendritic Cell Neoplasm: Progress in Cell Origin, Molecular Biology, Diagnostic Criteria and Therapeutic Approaches
Current Medical Science (2021)
