
Abstract
Recently reported mature survival data have confirmed the favorable prognosis in polycythemia vera (PV), with an estimated median survival of 24 years, in patients younger than age 60 years old. Currently available drugs for PV have not been shown to prolong survival or alter the natural history of the disease and are instead indicated primarily for prevention of thrombosis. Unfortunately, study endpoints that are being utilized in currently ongoing clinical trials in PV do not necessarily target clinically or biologically relevant outcomes, such as thrombosis, survival, or morphologic remission, and are instead focused on components of disease palliation. Even more discouraging has been the lack of critical appraisal from “opinion leaders”, on the added value of newly approved drugs. Keeping these issues in mind, at present, we continue to advocate conservative management in low-risk PV (phlebotomy combined with once- or twice-daily aspirin therapy) and include cytoreductive therapy in “high-risk” patients; in the latter regard, our first, second, and third line drugs of choice are hydroxyurea, pegylated interferon-α and busulfan, respectively. In addition, it is reasonable to consider JAK2 inhibitor therapy, in the presence of protracted pruritus or markedly enlarged splenomegaly shown to be refractory to the aforementioned drugs.
Similar content being viewed by others

Introduction
Polycythemia vera (PV) is currently classified by the World Health Organization (WHO) classification system under the major category of myeloproliferative neoplasms (MPN)1. Although the WHO MPN category includes seven subcategories, the term “MPN” usually refers to the three JAK2 mutation-enriched clinicopathologic entities: PV, essential thrombocythemia (ET) and primary myelofibrosis (PMF)1. PV and its sister diseases constitute stem cell-derived clonal myeloproliferation that is characterized by three mutually-exclusive “driver” mutations: JAK2, CALR, and MPL, with respective distribution frequency of ~99, 0, and 0% for PV, 55, 22, and 3% for ET and 65, 20 and 7% for PMF2. The most frequent MPN-associated JAK2 mutation is the exon 14 JAK2V617F, which is responsible for almost all the JAK2 mutations in ET and PMF, and 97% of those seen in PV; the remainder 3% of JAK2 mutations in PV are spread across exons 12, 13, and 143,4.
Diagnosis of PV often requires the presence of a JAK2 mutation, in addition to documentation of increased hemoglobin/hematocrit, to a threshold level established by the 2016 World Health Organization (WHO) revised criteria (>16.5 g/dL/49% for males and >16 g/dL/48% for females)1. In addition, bone marrow morphologic assessment is encouraged, in order to distinguish PV from JAK2-mutated ET5,6,7 and obtain cytogenetic information, which has recently been shown to be prognostically relevant8,9,10. Clinical features in PV include mild-to-moderate degree of splenomegaly, mild-to-moderate degree of constitutional symptoms, including fatigue and pruritus, symptoms of hyperviscosity, leukocytosis, thrombocytosis, microvascular symptoms (e.g., headaches, lightheadedness, visual disturbances, atypical chest pain, erythromelalgia, paresthesia), thrombotic and bleeding complications, and risk of leukemic transformation or fibrotic progression11.
Current treatment in PV has not affected the natural history of the disease in regards to overall, leukemia-free or myelofibrosis-free survival, but thrombosis-free survival has been positively affected by treatment with phlebotomy12, aspirin13 and cytoreductive drugs11. In the latter regard, the most popular and evidence-supported cytoreductive agent is hydroxyurea, while busulfan has been effectively and safely utilized for an even longer period11,14. More recently, interferon (IFN)-α and ruxolutinib (a JAK1 and JAK2 inhibitor) have been introduced to the therapeutic armamentarium, without controlled evidence of superiority over the older drugs and documentation of safety during long-term use. In the current review, we provide a risk-adapted treatment algorithm in PV, including critical assessment of the currently available cytoreductive agents.
Risk-adapted treatment algorithm in polycythemia vera
Survival and complications rates
Survival in PV is inferior to that of ET but superior to that of PMF, with estimated medians of 14, 20, and 6 years, respectively;15 the corresponding figures for patients younger than age 60 years are 24, 33, and 15 years15. Life-expectancy in all three MPN is significantly worse than that of the age- and sex-matched general population15. These observations are similar to those from a large population-based study of 9,384 patients16. The major life-threatening complications in PV are leukemic transformation, fibrotic progression and thrombosis, with incidence ranges of 5.5–18.7, 6–14, and 6–17%, respectively, over the course of 15, 15, and 3 years17,18.
Risk factors for survival and predictors of leukemic or fibrotic progression
In the largest international study of 1545 patients with PV, independent risk factors for overall survival included age > 61 years, leukocyte count > 10.5 × 10(9)/L, venous thrombosis and abnormal karyotype and for leukemia-free survival age >61 years, leukocyte count >15 × 109/L and abnormal karyotype;10 median survivals were 23 and 9 years, in the absence or presence of the first two risk factors; these observations were validated by another population-based study of 327 patients19.
The prognostic relevance of karyotype in PV was recently confirmed by subsequent Mayo Clinic and MD Anderson Cancer Center (MDACC) reports;8,9 in both studies, abnormal karyotype was reported in approximately 20% of patients (+9, +8, and 20q− being the most frequent) and adversely affected overall and transformation-free survival. Other genetic alterations, revealed by next generation sequencing, occur in the majority of patients with PV, and include TET2 (22% frequency), ASXL1 (12%), and SH2B3 (9%) mutations20. Some of these mutations, in particular ASXL1, SRSF2, or IDH2, have been shown to adversely impact overall and transformation-free survival; median survival of patients with and without adverse mutations was 7.7 vs. 16.9 years, respectively20.
Incidence of thrombosis and bleeding
The European Collaboration on Low-dose Aspirin in PV (ECLAP) recruited 1638 patients with Polycythemia Vera Study Group (PVSG)-defined PV, at variable times from initial diagnosis, and reported thrombosis history at time of recruitment in 39% (29% arterial and 14% venous) of the patients21,22. After a median follow-up of 2.8 years, 14% of the patients experienced cardiovascular events (incidence of 5.5 events/100 persons/year; 6.95 and 2.52 in high and low-risk patients, respectively) and thrombosis accounted for the main cause of death (41%). Bleeding history in the ECLAP study was 8.1% at time of study entry and 2.9% during follow-up.
In a more recent retrospective study of 1545 patients with WHO-defined PV, conducted by the International Working Group for MPN Research and Treatment (IWG-MRT), thrombosis history at diagnosis was documented in 23% of the patients and included 16% arterial and 7.4% venous events23. These figures were lower than those described above for the ECLAP study but similar to those reported by the Cytoreductive Therapy in PV (CYTO-PV) study (17% arterial and 12% venous), which included patients with WHO-defined PV12. The rate of post-diagnosis vascular events in the IWG-MRT study, after a median follow-up of 6.9 years, was 21% (2.62% patients/year), including 12% arterial and 9% venous events; additional analysis revealed 21% and 23% incidence of thrombosis history at diagnosis, in patients diagnosed before or after 2005, respectively;18 the corresponding thrombosis rates after median follow-up of 2.5–3.5 years from diagnosis were 10%/17% in low/high-risk patients diagnosed before 2005 and 6%/7% for those diagnosed after 200518.
Risk factors for thrombosis and current risk stratification
Risk factors for thrombosis, in the aforementioned ECLAP study, were age >65 years, thrombosis history, hypertension, tobacco use and congestive heart failure;21,22 subsequent observations from the same study also identified leukocyte count >15 × 109/L compared to <10 × 109/L, but not hematocrit level or platelet count24, as a risk factor for thrombosis, especially myocardial infarction25. The potential contribution of increased leukocyte count to thrombosis in PV was also highlighted in the context of the CYTO-PV study26 and recurrent thrombosis, especially in patients younger than age 60 years27,28. In the IWG-MRT study, arterial and venous thromboses were the main risk factors for recurrent arterial or venous vascular events, respectively23. In addition, history of hypertension predicted arterial thrombosis and advanced age (≥65 years) venous thrombosis. A more recent study has suggested that arterial hypertension might be a significant risk factor for thrombosis, even in low-risk patients29. Another study suggested that PV patients with bone marrow fibrosis might be at a lower risk for thrombosis30.
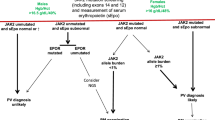
On the basis of the above, we consider PV patients with thrombosis history to be at a significantly higher risk for recurrent thrombosis; in this regard, it is therapeutically relevant to distinguish patients with arterial vs venous thrombosis history (Fig. 1). The above-described studies also identify advanced age as an independent risk factor for thrombosis in PV and, accordingly, patients with either thrombosis history or advanced age are currently classified as having “high-risk” disease, while the absence of both risk factors is required for “low-risk” disease classification (Fig. 1). In addition, although not included in our current risk stratification scheme, we take the presence of hypertension and leukocytosis into consideration, when deciding treatment in certain circumstances (Fig. 1).

Current treatment algorithm in Polycythemia vera
Risk-adapted therapy: low-risk disease
Prior to the introduction of phlebotomy as a treatment modality in PV, reported median survivals for untreated PB were less than 2 years and attributed to excess death from thrombotic complications31. The anti-thrombotic value of aggressive phlebotomy in PV was reinforced by a randomized study that showed the benefit of keeping the hematocrit below 45%12. Controlled studies have also confirmed the additional anti-thrombotic value of low-dose aspirin in PV, among all risk categories13. Accordingly, we recommend aspirin therapy (81–100 mg once-daily) + phlebotomy with a target hematocrit of 45%, in all male and female patients with PV, regardless of risk status (Fig. 1). In addition, the emerging data from laboratory studies and clinical observations suggest that the increased platelet turnover in MPN results in suboptimal 24-hour suppression of thromboxane-A2 synthesis by once-daily dosing;32,33 therefore, we consider twice-daily dosing in low-risk patients whose microvascular symptoms are not adequately controlled with once-daily dosing or who are at high-risk for arterial thrombosis, including those with cardiovascular risk factors (especially hypertension) and leukocytosis (Fig. 1).
Most recently, two studies have re-visited the issue of the frequency of phlebotomy and thrombosis risk in PV, based on older polycythemia vera study group data that suggested increased risk of thrombosis in the first 3 years of treatment, in patients treated with phlebotomy alone34,35. In the first observational study35, the authors showed an association between requiring 3 or more phlebotomies per year and increased risk of thrombosis, despite concomitant treatment with hydroxyurea. This observation was not confirmed by the more robust second study that utilized data from a controlled study and implicated uncontrolled hematocrit level instead of frequency of phlebotomy as the culprit;34 furthermore, there is additional evidence that suggests the contribution of leukocytosis to the increased risk of thrombosis in patients with inadequate control of hematocrit26.
Risk-adapted therapy: high-risk disease
Our current recommendations on the management of high-risk PV are based on both controlled and large retrospective and single arm prospective studies (Fig. 1). The PVSG conducted the first controlled study in PV (1967–1974) that compared treatment with phlebotomy alone or in combination with either oral chlorambucil or intravenous radioactive phosphorus (P32). The results revealed accelerated leukemic transformation and shortening of survival in patients receiving chlorambucil of P3236,37. During the same time period (1967–1978), the European Organization for Research on Treatment of Cancer (EORTC) conducted another randomized study in PV comparing oral busulfan with P32 and showed a survival advantage for busulfan38.
Other randomized studies in PV compared hydroxyurea with pipobroman (shorter survival and an increased risk of leukemic transformation with pipobroman therapy)39,40, P32 alone or with hydroxyurea (no difference in survival but the combination treatment was associated with increased leukemic transformation)41, and P32 + phlebotomy vs phlebotomy + high-dose aspirin (900 mg/day) in combination with dipyridamole (225 mg per day) (the addition of anti-platelet agents increased the risk of gastrointestinal bleeding)42. However, in a subsequent study by ECLAP, a lower dose of aspirin (100 mg daily), compared to placebo, was not associated with excessive bleeding and was shown to reduce thrombosis risk13.
The above-outlined observations on hydroxyurea treatment for PV were further supported by several other uncontrolled studies, including a PVSG study where the drug was associated with lower incidences of thrombosis, compared to a historical cohort treated with phlebotomy alone (6.6% vs 14% at 2 years), and leukemic transformation, compared to a historical control treated with either chlorambucil or P32 (5.9 vs. 10.6 vs. 8.3%, respectively, in the first 11 years of treatment)43. Several other uncontrolled studies have since confirmed the lack of association between hydroxyurea treatment and leukemic transformation with reported incidence range of 1–5.6%44,45,46. Pegylated IFN-α, as initial therapy for PV, has also been shown to be safe and effective, with reported complete hematologic and molecular response rates of 76–95 and 18%, respectively;47,48 overall and complete hematologic remission rates from an ongoing phase-3 study were 81% and 19%49.
On the bais of the above observations, our first-line cytoreductive drug of choice in PV is hydroxyurea (Fig. 1). This, however, does not undermine the possible value of IFN-α as first-line therapy, as an alternative to hydroxyurea, but follow-up time and number of patients treated so far with IFN-α are not adequate enough to justify such a measure at the present time. Our reservation, in this regard, is consistent with the results of interim analysis from an ongoing phase-3 study in PV and ET that suggested similar efficacy but higher toxicity events with pegylated IFN-α, compared to hydroxyurea49. Similarly, the relevance to disease outcome of molecular remissions seen in some PV patients treated with either IFN-α47,48,50 or busulfan51 is currently unclear.
Finally, there is evidence from observational studies that the use of oral anticoagulants, as well as that of aspirin therapy, prevents recurrent venous thrombosis in PV28,52. In one of the few studies that addressed the issue of recurrent thrombosis in PV, the authors were able to demonstrate the anti-thrombotic value of cytoreductive therapy in combination with either anti-platelet agents or systemic anticoagulation28. Furthermore, the study identified specific value for systemic anticoagulation, aspirin therapy and cytoreduction, in the prevention of venous thrombosis, cerebral and coronary events, respectively28. Interestingly, aspirin therapy was also associated with lower risk of venous thrombosis28. These observations were taken into consideration in formulating our treatment recommendations for high-risk disease (Fig. 1).
Treatment options for hydroxyurea intolerant or refractory disease
We currently consider three drugs, as second-line therapy for PV: pegylated IFN- α, busulfan and ruxolutinib. Among the three, long-term safety data are available and considered acceptable for pegylated IFN-α and busulfan (as discussed above). Also, these two drugs, compared to ruxolutinib, display broader activity against clonal myeloproliferation and display better quality of response, including the ability to induce molecular remission47,48,50,51, although the clinical relevance of the latter is debatable. Furthermore, there is already extensive experience in the use of both drugs as initial therapy for PV, with results that appear to be comparable to that of hydroxyurea, as discussed above in the previous section.
In addition to the experience from the aforementioned randomized studies (see above), favorable outcome has also been reported in single arm studies using busulfan as initial therapy;53,54 in 65 busulfan-treated patients with PV followed between 1962 and 1983, median survival was 19 years in patients whose disease was diagnosed before age 60 years and only 2 patients (3.5%) developed acute leukemia53. Busulfan has also been shown to induce durable hematologic response in the majority and molecular response in a minority of patients who are intolerant or resistant to hydroxyurea therapy55,56,57. The concern regarding leukemogenicity of busulfan is unfounded and not supported by controlled evidence; in a large international study of over 1500 patients with PV, neither busulfan nor IFN- α were implicated as being leukemogenic10. Incidentally, busulfan expresses less DNA/RNA binding, compared to other alkylating agents, no inter- or intra-strand DNA binding and no immunosuppression14.
In a randomized study, ruxolitinib was compared to best available therapy, in hydroxyurea-resistant or intolerant PV with splenomegaly;58 treatment with ruxolutinib produced higher rates of hematocrit control (60 vs 20%), ≥ 35% reduction in spleen volume (38 vs 1%) and symptom control (49 vs 5%). However, ruxolutinib therapy was associated with higher rate of herpes zoster infection (6 vs 0%) and showed limited evidence of disease-modifying activity, with complete hematologic remission rate of only 24% and complete molecular remission rate of <2%59. Furthermore, the study was not designed to address clinically relevant endpoints in PV such as thrombosis-free, leukemia-free or myelofibrosis-free survival or bone marrow morphologic remission. Also, resistance to hydroxyurea treatment in PV might reflect more aggressive disease biology that warrants disease-modifying rather than palliative treatment strategy60. Finally, because of their relatively long survival, ruxolutinib-treated patients with PV are vulnerable to drug-induced immunosuppression and opportunistic infections, including reactivation of viral diseases61.
Management of pruritus
Pruritus, sometimes aquagenic, is one of the most annoying, but not necessarily life-threatening complications of PV. In fact, in a recent large international study of 1545 patients with PV, presence of pruritus was independently associated with longer survival10. Among 441 German patients with PV, 301 (68%) reported pruritus (severe and unbearable in 15%), occurring in the majority prior to the diagnosis of PV62. In our own experience involving 418 patients seen at the Mayo Clinic, pruritus at diagnosis was documented in 31% and was associated with a lower rate of arterial thrombosis and higher JAK2V617F allele burden63. The latter observation was confirmed by others as well. Treatment options for PV-associated pruritus include antihistamines64, selective serotonin reuptake inhibitors (SSRIs)65, danazol66, IFN-α67, narrow-band ultraviolet B phototherapy68, photochemotherapy with psoralen and ultraviolet A light (PUVA)69, and JAK270 or mTOR inhibitors71. Amongst these, we recommend initial therapy with antihistamines and SSRIs, followed by IFN-α therapy for more severe and refractory cases, and reserve treatment with JAK2 inhibitors for high-risk patients with IFN-α-resistant pruritus.
Management during pregnancy
There are few published data on the occurrence and outcome of pregnancy in PV patients72,73,74. Therefore, our recommendations in this regard are mostly extrapolated from the experience in ET, which is consistent with the management strategy outlined in the limited series of anecdotal reports on PV. As is the case with ET, there appears to be increased miscarriage rates but otherwise mostly uneventful course and successful outcome72,73,74. Accordingly, we do not consider pregnancy to be contraindicated in women with PV and advise conservative management with once-daily aspirin therapy and phlebotomy to be adequate in “low risk” women, while we recommend the addition of pegylated IFN- α for high-risk disease75. In addition, patients should be closely monitored for pregnancy-induced hypertension.
Conclusions
Patients with PV should look forward to long and productive life and avoid exposure to new drugs whose long-term consequences are not known and might include acceleration of clonal degeneration into acute myeloid leukemia or myelofibrosis. This is not only of theoretical concern and has happened before to PV patients treated with chlorambucil, radiophosphorus or pipobroman37,39 and to ET patients treated with anagrelide76. Decades of experience with hydroxyurea and busulfan, for the treatment of PV or ET, has not produced controlled evidence that implicates either one of these two drugs as being leukemogenic or immunosuppressive. Our concern lies with the newer drugs, including IFN-α77 and ruxolutinib78,79, neither of which has been shown to be superior or safer than conventional therapy, in a controlled study with clinically relevant endpoints (e.g., survival, thrombosis or bone marrow morphologic remission). Furthermore, neither IFN-α nor ruxolutinib induces morphologic or cytogenetic remission in PV or has been shown to alter the natural history of the disease; of note, the clinical relevance of IFN-α-induced suppression of JAK2V617F allele burden, which is seen in a small minority of patients47, and also documented with busulfan therapy51, is unclear. Therefore, it is incumbent upon the MPN community of physicians and scientists to dig deeper into more precise pathogenetic mechanisms and identify targetable disease-specific pathways. In other words, our patients need drugs with anti-tumor activity and do not have to settle for symptomatic relief.
References
Arber, D. A. et al. The2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Tefferi, A. & Pardanani, A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 1, 97–105 (2015).
Tefferi, A. Somatic JAK2 mutations and their tumor phenotypes. Blood 128, 748–749 (2016).
Alghasham, N., Alnouri, Y., Abalkhail, H. & Khalil, S. Detection of mutations in JAK2 exons 12-15 by Sanger sequencing. Int. J. Lab. Hematol. 38, 34–41 (2016).
Barbui, T., Thiele, J., Vannucchi, A. M. & Tefferi, A. Rationale for revision and proposed changes of the WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Blood Cancer J. 5, e337 (2015).
Kvasnicka H. M. et al. European LeukemiaNet study on the reproducibility of bone marrow features in masked polycythemia vera and differentiation from essential thrombocythemia. Am. J. Hematol. 92, 1062–1067 (2017).
Barbui, T. et al. Diagnostic impact of the2016 revised who criteria for polycythemia vera. Am. J. Hematol. 92, 417–419 (2017).
Tang G. et al. Characteristics and clinical significance of cytogenetic abnormalities in polycythemia vera. Haematologica 102, 1511–1518 (2017).
Barraco D. et al. Cytogenetic findings in WHO-defined polycythaemia vera and their prognostic relevance. Br. J. Haematol 2017 (in press).
Tefferi, A. et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 27, 1874–1881 (2013).
Tefferi, A. & Barbui, T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 92, 94–108 (2017).
Marchioli, R. et al. Cardiovascular events and intensity of treatment in polycythemia vera. N. Engl. J. Med. 368, 22–33 (2013).
Landolfi, R. et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 350, 114–124 (2004).
Brodsky, I. Busulfan versus hydroxyurea in the treatment of polycythemia vera (PV) and essential thrombocythemia (ET). Am. J. Clin. Oncol. 21, 105–106 (1998).
Tefferi, A. et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124, 2507–2513 (2014). quiz 2615.
Hultcrantz, M. et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J. Clin. Oncol. 30, 2995–3001 (2012).
Cerquozzi, S. & Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 5, e366 (2015).
Barbui, T. et al. Patterns of presentation and thrombosis outcome in patients with polycythemia vera strictly defined by WHO-criteria and stratified by calendar period of diagnosis. Am. J. Hematol. 90, 434–437 (2015).
Bonicelli, G. et al. Leucocytosis and thrombosis at diagnosis are associated with poor survival in polycythaemia vera: a population-based study of 327 patients. Brit J. Haematol. 160, 251–254 (2013).
Tefferi, A. et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 1, 21–30 (2016).
Finazzi, G. low-dose aspirin in p. A prospective analysis of thrombotic events in the European collaboration study on low-dose aspirin in polycythemia (ECLAP). Pathol. Biol. (Paris). 52, 285–288 (2004).
Marchioli, R. et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J. Clin. Oncol. 23, 2224–2232 (2005).
Barbui, T. et al. In contemporary patients with polycythemia vera, rates of thrombosis and risk factors delineate a new clinical epidemiology. Blood 124, 3021–3023 (2014).
Di Nisio, M. et al. The haematocrit and platelet target in polycythemia vera. Br. J. Haematol. 136, 249–259 (2007).
Landolfi, R. et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 109, 2446–2452 (2007).
Barbui, T. et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood 126, 560–561 (2015).
De Stefano, V. et al. Leukocytosis is a risk factor for recurrent arterial thrombosis in young patients with polycythemia vera and essential thrombocythemia. Am. J. Hematol. 85, 97–100 (2010).
De Stefano, V. et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 93, 372–380 (2008).
Barbui, T. et al. The effect of arterial hypertension on thrombosis in low-risk polycythemia vera. Am. J. Hematol. 92, E5–E6 (2017).
Barbui, T. et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood 119, 2239–2241 (2012).
Chievitz, E. & Thiede, T. Complications and causes of death in polycythaemia vera. Acta Med. Scand. 172, 513–523 (1962).
Dillinger, J. G. et al. Twice daily aspirin to improve biological aspirin efficacy in patients with essential thrombocythemia. Thromb. Res. 129, 91–94 (2012).
Pascale, S. et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 119, 3595–3603 (2012).
Barbui, T. et al. No correlation of intensity of phlebotomy regimen with risk of thrombosis in polycythemia vera: evidence from European Collaboration on low-dose aspirin in polycythemia vera and cytoreductive therapy in Polycythemia vera clinical trials. Haematologica 102, e219–e221 (2017).
Alvarez-Larran, A. et al. Risk of thrombosis according to need of phlebotomies in patients with polycythemia vera treated with hydroxyurea. Haematologica 102, 103–109 (2017).
Berk, P. D, Wasserman, L. R, Fruchtman, S. M. & Goldberg, J. D. In: Polycythemia Vera and the Myeloproliferative Disorders. (eds Wasserman, L. R; Berk, P. D & Berlin, N. I) 166–194 (W.B. Saunders: Philadelphia, 1995).
Berk, P. D. et al. Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil therapy. N. Engl. J. Med. 304, 441–447 (1981).
Treatment of polycythaemia vera by radiophosphorus or busulphan: a randomized trial. “Leukemia and hematosarcoma” Cooperative Group, European Organization for Research on Treatment of Cancer (E.O.R.T.C.). Br. J. Cancer 44, 75–80 (1981).
Kiladjian, J. J., Chevret, S., Dosquet, C., Chomienne, C. & Rain, J. D. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J. Clin. Oncol. 29, 3907–3913 (2011).
Najean, Y. & Rain, J. D. Treatment of Polycythemia Vera - the Use of Hydroxyurea and Pipobroman in 292 Patients Under the Age of 65 Years. Blood 90, 3370–3377 (1997).
Najean, Y. & Rain, J. D. Treatment of polycythemia vera: use of 32P alone or in combination with maintenance therapy using hydroxyurea in 461 patients greater than 65 years of age. The French Polycythemia Study Group. Blood 89, 2319–2327 (1997).
Tartaglia, A. P., Goldberg, J. D., Berk, P. D. & Wasserman, L. R. Adverse effects of antiaggregating platelet therapy in the treatment of polycythemia vera. Semin. Hematol. 23, 172–176 (1986).
Fruchtman, S. M. et al. From Efficacy to Safety - a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin. Hematol. 34, 17–23 (1997).
West, W. O. Hydroxyurea in the treatment of polycythemia vera: a prospective study of 100 patients over a 20-year period. South. Med. J. 80, 323–327 (1987).
Tatarsky, I. & Sharon, R. Management of polycythemia vera with hydroxyurea. Semin. Hematol. 34, 24–28 (1997).
Finazzi, G. et al. Acute leukemia in polycythemia vera. An analysis of 1,638 patients enrolled in a prospective observational study. Blood 105, 2664–2670 (2005).
Quintas-Cardama, A. et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon alpha-2a. Blood 122, 893–901 (2013).
Kiladjian, J. J. et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 112, 3065–3072 (2008).
Mascarenhas, J. O. et al. Interim Analysis of the Myeloproliferative Disorders Research Consortium (MPD-RC) 112 global phase iii trial of front line pegylated interferon alpha-2a vs. hydroxyurea in high risk polycythemia vera and essential thrombocythemia. Blood 128, 479–479 (2016).
Them, N. C. et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am. J. Hematol. 90, 288–294 (2015).
Kuriakose, E. T. et al. JAK2V617F allele burden is reduced by busulfan therapy: a new observation using an old drug. Haematologica 98, e135–e137 (2013).
Hernandez-Boluda, J. C. et al. Oral anticoagulation to prevent thrombosis recurrence in polycythemia vera and essential thrombocythemia. Ann. Hematol. 94, 911–918 (2015).
Messinezy, M., Pearson, T. C., Prochazka, A. & Wetherley-Mein, G. Treatment of primary proliferative polycythaemia by venesection and low dose busulphan: retrospective study from one centre. Br. J. Haematol. 61, 657–666 (1985).
D’Emilio, A., Battista, R. & Dini, E. Treatment of primary proliferative polycythaemia by venesection and busulphan. Br. J. Haematol. 65, 121–122 (1987).
Alvarez-Larran, A. et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann. Hematol. 93, 2037–2043 (2014).
Begna, K. et al. Busulfan for the treatment of myeloproliferative neoplasms: the Mayo Clinic experience. Blood Cancer J. 6, e427 (2016).
Douglas, G. et al. Busulfan is effective second-line therapy for older patients with Philadelphia-negative myeloproliferative neoplasms intolerant of or unresponsive to hydroxyurea. Leuk. Lymphoma 58, 89–95 (2017).
Vannucchi, A. M. et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Eng. J. Med. 372, 426–435 (2015).
Vannucchi, A. M. et al. Ruxolitinib reduces JAK2p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann. Hematol. 96, 1113–1120 (2017).
Alvarez-Larran, A. et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 119, 1363–1369 (2012).
Kirito, K., Sakamoto, M. & Enomoto, N. Elevation of the Hepatitis B Virus DNA during the Treatment of Polycythemia Vera with the JAK Kinase Inhibitor Ruxolitinib. Intern. Med. 55, 1341–1344 (2016).
Siegel, F. P., Tauscher, J. & Petrides, P. E. Aquagenic pruritus in polycythemia vera: characteristics and influence on quality of life in 441 patients. Am. J. Hematol. 88, 665–669 (2013).
Gangat, N. et al. Pruritus in polycythemia vera is associated with a lower risk of arterial thrombosis. Am. J. Hematol. 83, 451–453 (2008).
Weick, J. K. et al. The use of cimetidine for the treatment of pruritus in polycythemia vera. Arch. Intern. Med. 142, 241–242 (1982).
Tefferi, A. & Fonseca, R. Selective serotonin reuptake inhibitors are effective in the treatment of polycythemia vera-associated pruritus. Blood 99, 2627 (2002).
Kolodny, L., Horstman, L. L., Sevin, B. U., Brown, H. & Ahn, Y. S. Danazol relieves refractory pruritus associated with myeloproliferative disorders and other diseases. Am. J. Hematol. 51, 112–116 (1996).
Finelli, C. et al. Relief of intractable pruritus in polycythemia vera with recombinant interferon alfa. Am. J. Hematol. 43, 316–318 (1993).
Madkan, V. K., Bandow, G. D. & Koo, J. Y. Resolution of pruritus secondary to polycythemia vera in a patient treated with narrow-band ultraviolet B phototherapy. J. Dermatol. Treat. 16, 56–57 (2005).
Jeanmougin, M., Rain, J. D. & Najean, Y. Efficacy of photochemotherapy on severe pruritus in polycythemia vera. Ann. Hematol. 73, 91–93 (1996).
Vaa, B. E. et al. Pruritus in primary myelofibrosis: management options in the era of JAK inhibitors. Ann. Hematol. 95, 1185–1189 (2016).
Guglielmelli, P. et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood 118, 2069–2076 (2011).
Aggarwal, N. et al. Polycythemia vera and pregnancy: experience of four pregnancies in a single patient. Arch. Gynecol. Obstet. 283, 393–395 (2011).
Pata, O. et al. Polycythemia vera and pregnancy: a case report with the use of hydroxyurea in the first trimester. Am. J. Perinatol. 21, 135–137 (2004).
Ferguson, J. E. 2nd, Ueland, K. & Aronson, W. J. Polycythemia rubra vera and pregnancy. Obstet. Gynecol. 62, 16s–20s (1983). 3 Suppl.
Tefferi, A. & Passamonti, F. Essential thrombocythemia and pregnancy: Observations from recent studies and management recommendations. Am. J. Hematol. 84, 629–630 (2009).
Harrison, C. N. et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 353, 33–45 (2005).
Kovacsovics-Bankowski, M. et al. Changes in peripheral blood lymphocytes in polycythemia vera and essential thrombocythemia patients treated with pegylated-interferon alpha and correlation with JAK2V617F allelic burden. Exp. Hematol. Oncol. 5, 28 (2015).
Kremyanskaya, M., Mascarenhas, J., Rampal, R. & Hoffman, R. Development of extramedullary sites of leukaemia during ruxolitinib therapy for myelofibrosis. Br. J. Haematol. 167, 144–146 (2014).
Heine, A., Brossart, P. & Wolf, D. Ruxolitinib is a potent immunosuppressive compound: is it time for anti-infective prophylaxis? Blood 122, 3843–3844 (2013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tefferi, A., Vannucchi, A.M. & Barbui, T. Polycythemia vera treatment algorithm 2018. Blood Cancer Journal 8, 3 (2018). https://doi.org/10.1038/s41408-017-0042-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-017-0042-7
This article is cited by
-
Analysis of real-world data demonstrating the efficacy of current management of polycythaemia vera in attaining and maintaining therapeutic haematocrit
Irish Journal of Medical Science (1971 -) (2024)
-
JAK2V617F variant allele frequency, non-driver mutations, single-nucleotide variants and polycythemia vera outcome
Journal of Cancer Research and Clinical Oncology (2023)
-
Efficacy and safety of ropeginterferon alfa-2b in Japanese patients with polycythemia vera: an open-label, single-arm, phase 2 study
International Journal of Hematology (2022)
-
Polycythemia vera: historical oversights, diagnostic details, and therapeutic views
Leukemia (2021)
-
MTAP-related increased erythroblast proliferation as a mechanism of polycythaemia vera
Scientific Reports (2021)
