
Abstract
Polycythemia vera is characterized by the acquisition of the JAK2V617F mutation. Recommended treatments include hydroxyurea and interferon-alpha. Several groups have reported a reduction in the JAK2 mutant allele burden in interferon-treated patients, but significance of this observation is questioned. We characterized the activity of ropeginterferon alpha-2b, a novel form of interferon-alpha recently shown to be safe and efficacious in polycythemia vera. Ropeginterferon was able to inhibit the proliferation of the HEL, UKE-1, and UT-7 JAK2-mutant cell lines while sparing JAK2-wild-type UT-7 and normal CD34+ cells growth. In vitro treatment of erythroid progenitors derived from PV patients showed that ropeginterferon could considerably inhibit the growth of endogenous erythroid colonies, a hallmark of polycythemia vera. Finally, we could study in sequential samples the clonal architecture of erythroid progenitors derived from patients included in a randomized study comparing hydroxyurea to ropeginterferon. After 1 year of treatment with ropeginterferon, the ratio of JAK2-mutated to wild-type colonies grown from bone marrow progenitors was reduced by 64%, compared to 25% in patients receiving hydroxyurea. This study shows that ropeginterferon has a potent targeted activity against JAK2-mutant cells and is able to drastically reduce the proportion of malignant progenitors in patients treated with this drug.
Similar content being viewed by others


Introduction
Polycythemia vera (PV) is characterized by a deregulated erythropoiesis related to the presence of the JAK2V617F mutation1, which induces a constitutive activation of the JAK/STAT intracellular signaling. More recently, additional mutations in various genes have been discovered, including mutations in genes affecting the epigenome (TET2, ASXL1, DNMT3a, or EZH2), or contributing to transformation to acute leukemia (NRAS, KRAS, TP53, or IDH1/2)2,3. The presence of such additional mutations may also influence the response to therapy4. Current first-line recommended cytoreductive treatment of PV include hydroxyurea (HU) or interferon-alfa (IFNα)5,6,7. The clinical efficacy of IFNα has been reported since 30 years8 and was improved with the development of pegylated forms9,10. Furthermore, we and others have observed significant reductions of the JAK2V617F allele burden (%JAK2V617F) in IFNα-treated patients11,12 suggesting that IFNα is able to specifically target the malignant clone. The mechanism of action of IFNα in myeloproliferative neoplasms is not clearly elucidated, but several studies confirmed a targeted effect against JAK2V617F mutant clones in both patients13 and animal models14,15. In clinical practice, a significant proportion of patients still experience adverse events leading to treatment discontinuation with current formulations of IFNα16. RopegInterferon alpha-2b (Ropeg) is a long-acting pegylated-IFNα-2b, recently shown to be safe and well tolerated in phase 1–2 studies in PV patients17,18. Both hematological and molecular responses have been reported in a phase 2 trial including 51 patients.
In this study, we characterized Ropeg activity against JAK2V617F mutated cells. We tested this new drug in vitro against JAK2-mutant and wild-type cell lines and patients’ primary cells. We could also assess the impact of HU and Ropeg treatments in vivo by sequential studies of bone marrow (BM) progenitors of PV patients treated for 12 months with both drugs in a prospective, randomized study.
Patients and methods
Cell lines and reagents
Ropeg (provided by AOP Orphan) was used at two different concentrations consistent with assumed exposure in patients (0.5 and 2 µg/ml), and commercially available standard recombinant interferon-alpha-2a (rIFNα-2a) (Roferon, Roche) was used at 700 U/ml as control. The JAK2V617F positive HEL and UKE-1 cell lines were grown in RPMI + 10% FBS + 1% Glutamax, and IMDM + 10% horse serum + 10% FBS and 1% Glutamax, respectively. We also used the megakaryoblastic leukemia UT-7 cell line expressing the EPO receptor in which either a wild type or a JAK2V617F mutant JAK2 were retrovirally transduced. These cells were cultured in DMEM + 10% FBS + 2 U/mL EPO. Drug efficacy was assessed by counting live cells after Trypan blue staining. All cell lines were tested and found negative for mycoplasma contamination.
Patients
Normal hematopoietic progenitors derived from cord blood and primary cells from PV patients obtained after informed consent were studied in clonogenic assays with or without EPO according to manufacturer’s instructions (Methocult, Stemcell technologies©). The study was approved by the local ethics committee (IRB0006477). The presence of endogenous erythroid colonies (EECs) was determined in cultures without erythropoietin (EPO). Genotyping of the colonies (at least 60 colonies were tested in each condition) was performed by picking, extracting the DNA and testing for the presence of JAK2V617F mutation using the JAK2 Mutascreen kit (Qiagen©). We studied untreated patients and patients included in the PROUD-PV phase 3 clinical trial (NCT01949805) randomly treated with Ropeg or HU. A total of 13 patients have been included in the PROUD-PV study in France for which BM samples were sent to our laboratory before the initiation of treatment and after 1 year. Informed consent was obtained from all subjects.
Mutant allele burden assessment
The allelic frequency of the JAK2V617F mutation was assessed on DNA extracted from whole blood (QiaAmp DNA blood mini kit, Qiagen) using the JAK2 MutaQuant kit (Qiagen) according to the manufacturer’s instruction. Search for additional mutations was performed using a targeted next generation sequencing (NGS) assay as previously described4.
Results
We first intended to evaluate the action of Ropeg on the proliferation of JAK2V617F-mutated cell lines. In both HEL and UKE-1 cell lines Ropeg exhibited a dose-dependent anti-proliferative effect, comparable to that of standard rIFNα-2a (Fig. 1A, B; Supplementary figure 1). Ropeg at 0.5 µg/ml and 2 µg/ml induced 9%, and 41% inhibition of HEL cells proliferation, and 18 and 35% inhibition of UKE-1 cells proliferation, respectively (compared to 38%, and 36% inhibition of HEL, and UKE-1 cells proliferation, respectively, with rIFNα-2a). To test for a more-specific action of Ropeg against the JAK2V617F mutant form compared to JAK2 wild type, we used the UT-7 model in which both forms of the JAK2 gene have been transduced. We observed a modest impact of AOP treatment on the JAK2 wild-type UT-7 cells proliferation (23% of inhibition at day 3) while a marked decrease in the proliferation of JAK2V617F positive cells was observed (40% reduction at day 3 with 2 µg/ml) (Fig. 1C, D; Supplementary Figure 1).

A and B The JAK2V617F positive UKE1 and HEL cell lines. C and D The UT-7 cell line expressing a wild type or a mutant form of JAK2. Cells were treated with the indicated drugs and the living cells were counted every day. Results are expressed as the fold increase compared to day 0
We then studied the impact of an in vitro treatment with Ropeg on the clonogenic potential of erythroid progenitors derived from four untreated PV patients. Ropeg reduced the numbers of colonies grown with or without EPO in all samples. The mean reduction of the numbers of EPO-stimulated colonies was 29 and 59% with Ropeg at 0.5 µg/ml, and 2 µg/ml, respectively. A more striking effect was observed on EECs, the number of colonies being reduced by 90% with Ropeg 2 µg/ml (Fig. 2A). In contrast, Ropeg didnot significantly change clonogenic properties of normal (JAK2 wild type) hematopoietic progenitors isolated from three different cord blood samples (Supplementary figure 2), suggesting that Ropeg inhibits JAK2-mutated hematopoietic progenitors while sparing wild-type cells. To confirm this hypothesis we studied the JAK2 mutational status at the clonogenic level by genotyping individual colonies grown after in vitro treatment with Ropeg. Single colonies from the 4 PV patients were picked (at least 60 colonies per condition) and genotyped. The proportion of mutant colonies was reduced in every patient (Supplementary figure 3) with a median twofold increase in the ratio of wild type to mutant colonies. In addition, in two patients with both homozygous and heterozygous JAK2V617F colonies, eradication of homozygous colonies was achieved with Ropeg, suggesting that JAK2V617F homozygous progenitors are more sensitive to Ropeg, in agreement with previous findings in MPN patients treated with pegylated-IFNα−2a.

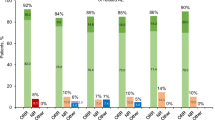
A Clonogenic assays on primary peripheral blood mononuclear cells from 4 PV patients. Median percentages and standard deviations of residual erythroid colonies in treated conditions compared to untreated are presented. B and C JAK2V617F allele burden evolution in patients included in the PROUD-PV trial in France. B median of the %JAK2V617F in HU (N = 5) or Ropeg (N = 3) treatment arms. C Comparison of the % reduction of JAK2V617F allele burden in patients after 1 year of treatment in each arm. Median and standard deviations in both groups of treatment are represented. (*p < 0.02; unpaired t-test). D and E Clonogenic assays performed on bone marrow samples from patients included in the PROUD-PV trial in France. Percentages of JAK2V617F positive erythroid colonies before and after 1 year of treatment in patients treated with Ropeg or HU. UPN of each individual patient is given
In addition to these in vitro studies, we took advantage of the PROUD-PV randomized study to search for any correlation between the evolution of the %JAK2V617F in peripheral blood and the impact of therapy on malignant clones assessed by clonogenic assays on BM progenitors. Thirteen patients were included in this study in France, who accepted to participate in an ancillary study of the evolution of their BM progenitors before and after 12 months of treatment in both randomization arms. In this group of patients the mean %JAK2V617F at baseline were 39.4% and 46.5% in the Ropeg and HU arms, respectively. Median %JAK2 V617F was reduced to 29% and 25.8% after 6 months, and to 13.8% and 33.2% after 12 months (Fig. 2B) of treatment with Ropeg and HU, respectively. These data suggested a sustained activity of Ropeg, but not of HU, against JAK2V617F mutant cells. Indeed, we observed a difference between the two drugs considering the median reduction of the %JAK2V617F (p < 0.02) and the responses were more homogeneous in Ropeg compared to HU-treated patients (Fig. 2C).
To better assess the impact of both drugs on JAK2-mutated cells in vivo we performed clonogenic assays on BM mononuclear cells taken before treatment initiation and after 1 year in order to analyze the changes in clonal architecture. As expected in PV, we observed the presence of EECs in every patient. The ratio of JAK2-mutant to JAK2 wild-type colonies decreased in all of the three patients treated with Ropeg (Fig. 2D) while it decreased in only one out of four patients receiving HU (Fig. 2E). Of note, the poorer clonal response measured by %JAK2V617F in the HU arm wasn't explained by the presence of additional mutations. Using targeted NGS4, we could detect additional mutations in only two patients: one TET2 mutation (p.S393Lfs*34; VAF 27%) in a patient randomized in the HU arm, and one DNMT3A mutation (p.S837*; VAF 7%) in a patient included in the Ropeg arm. Interestingly, the only patient in whom the percentage of JAK2-mutant colonies decreased during HU therapy was the patient with concomitant JAK2 and TET2 mutations.
Discussion
IFNα is a cytokine with a wide range of biological properties including antitumor activity used for decades to treat several types of cancers like melanoma, renal cancer, and hematological malignancies, including MPNs9. In this study, we assessed the ability of a new form of IFNα to specifically target JAK2-mutant cells. We first showed that Ropeg has an antiproliferative effect on JAK2V617F mutant cell lines similar to that of standard recombinant IFNα. In the UT-7 cell line model we could further show that Ropeg has a twofold greater inhibitory effect against JAK2V617F-mutated cells compared to their wild-type counterpart. When tested in cultures of primary hematopoietic progenitors derived from PV patients, Ropeg showed an important inhibitory effect on the growth of erythroid colonies, when it had no particular impact on the growth of erythroid colonies when tested at therapeutic concentrations against normal JAK2-wild-type progenitors. Altogether, these results suggest that Ropeg preferentially targets progenitors harboring the JAK2V617F mutation while sparing wild type cells.
In the subset of patients included in France in the PROUD-PV trial, we observed deeper and more prolonged molecular responses (assessed by the reduction of %JAK2V617F in peripheral blood) in patients randomly assigned to Ropeg versus those receiving HU. In chronic myeloid leukemia, achievement of molecular remission has been clearly associated with better clinical outcome and possible treatment free remissions after discontinuation of tyrosine-kinase inhibitor treatment19. Such correlation between reduction of the JAK2 mutant allele burden in PV with IFNα and clinically relevant outcomes has not been demonstrated yet. To better understand the biological significance of sustained circulating mutant allele burden reduction, we studied the evolution of clonal architecture of hematopoietic progenitors in PV patients randomly treated with IFNα or HU in the PROUD-PV study. Clonal studies of bone marrow progenitors of these patients showed that after 12 months of treatment with IFNα the proportion of wild type to mutant JAK2 colonies was clearly increased, an effect not seen in patients receiving HU. This is to our knowledge the first evidence suggesting that a sustained decrease in the circulating JAK2V617F allele burden could reflect a diminution of the proportion of malignant progenitors in the bone marrow. In that perspective, such results suggest that achievement of deep molecular response in PV could result in the exhaustion of MPN progenitors and open the way for safe cytoreductive treatment discontinuation as currently proposed in CML patients achieving deep molecular response.
Altogether, these preliminary results obtained in a small subset of patients included in the PROUD-PV study must be confirmed in larger numbers, but they suggest that IFNα could be a good candidate for achieving long-term eradication of JAK2-mutant clones and restoring normal hematopoiesis in PV patients.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005).
Vainchenker, W. & Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 129, 667–679 (2017).
Tefferi, A. Myeloproliferative neoplasms: a decade of discoveries and treatment advances. Am. J. Hematol. 91, 50–58 (2016).
Verger, E. et al. Clinical and molecular response to interferon alpha therapy in essential thrombocythemia patients with CALR mutations. Blood 126, 2585–91 (2015).
Vannucchi, A. M. et al. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 26(suppl 5), v85–v99 (2015).
Barbui, T. et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European Leukemia Net. Leukemia 32, 1057–1069 (2018).
Tefferi, A., Vannucchi, A. M. & Barbui, T. Polycythemia vera treatment algorithm 2018. Blood Cancer J. 8, 3 (2018).
Silver, R. T. Recombinant interferon-alpha for treatment of polycythaemia vera. Lancet 2, 403 (1988).
Kiladjian, J. J., Giraudier, S. & Cassinat, B. Interferon-alpha for the therapy of myeloproliferative neoplasms: targeting the malignant clone. Leukemia 30, 776–781 (2016).
Kiladjian, J. J. Long-term treatment with interferon alfa for myeloproliferative neoplasms. Lancet Haematol. 4, e150–e151 (2017).
Kiladjian, J. J. et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 112, 3065–3072 (2008).
Quintás-Cardama, A. et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J. Clin. Oncol. 27, 5418–5424 (2009).
Hasan, S. et al. Use of the 46/1 haplotype to model JAK2(V617F) clonal architecture in PV patients: clonal evolution and impact of IFNα treatment. Leukemia 28, 460–463 (2014).
Mullally, A. et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood 121, 3692–3702 (2013).
Hasan, S. et al. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNα. Blood 122, 1464–1477 (2013).
Masarova, L. et al. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 4, e165–e175 (2017).
Them, N. C. et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am. J. Hematol. 90, 288–294 (2015).
Gisslinger, H. et al. Ropeginterferon alfa-2b, a novel IFNα-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 126, 1762–9 (2015).
Mahon, F. X. Treatment-free remission in CML: who, how, and why? Hematol. Am. Soc. Hematol. Educ. Program. 2017, 102–109 (2017).
Acknowledgements
We are indebted to the Laboratoire de Biologie Cellulaire lab routine team and the Centre d’Investigations Cliniques staff members for their excellent technical support. We are grateful to Dr Norio Komatsu (Juntendo University, Tokyo, Japan) for sharing the UT-7 cell line. This study was supported in part by the Canceropole Ile de France, the French National Cancer Institute (INCa: grant 2014-1-PL BIO-12 and “CTIM3” grant TRANSLA 13-140) and AOP Orphan who also gave Ropeginterferon for in vitro testing.
Authors contributions
J.-J.K. and B.C. designed and performed the study and analyzed the data; E.V., N.M., and R.K. performed the proliferation assays, clonal architecture, and molecular studies; J.S.-D., L.R., J.R., H.G., and J.-J.K. enrolled patients in the study and collected data; B.G.-I., C.K., Z.G., C.C., and B.C. designed the in vitro assays; J.-J.K. and B.C. drafted the manuscript; all authors reviewed and gave final approval of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr Cassinat and Dr Kiladjian received institutional research grants from Novartis and AOP Orphan; Dr Kiladjian participated in advisory boards for Novartis and AOP Orphan.
Ethics approval and consent to participate
The study was approved by the local ethics committee (IRB0006477). All patients participating in the PROUD-PV clinical trial (NCT01949805) provided informed consent.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verger, E., Soret-Dulphy, J., Maslah, N. et al. Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer Journal 8, 94 (2018). https://doi.org/10.1038/s41408-018-0133-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-018-0133-0
This article is cited by
-
ROP-ET: a prospective phase III trial investigating the efficacy and safety of ropeginterferon alfa-2b in essential thrombocythemia patients with limited treatment options
Annals of Hematology (2024)
-
Clonal architecture evolution in Myeloproliferative Neoplasms: from a driver mutation to a complex heterogeneous mutational and phenotypic landscape
Leukemia (2023)
-
Treatment of hydroxyurea-resistant/intolerant polycythemia vera: a discussion of best practices
Annals of Hematology (2023)
-
Long-term outcomes of polycythemia vera patients treated with ropeginterferon Alfa-2b
Leukemia (2022)
-
Polycythemia vera: historical oversights, diagnostic details, and therapeutic views
Leukemia (2021)
