
Abstract
Follicular lymphoma (FL) is a heterogeneous disease whose pathogenesis remains partially unknown. Around 20% of FL patients experience early progression or treatment-refractory disease and 2–3% of patients per year experience histological transformation (HT) into a more aggressive lymphoma (tFL). Here, we evaluate the immunoglobulin heavy chain variable (IGHV) gene usage and mutational status in 187 FL cases to assess its impact on clinical outcome and histological transformation. The IGHV gene repertoire was remarkably biased in FL. The IGHV4-34 (14%), IGHV3-23 (14%), IGHV3-48 (10%), IGHV3-30 (9%) and IGHV3-21 (7%) genes accounted for more than half of the whole cohort. IGHV3-48 was overrepresented in cases of tFL (19%) compared with non-transformed FL at 5 years (5%, P = 0.05). Patients with the IGHV3-48 gene were significantly more likely to have had HT after 10 years than those who used other genes (71% vs. 25%, P < 0.05), irrespective of the therapy they received. Moreover, IGHV3-30 was also overrepresented in cases of FL (9%) and tFL (13%) compared with diffuse large B-cell lymphoma in which it was nearly absent. In conclusion, our results indicate a role for antigen selection in the development of FL, while the use of IGHV3-48 could help predict histological transformation.
Similar content being viewed by others

Introduction
Follicular lymphoma (FL) is the most common indolent non-Hodgkin lymphoma (NHL, 20–30%), with an incidence of 12.5 new cases per 100 000 inhabitants/year. FL is typically characterized by a non-aggressive nature, usually responds successfully to first-line therapy, and has a median survival of 15 to 20 years1,2,3.
FL is considered an incurable disease that is characterized by a pattern of multiple relapses, a decreasing duration of response, and a gradual acquisition of resistance to different drugs. Despite the improvement in treatment effectiveness due to the incorporation of immunochemotherapy regimens, including anti-CD20 monoclonal antibody, 20% of patients experience early progression or develop a treatment-resistant disease within 2 years of receiving first-line therapy2,4,5. Furthermore, FL can undergo histological transformation (HT) into a more aggressive lymphoma, most commonly to diffuse large B-cell lymphoma (DLBCL), with a transformation rate of 2–3% per year6,7,8. Transformed follicular lymphoma (tFL) involves a clonal relationship between the initial FL and the aggressive form and it is considered to be one of the most unfavorable events in the natural history of FL9, because transformation has customarily been associated with low cure rates with conventional therapies and short survival10. Several prognostic indices for FL, such as the Follicular Lymphoma International Prognostic Index (FLIPI)11, FLIPI212 or the recently described complete response rate at 30 months (CR30)4 have been defined because of the need to identify high-risk group of patients. However, FLIPI perform poorly in terms of specificity to predict transformation13, and the remaining indexes are not useful to predict histological transformation. Unraveling the biology and pathogenesis of this disease would improve the identification of poor-prognosis groups at the time of diagnosis and might enable risk-adapted treatment strategies to be employed.
FL arises from the clonal proliferation of germinal center (GC) B-cells, which have experienced malignant transformation, resulting in a follicular pattern with architectural features typical of normal germinal centers3,14. These GC B-cells harbor a clonal rearrangement of the immunoglobulin heavy chain gene (IGH), which occurs during B-cell differentiation to generate a completely functional variable (IGHV)-diversity (IGHD)-joining (IGHJ) junction in the IGH gene. The IGHV gene repertoire and somatic hypermutation status has been extensively analyzed in B-cell lymphomas, showing a biased use of IGHV genes in chronic lymphocytic leukemia (CLL), DLBCL, splenic marginal zone lymphoma (SMZL), Waldenström macroglobulinemia (WM), mantle cell lymphoma (MCL) and primary central nervous system lymphoma15,16,17,18,19,20,21,22. Some IGHV genes have also been associated with clinical outcome. Thus, the unmutated status of the IGHV rearrangement, or the presence of those IGHV3-21 belonging to the stereotyped B-cell receptor subset #2 are associated with unfavorable prognosis in CLL16,17,23,24.
In the case of FL, the IGHV repertoire has only occasionally been reported in small series, and without any clinical associations25,26,27. Recently, Berget et al. reported an association in a series of 106 FL patients between the presence of IGHV5 or to use more than one IGHV subgroup and poor survival14. However, there is no information about the role of IGHV rearrangements in the treatment response or the risk of HT.
The aim of the present study was to analyze in detail the use of IGHV, IGHD and IGHJ genes and the somatic hypermutation (SHM) rate in the largest FL series to date, to improve our understanding of the biology of FL and its impact on clinical outcome and the risk of histological transformation.
Patients and methods
Patient characteristics
The study included a total of 187 consecutive patients with histologically confirmed FL grade I-IIIA according to the 2017 WHO classification3 diagnosed between January 1995 and May 2017 (Supplemental Fig. S1). FL grade IIIB and composite cases of FL+DLBCL were excluded28. Cases without biological sample and/or clinical data were also excluded.
Clinical characteristics of the cohort are described in Table 1. The median age at diagnosis was 58 years (range 19–87 years) and 53% of the patients were females. Within our series, 36% of the patients were high-risk according to FLIPI criteria. One hundred sixty-three patients received treatment, of whom 98 (60%) received rituximab-based immunochemotherapy (R-ICT) -R-CHOP therapy in most of them (68/98 cases, 69%)-, and 65 (40%) received other schemes without rituximab. Seventy-three patients (39%) also received rituximab as maintenance. The other 24 patients (13%) never were treated until the time of last follow-up or HT into aggressive lymphoma.
This study was approved by the Ethical Committee of the University Hospital of Salamanca in accordance with Spanish law and the Declaration of Helsinki. Written informed consent was obtained from all participants.
DNA extraction
DNA was isolated on samples at the time of diagnosis of FL, and in the biopsy of those cases with documented HT. gDNA from fresh-frozen tissue (65% of cases) was isolated by the phenol-chloroform method29 or the Maxwell® 16 System (Promega, Madison, WI, USA). In cases in which DNA was extracted from bone marrow (20%), peripheral blood (2%) and other tissues (3%), the Maxwell® 16 System (Promega) or DNAzol reagent (MRC, Cincinnati, OH, USA) was used30. Finally, gDNA extraction from formalin-fixed paraffin-embedded (FFPE) tissue (10%) was isolated using the RecoverAll Total Nucleic Acid Isolation Kit (Ambion/Applied Biosystems, Foster City, CA, USA) or the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany).
DNA was quantified in a NanoDrop 1000TM Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The quality and purity of the gDNA extracted from FFPE tissue, were assessed with the 4200 TapeStation (Agilent Technologies, Santa Clara, CA, USA) system, using the Genomic DNA ScreenTape assay.
IGH rearrangements amplification
IGH rearrangements were amplified according to the BIOMED-2 Concerted Action protocols31, in which the complete V-D-J rearrangement was amplified by multiplex PCR with a set of six family-specific IGHV primers of the framework region 1 (FR1), and one IGHJ consensus primer. For the samples with no detectable amplification from FR1, PCR was performed from Leader or FR2 region. The presence of the monoclonal rearrangement was then confirmed by GeneScan with an ABI 3500xL DNA Sequencer (Applied Biosystems, Foster City, CA, USA).
IGH rearrangement sequencing and identification
PCR products were sequenced in forward and reverse reads, using the same primers as for PCR amplification and the Big-Dye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems)31. Sequencing was carried out with an ABI 3500xL DNA Sequencer (Applied Biosystems).
Complete V-D-J rearrangements and the percentage of germline identity were identified using the IMGT/V-QUEST software (http://www.imgt.org). Sequences with germline identity less than or equal to 98% were considered to be mutated, while those with >98% identity were considered to be sequences without somatic hypermutation (SHM) or to be unmutated.
t(14;18) Nested PCR
The t(14;18) was amplified by nested PCR adapting the method of Gribben et al.32 using two BCL2 primers directed at major breakpoint region (MBR) and minor cluster region (mcr) in chromosome 18 and a consensus primer directed to JH in chromosome 14. The JH internal primer was marked with a fluorochrome for detecting BCL2 translocation size by GeneScan with an ABI 3500xL DNA Sequencer (Applied Biosystems).
Definitions and statistical analysis
We explored the following clinical endpoints: (i) response to induction treatment according to International Working Group criteria, based on computed tomography (CT) scans (n = 130) or a 18-fluorodeoxyglucose-positron emission tomography (PET)/CT scan (n = 16);33,34,35 (ii) complete response rate at 30 months (CR30), defined as complete response 30 months after the date induction treatment began;36 (iii) failure-free survival (FFS), defined as less than a partial response at the end of induction, relapse, progression or death;37 (iv) time to transformation (TTT), measured as the time from the date of FL diagnosis to that of histologically confirmed transformation into aggressive lymphoma (tFL)38, defined solely on the basis of pathological criteria, getting biopsies at the time of FL diagnosis and when there was a clinical suspicion of tFL; (v) histological transformation at 10 years (10y-HT), defined as the probability that HT occurred at any time within a given 10-year period; and (vi) overall survival (OS), which is the time from the date of diagnosis of FL to that of death from any cause.
Possible associations between variables were analyzed by the χ2-square test for categorical variables, and by Student’s unpaired-samples t-test or the Mann-Whitney U test for continuous variables. P values were Bonferroni corrected (Pc) to take into account multiple testing. Survival was estimated by the Kaplan–Meier method and the differences were assessed by the log-rank test. Subsequently, all variables for which there was some indication of a significant association with clinical features in the univariate test (P < 0.1) were considered in a multivariate Cox regression model. Differences were considered to be statistically significant for values of P < 0.05. All analyses were performed using SPSS (IBM SPSS 23.0, IBM Corp, Armonk, NY, USA).
Results
IGHV, IGHD and IGHJ Gene Repertoire in FL
A total of 138 clonal V–D–J rearrangements out of 187 cases (74%) were detected in our study cohort. No clonal rearrangements were identified in the other 49 cases due to their highly polyclonal background (82%) or the absence of amplification arising from the small quantity or poor quality of the available DNA (18%). Most sequences were obtained from FR1 (80%). Clinical characteristics of the cohort with clonal IGH rearrangements (n = 138) in comparison with the global cohort are described in Supplemental Table S1.
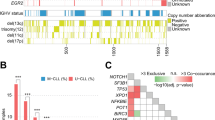
IGHV3 (63%) and IGHV4 (27%) were the most frequent subgroups of clonal V-D-J rearrangements (Supplemental Table S2). A total of 35 functional IGHV genes were identified, being IGHV4-34 (14%), IGHV3-23 (14%), IGHV3-48 (10%), IGHV3-30 (9%) and IGHV3-21 (7%) the most represented (Fig. 1 and Supplemental Table S3). Of the IGHD genes, IGHD3 (37%) and IGHD2 (25%) were the most frequent subgroups (Supplemental Table S2), with IGHD3-10 (12%) and IGHD3-22 (10%) being the most frequently expressed (Supplemental Table S4). Finally, of the IGHJ genes, IGHJ4 (48%) was the most prevalent gene in this FL series (Supplemental Table S2). Summary list of the IGHV, IGHD and IGHJ subgroups and gene usage in the present series are provided in Supplemental Tables S2–S4.

The 35 IGHV genes expressed in our series are presented along the X-axis
Analysis of IGHV mutational status in FL and VH CDR3 length
Somatic hypermutation (SHM) was feasibly studied in 130 of 138 (94%) clonal V–D–J rearrangements. SHM was detected in 126 (97%) of them. The percentage of germline identity to the closest IGHV gene ranged from 67.2 to 100%, with a mean of 87.2%. No differences were observed in the proportions of patients with SHM between the IGHV subgroups (Supplemental Fig. S2).
Only four patients (3%) in our series were unmutated, three of them with a 100% germline identity and 99.8% in the other patient. Unmutated sequences were observed in the IGHV1 and IGHV3 subgroups (Supplemental Fig. S2); the affected genes were IGHV1-69, IGHV3-21 and IGHV3-30-3.
The VH CDR3 region was identified in 113/138 (82%) cases with clonal V–D–J rearrangement. The median VH CDR3 length was 16 amino acids (range 8 to 28). There were no statistically significant differences in mean length between the subgroups or genes (Supplemental Table S5).
Comparative Analysis of IGH Rearrangements in FL with Other B-LPDs and Normal B Cells
In order to identify unique characteristics of FL, we compared our FL dataset with the results reported for CD5-/IgM+normal B cells and other B-cell lymphoproliferative disorders (B-LPDs), namely, de novo DLBCL, MCL, CLL, SMZL, hairy cell leukemia (HCL), multiple myeloma (MM) and WM (Table 2 and Supplemental Table S6)19,21,39,40,41,42,43,44. The IGHV1 subgroup was significantly underrepresented in FL relative to all B-LPDs (5% vs. 15 to 30%, P < 0.05, Pc < 0.05) except WM (7%). Conversely, IGHV3 subgroup was overrepresented in FL relative to DLBCL (63% vs. 44%, P < 0.01, Pc < 0.05), CLL (48%, P < 0.01, Pc < 0.05), MCL (52%, P < 0.05, Pc < 0.1), and MM (49%, P < 0.01, Pc < 0.1), although statistical differences were lost after Bonferroni correction in the two latter cases.
IGHV3-21 (7%), IGHV genes that are frequently used in FL, was completely absent from CD5-/IgM+normal B cells (P < 0.01, Pc < 0.05) and relatively underrepresented in other B-LPDs19,21,39–44. There were significant differences in the frequency of the IGHV3-30 gene, which was absent from DLBCL (P < 0.01, Pc < 0.05)21. IGHV4-34 was more frequent than in FL (14%) than in CD5-/IgM+normal B cells (4%, P < 0.01, Pc < 0.05), MM (1%, P < 0.01, Pc < 0.05) or WM (0%, P < 0.01, Pc = 0.06)19,39,44.
Comparative Analysis of IGH Rearrangements in tFL with FL and DLBCL
In the FL series, 47 patients (25%) underwent HT into aggressive lymphoma (tFL). The clonal relationship between the paired samples (FL/tFL) was confirmed in 45/47 cases (96%) by BIOMED-2 or BCL2-IGH breakpoint analysis. Two patients were excluded because they expressed different clones at diagnosis and transformation. A total of 31 clonal V-D-J rearrangements out of 45 tFL cases (69%) were detected (31/138 (22%) total cases with IGHV gene available).
With the purpose of identifying singular characteristics of tFL, we compared these cases with those of FL without HT for a minimum of 5 years follow-up (n = 63), and germinal (GCB, n = 32) and non-germinal (non-GCB, n = 48) center B-cell-like de novo DLBCL previously analyzed by our group21. The IGHV1 subgroup was significantly underrepresented in tFL relative to the non-GCB DLBCL (3% vs. 23%, P < 0.05) while the IGHV3 subgroup was overrepresented in tFL compared with non-GCB DLBCL cases (74% vs. 40%, P < 0.01) (Supplemental Tables S7–S10).
The IGHV3-48 gene was more frequent in tFL (19%) than in FL (5%, P = 0.05), and in non-GCB DLBCL (4%, P < 0.05) (Fig. 2 and Supplemental Tables S10-S11). Finally, the IGHV3-30 gene, which was frequently used in tFL (13%), was completely absent from both types of DLBCL (P < 0.05). Similarly, the IGHV4-34 gene was overrepresented in tFL relative to GCB DLBCL, in which it was never present (16 vs. 0%, P = 0.05) (Fig. 2 and Supplemental Tables S10–S11). Statistical significances were lost after Bonferroni correction due to the relative low number of cases in each of the groups.

The five IGHV genes most frequently expressed in our series are shown along the X-axis. *P < 0,05. GCB, germinal center B-cell-like; non-GCB, non-germinal center B-cell-like
Effect of IGHV gene usage and mutational status on clinical outcomes
Clinical outcomes in whole FL series
In the whole series, after the median follow-up of 7.9 years (range: 0.6–21.4 years) for surviving patients, 48 patients had died. The 5- and 10-year OS were 82 and 70%, respectively, and the 5-year OS for low/intermediate-risk FLIPI was 92% (P < 0.01). Most of the treated patients, received R-ICT (60%) and the 5- and 10-year FFS were 51 and 29%, respectively. After induction therapy, 39 patients (40%) had achieved CR and 48 patients (54%) were in CR at 30 months (CR30).
We explored the use of IGHV subgroups and genes with clinical outcome. Two FL patients with IGHV5 subgroup had poorer clinical course that the remaining patients and both died before the time of median follow-up. No correlations were observed between the IGHV gene usage and response or CR30, even taking into account only the 130 FL patients in whom the response was assessed by CT scan. No statistically significant differences were observed between the IGHV gene and FFS or OS.
There were four unmutated patients, all of whom had an intermediate/high-risk FLIPI status, although β2-microglobulin was always normal. Three of these patients received R-ICT but none was in CR at 30 months (data not shown). The three cases experienced an early treatment failure, leading to a shorter 5-year FFS than found in mutated FL cases (0% vs. 57%, P < 0.001). Two of these cases had died due to progression of their disease 29 and 40 months after initial diagnosis.
Risk of Histological Transformation
Considering only the 45 patients who developed clonally related HT, 18 patients had died after a median follow-up of 10.2 years for surviving patients (range: 1.2–21.4 years), giving a 5 and 10-year OS of 82 and 59%, respectively. The median TTT was 4.4 years after diagnosis (range: 0.1–19.6 years), with an estimated 84% of 10-year HT. Thirty-three (73%) patients received treatment as symptomatic FL. The other patients had never been treated until the date of HT into aggressive lymphoma. In this context, those who had never been treated (n = 12) had a higher 10-year HT (100 vs. 79%, P < 0.01) and shorter median TTT (2.9 vs. 6.6 years, P < 0.01) than those who received treatment as symptomatic FL (n = 33).
In the global series (n = 187), the 10-year HT was significantly higher for FL patients who never received treatment than for those who were treated (85 vs. 24%, P < 0.01). With respect to the IGHV gene, six out of 14 (43%) patients harboring the IGHV3-48 gene at diagnosis experienced HT, resulting in a significantly higher 10-year HT than for those FL patients who had any other IGHV genes (71 vs. 25%, P < 0.05; Fig. 3). This result did not differ after excluding those cases with limited follow-up (n = 7 below 30 months, 5%; data not shown).

The absence and presence of IGHV3-48 are depicted by black and grey lines, respectively. The vertical dashed line indicates 10-year follow-up
In the multivariate analysis, variables independently associated with longer 10-year HT were never treatment received (HR: 21.2, 95% CI: 6.3–70.7) and IGHV3-48 gene usage (HR: 5.0, 95% CI: 1.5–17.2) (Table 3).
Discussion
Although FL is the most frequent variant of indolent non-Hodgkin lymphoma, its ontogeny is still not well understood. Our study reports the largest FL series examined to date (n = 187) in which the use of IGHV, IGHD and IGHJ genes in the clonal BCR rearrangement has been extensively characterized. We also analyze of the potential association with clinical outcome and, for the first time, the risk of histological transformation (HT). We observed a biased use of certain IGHV genes, which provides new insight into the ontogeny of FL as part of B-cell differentiation. Thus, FL shows clear differences from other B-LPDs with regard to IGHV usage and SHM. Our findings suggest that certain antigens are involved in FL development through the stimulation of B-cells proliferation in cells expressing surface IG encoded by specific IGHV genes.
No differences were observed in the frequencies of IGHV subgroups between FL cells and CD5-/IgM+normal B-cells, although IGHV1 frequency was slightly lower in the patient cohort39. These numbers mirrored those of other smaller series14,25,26,27,45,46,47. Conversely, significant differences were observed in the IGHV use compared with CD5-/IgM+normal B cells39. Thus, there was a strong bias towards higher gene usage of IGHV4-34, IGHV3-23, IGHV3-48, IGHV3-30 and IGHV3-21 in FL, these cases involving around half of the patient cohort. These results are similar to those previously reported, although IGHV4-34 gene was more frequent in our series than in other FL series, probably due to differences in sample size or geographic distribution48. Compared with other B-LPDs, we also note a bias in IGHV gene usage in FL. The most interesting finding may be the high frequency of the IGHV3-30 and IGHV4-34 genes in tFL compared with their very low frequency or complete absence in GCB or non-GCB DLBCL21,49,50. The increased selection of IGHV4-34 in tFL would favor a more immature cell of origin or with an extra-germinal center development. This information could be very helpful at diagnosis for distinguishing between DLBCL originating from histological transformation and true de novo DLBCL. Nevertheless, an analysis of a larger series is desirable in order to confirm this relationship. Taken together, these findings suggest that certain antigens participate in the lymphomagenesis of FL by stimulating the expression of surface IGs encoded by specific IGHV genes.
We analyzed the influence of the IGHV gene usage and SHM on clinical outcomes. None of the IGHV subgroups or genes appeared to be associated with clinical outcome, indicating that there is no relationship between IGHV usage and treatment requirement or response to therapy. The only relevant finding was that the two FL patients in the IGHV5 subgroup had a short survival, which is consistent with a previous report14, although this is not sufficient to allow a definitive conclusion to be drawn. For the other IGHV genes, the low selection rate seen in FL precludes any reliable interpretation about their clinical significance. A larger series (probably of > 1000 cases) needs to be analyzed, as has recently been done with CLL and MM23,51. Something similar can be said in regard to IGHV mutational status, since only four of our cases had unmutated IGHV, consistent with the proportions in previous reports46,52. Nevertheless, all four unmutated cases had poor prognostic findings and short survival. A careful review of these four cases ruled out the presence of concomitant B-LPDs. The inclusion of several standard treatments with R-ICT has to be taken in consideration as a potential limitation of the present study.
In this report, we have analyzed for the first time the role of IGHV gene in histological transformation by analyzing 47 paired FL and tFL samples from the same patients. First, we confirmed the clonal relationship between each sample pair. Only two patients (4%) had a different clonal rearrangement between the two samples and were excluded from subsequent analyses. Consequently, 45 patients (24%) underwent clonally related HT into aggressive DLBCL. We observed that those FL patients in our cohort who were never treated had significantly higher 10y-HT than FL patients who received treatment, suggesting that early treatment is associated with a lower transformation risk, that is consistent with those of other studies9,53.
The IGHV3-48 gene was associated with the risk of HT in the multivariate analysis, suggesting that it has a biological role in the transformation. Our FL series did not display a biased usage of IGHV3-48 compared with other published FL series14,27,46, although IGHV3-48 was significantly more frequent in HT than in non-transformed FL at 5 years (19 vs. 5%). Recently, the use of rituximab as first-line treatment in FL has been associated with a reduced risk of HT53. Since most IGHV3-48 FL cases received R-ICT, the protective effect of rituximab on HT seems not to work so well for them. In addition, IGHV3-48 FL cases were not associated to outcome probably due to some patients could be rescued by using rituximab-based therapy followed by auto-SCT to treat the transformation. Therefore, this finding implies that the IGHV3-48 gene functions as a pathogenic factor involved in transformation, as seems to happen in CLL that bear IGHV4-39 gene54.
In summary, in the largest FL series examined to date, a particular bias in IGHV gene selection towards the clonal BCR rearrangement was observed. The selection of the IGHV3-48 gene seems to have a role in malignant transformation from FL to aggressive lymphoma. This argues in favor of the close monitoring of FL patients, examining IGHV3-48 in anticipation of a potential transformation. We also found that IGHV3-30 could contribute to the differential diagnostic process. Taken together, these results indicate a role for antigen selection in the development and transformation of FL, and suggest possibilities for future research into the biology of tFL.
References
Lackraj, T., Goswami, R. & Kridel, R. Pathogenesis of follicular lymphoma. Best Pract. Res. Clin. Haematol. 31, 2–14 (2018).
Salles, G. et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet 377, 42–51 (2011).
Swerdlow, S. H. et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 4th edn, (IARC Press, 2017).
Casulo, C. et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the national lymphocare study. J. Clin. Oncol. 33, 2516–2522 (2015).
Flinn, I. W. et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 123, 2944–2952 (2014).
Alcoceba M., Alonso-Alvarez S., García-Álvarez M., Martín A. & Caballero M. D. Unmet needs in histological transformation of follicular lymphoma: a clinical and biological review. Ann. Lymphoma 1, 11 2017.
Fischer, T., Zing, N. P. C., Chiattone, C. S., Federico, M. & Luminari, S. Transformed follicular lymphoma. Ann. Hematol. 97, 17–29 (2018).
Casulo, C., Burack, W. R. & Friedberg, J. W. Transformed follicular non-Hodgkin lymphoma. Blood 125, 40–47 (2015).
Alonso-Alvarez, S. et al. Risk of, and survival following, histological transformation in follicular lymphoma in the rituximab era. A retrospective multicentre study by the Spanish GELTAMO group. Br. J. Haematol. 178, 699–708 (2017).
Montoto, S. et al. Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J. Clin. Oncol. 25, 2426–2433 (2007).
Solal-Celigny, P. et al. Follicular lymphoma international prognostic index. Blood 104, 1258–1265 (2004).
Federico, M. et al. Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J. Clin. Oncol. 27, 4555–4562 (2009).
Kridel, R., Sehn, L. H. & Gascoyne, R. D. Can histologic transformation of follicular lymphoma be predicted and prevented? Blood 130, 258–266 (2017).
Berget, E., Molven, A., Lokeland, T., Helgeland, L. & Vintermyr, O. K. IGHV gene usage and mutational status in follicular lymphoma: correlations with prognosis and patient age. Leuk. Res. 39, 702–708 (2015).
Damle, R. N. et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 94, 1840–1847 (1999).
Hamblin, T. J., Davis, Z., Gardiner, A., Oscier, D. G. & Stevenson, F. K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94, 1848–1854 (1999).
Tobin, G. et al. Somatically mutated Ig V(H)3-21 genes characterize a new subset of chronic lymphocytic leukemia. Blood 99, 2262–2264 (2002).
Camacho, F. I. et al. Molecular heterogeneity in MCL defined by the use of specific VH genes and the frequency of somatic mutations. Blood 101, 4042–4046 (2003).
Martin-Jimenez, P. et al. Molecular characterization of heavy chain immunoglobulin gene rearrangements in Waldenstrom’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Haematologica 92, 635–642 (2007).
Zuckerman, N. S. et al. Ig gene diversification and selection in follicular lymphoma, diffuse large B cell lymphoma and primary central nervous system lymphoma revealed by lineage tree and mutation analyses. Int. Immunol. 22, 875–887 (2010).
Sebastian, E. et al. Molecular characterization of immunoglobulin gene rearrangements in diffuse large B-cell lymphoma: antigen-driven origin and IGHV4-34 as a particular subgroup of the non-GCB subtype. Am. J. Pathol. 181, 1879–1888 (2012).
Brisou, G. et al. A restricted IGHV gene repertoire in splenic marginal zone lymphoma is associated with autoimmune disorders. Haematologica 99, e197–e198 (2014).
Ghia, E. M. et al. Use of IGHV3-21 in chronic lymphocytic leukemia is associated with high-risk disease and reflects antigen-driven, post-germinal center leukemogenic selection. Blood 111, 5101–5108 (2008).
Baliakas, P. et al. Not all IGHV3-21 chronic lymphocytic leukemias are equal: prognostic considerations. Blood 125, 856–859 (2015).
Bahler, D. W. et al. gene expression among human follicular lymphomas. Blood 78, 1561–1568 (1991).
Noppe, S. M. et al. The genetic variability of the VH genes in follicular lymphoma: the impact of the hypermutation mechanism. Br. J. Haematol. 107, 625–640 (1999).
Aarts, W. M. et al. Variable heavy chain gene analysis of follicular lymphomas: correlation between heavy chain isotype expression and somatic mutation load. Blood 95, 2922–2929 (2000).
Lynch, R. C., Gratzinger, D. & Advani, R. H. Clinical Impact of the 2016 update to the WHO lymphoma classification. Curr. Treat. Options Oncol. 18, 45 (2017).
Casaril, A. E. et al. Standardization of DNA extraction from sand flies: application to genotyping by next generation sequencing. Exp. Parasitol. 177, 66–72 (2017).
Gonzalez, M. et al. Heteroduplex analysis of VDJ amplified segments from rearranged IgH genes for clonality assessments in B-cell non-Hodgkin’s lymphoma. A comparison between different strategies. Haematologica 84, 779–784 (1999).
Van Dongen, J. J. et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 17, 2257–2317 (2003).
Gribben, J. G. et al. All advanced stage non-Hodgkin’s lymphomas with a polymerase chain reaction amplifiable breakpoint of bcl-2 have residual cells containing the bcl-2 rearrangement at evaluation and after treatment. Blood 78, 3275–3280 (1991).
Cheson, B. D. et al. Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J Clin. Oncol. 17, 1244 (1999).
Cheson, B. D. et al. Revised response criteria for malignant lymphoma. J. Clin. Oncol. 25, 579–586 (2007).
Cheson, B. D. et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J. Clin. Oncol. 32, 3059–3068 (2014).
Shi, Q. et al. Thirty-month complete response as a surrogate end point in first-line follicular lymphoma therapy: an individual patient-level analysis of multiple randomized trials. J. Clin. Oncol. 35, 552–560 (2016).
Pastore, A. et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 16, 1111–1122 (2015).
Blaker, Y. N. et al. The tumour microenvironment influences survival and time to transformation in follicular lymphoma in the rituximab era. Br. J. Haematol. 175, 102–114 (2016).
Brezinschek, H. P. et al. Analysis of the human VH gene repertoire. Differential effects of selection and somatic hypermutation on human peripheral CD5(+)/IgM+and CD5(-)/IgM+B cells. J. Clin. Invest. 99, 2488–2501 (1997).
Hadzidimitriou, A. et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood 118, 3088–3095 (2011).
Agathangelidis, A. et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood 119, 4467–4475 (2012).
Zibellini, S. et al. Stereotyped patterns of B-cell receptor in splenic marginal zone lymphoma. Haematologica 95, 1792–1796 (2010).
Arons, E. et al. Evidence of canonical somatic hypermutation in hairy cell leukemia. Blood 117, 4844–4851 (2011).
Gonzalez, D. et al. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood 110, 3112–3121 (2007).
Hsu, F. J. & Levy, R. Preferential use of the VH4 Ig gene family by diffuse large-cell lymphoma. Blood 86, 3072–3082 (1995).
Catherwood, M. A. et al. Improved clonality assessment in germinal centre/post-germinal centre non-Hodgkin’s lymphomas with high rates of somatic hypermutation. J. Clin. Pathol. 60, 524–528 (2007).
Rosenquist, R., Lindstrom, A., Holmberg, D. & Lindh, J. & Roass G. V. gene family utilization in different B-cell lymphoma subgroups. Eur. J. Haematol. 62, 123–128 (1999).
Ghia, P. et al. Geographic patterns and pathogenetic implications of IGHV gene usage in chronic lymphocytic leukemia: the lesson of the IGHV3-21 gene. Blood 105, 1678–1685 (2005).
Lossos, I. S. et al. Ongoing immunoglobulin somatic mutation in germinal center B cell-like but not in activated B cell-like diffuse large cell lymphomas. Proc. Natl Acad. Sci. USA 97, 10209–10213 (2000).
Ruminy, P. et al. The isotype of the BCR as a surrogate for the GCB and ABC molecular subtypes in diffuse large B-cell lymphoma. Leukemia 25, 681–688 (2011).
Medina, A. et al. VDJH gene repertoire analysis in multiple myeloma (MM) patients: correlation with clinical data. Blood 132, 4446 (2018).
Stamatopoulos, K. et al. Follicular lymphoma immunoglobulin kappa light chains are affected by the antigen selection process, but to a lesser degree than their partner heavy chains. Br J. Haematol. 96, 132–146 (1997).
Federico, M. et al. Rituximab and the risk of transformation of follicular lymphoma: a retrospective pooled analysis. Lancet Haematol. 5, e359–e367 (2018).
Rossi, D. et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br. J. Haematol. 142, 202–215 (2008).
Acknowledgements
This work was partially supported by the Instituto de Salud Carlos III (ISCIII), Spanish Ministry of Economy and Competitiveness PI15/01393, PI18-00410, RD12/0036/0069, CIBERONC-CB16/12/00233, and “Una manera de hacer Europa” (Innocampus; CEI-2010-1-0010)”, the Education Council or Health Council of the Junta de Castilla y León (CAS102P17, GRS 1180/A/15), and Gilead Sciences (GLD17/00334). M.G.A., I.P.C. and C.J. are supported by the Fundación Española de Hematología y Hemoterapia (FEHH, co-funded by Fundación Cris in the latter case), and M.E.S. by the ISCIII (CPII18/00028). All Spanish funding is co-sponsored by the European Union FEDER program. We wish to thank Felicitación García, Montserrat Hernández-Ruano, Estrella Arnés, Mercedes Jiménez, Alejandra Martín, Isabel Sánchez, Rocío Corral and Francisco Boix (University Hospital of Salamanca, Spain) for their technical support, and Phil Mason, for checking the English grammar of the manuscript. The authors are also very grateful to the patients who participated in this study.
Author information
Authors and Affiliations
Contributions
M.G-A., S.A-A., R.G-S. and M.A. conceived and designed the study. M.G-A., S.A-A., I.P-C., C.J., A.M., A.B., R.M. and A.A. produced the molecular results. M.R. and O.B. were the pathologists involved in the pathological and ICH review of the cases. M.A. and M.G-A. analysed the data, performed the statistical analyses, and wrote the first draft of the manuscript. V.G-C. incorporated the clinical data into the database, and checked data for inconsistencies. M.E.S. and M.C.C. provided statistical support. N.P., A.M., R.G-S., M.G. and M.D.C. provided patient samples and/or clinical data. V.G.C., A.M., R.G-S. and M.G. reviewed the final manuscript. M.G., M.A. and M.D.C. obtained funding for the study. R.G-S., the head of the group, produced the final revision of the manuscript and gave final approval its publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors reports grants PI15/01393, PI18-00410, CAS102P17, GRS 1180/A/15, GLD17/00334, CIBERONC-CB16/12/00233 during the conduct of the study, and from Gilead Sciences (GLD15/00348, GLD16/00162), Incyte, Janssen, and Amgen outside the submitted work. M.G.A., I.P.C., C.J., V.G.C. and M.E.S. are or were supported by the Spanish government (ISCIII and/or FEHH). Consulting fees and/or non-financial support were reported from Abbvie (M.G.), Amgen (N.P., M.D.C), BMS (R.G-S., M.D.C.), Celgene (N.P., A.M., M.D.C), Gilead (A.M., M.G., M.D.C.), Incyte (R.G-S.), Janssen (N.P., A.M., R.G-S., M.G., M.D.C., M.A.), Kite (M.D.C.), MSD (M.D.C.), Novartis (S.A-A., M.D.C.), Prothena (V.G-C.), Roche (A.M., M.G., M.D.C.), Servier (A.M.), Takeda (N.P., R.G-S., M.D.C.), and Weber (N.P.). R.G-S. is the president of the Spanish Society of Haematology. The remaining authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
García-Álvarez, M., Alonso-Álvarez, S., Prieto-Conde, I. et al. Immunoglobulin gene rearrangement IGHV3-48 is a predictive marker of histological transformation into aggressive lymphoma in follicular lymphomas. Blood Cancer J. 9, 52 (2019). https://doi.org/10.1038/s41408-019-0213-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-019-0213-9
This article is cited by
-
Next-generation sequencing for MRD monitoring in B-lineage malignancies: from bench to bedside
Experimental Hematology & Oncology (2022)
