
Abstract
The fight against coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 infection is still raging. However, the pathophysiology of acute and post-acute manifestations of COVID-19 (long COVID-19) is understudied. Endothelial cells are sentinels lining the innermost layer of blood vessel that gatekeep micro- and macro-vascular health by sensing pathogen/danger signals and secreting vasoactive molecules. SARS-CoV-2 infection primarily affects the pulmonary system, but accumulating evidence suggests that it also affects the pan-vasculature in the extrapulmonary systems by directly (via virus infection) or indirectly (via cytokine storm), causing endothelial dysfunction (endotheliitis, endothelialitis and endotheliopathy) and multi-organ injury. Mounting evidence suggests that SARS-CoV-2 infection leads to multiple instances of endothelial dysfunction, including reduced nitric oxide (NO) bioavailability, oxidative stress, endothelial injury, glycocalyx/barrier disruption, hyperpermeability, inflammation/leukocyte adhesion, senescence, endothelial-to-mesenchymal transition (EndoMT), hypercoagulability, thrombosis and many others. Thus, COVID-19 is deemed as a (micro)vascular and endothelial disease. Of translational relevance, several candidate drugs which are endothelial protective have been shown to improve clinical manifestations of COVID-19 patients. The purpose of this review is to provide a latest summary of biomarkers associated with endothelial cell activation in COVID-19 and offer mechanistic insights into the molecular basis of endothelial activation/dysfunction in macro- and micro-vasculature of COVID-19 patients. We envisage further development of cellular models and suitable animal models mimicking endothelial dysfunction aspect of COVID-19 being able to accelerate the discovery of new drugs targeting endothelial dysfunction in pan-vasculature from COVID-19 patients.
Similar content being viewed by others
Introduction
Coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 infection represents an ongoing public health burden leading to extensive morbidity and mortality worldwide [1]. SARS-CoV-2 targets the pulmonary system, as well as the extrapulmonary system [2]. COVID-19 can manifest with myocardial injury (ischemic heart disease, arrhythmias and cardiomyopathies), arterial stiffness, liver injury and kidney injury [3].
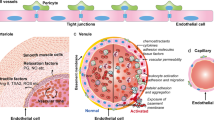
The vascular endothelium, the innermost layer of blood vessels, provides a dynamic interface between the circulating blood and various tissues/organs and thereby maintaining tissue homeostasis. Physiological functions of the endothelium include fine control of vascular tone, tissue hemostasis, barrier integrity, inflammation, oxidative stress, vascular permeability, and structural and functional integrity [4]. A number of viral species, such as dengue, ebola and cytomegalovirus can infect endothelial cells (ECs) and cause endothelial dysfunction [5]. To date, growing evidence supports endothelial dysfunction as a unified key mechanism in the pathogenesis of COVID-19 [6, 7]. Thus, the endothelium is regarded as the Achilles’ heel in COVID-19 patients [8]. Histopathological observations demonstrate that COVID-19 is a (micro)vascular and endothelial disease in which endothelial dysfunction plays a fundamental role [4, 6, 9,10,11]. Direct or indirect mechanism after SARS-CoV-2 infection and the consequent endotheliitis/endotheliopathy incites multiple instances of endothelial dysfunction, including altered vascular tone, oxidative stress, inflammation/leukocyte adhesion, endothelial mesenchymal transition (EndoMT), mitochondria dysfunction, virus-induced senescence, cytokine storm, and coagulopathy [12, 13]. Taken together, the concerted actions of above factors lead to dysfunctional status of the vascular endothelium (endothelial dysfunction) (Fig. 1) [14].

SARS-CoV-2 infection alters the balance of endothelial protective molecules and endothelial damaging molecules, leading to endothelial dysfunction. ADMA asymmetrical dimethylarginine, AngII angiotensin II, Angpt-2 angiopoietin-2, CAT catalase, EDHF endothelium-derived hyperpolarizing factor, eNOS endothelial nitric oxide synthase, ET-1 endothelin 1, GCH1 GTP cyclohydrolase 1, H2S hydrogen sulfide, HO-1 heme oxygenase-1, ICAM1 intercellular adhesion molecule 1, KLF2 krüppel-like factor 2, NO nitric oxide, Nrf2 nuclear factor erythroid 2-related factor 2, PAI-1 plasminogen activator inhibitor 1, PGI2 prostaglandin I2, ROS reactive oxygen species, SOD superoxide dismutase, TF tissue factor, Thbd thrombomodulin, Tie-2 tyrosine-protein kinase receptor, tPA tissue plasminogen activator, Tx-A2 thromboxane A2, uPA urokinase plasminogen activator, VCAM1 vascular cell adhesion molecule 1, vWF von Willebrand factor.
In light of the important contribution of endothelial dysfunction to COVID-19 and its sequelae, we overviewed, in this article, the pivotal role and mechanistic basis of endothelial dysfunction in COVID-19 and its multi-organ complications and markers of endothelial activation. We also present emerging therapeutic agents and therapeutic targets which are directed at reducing the consequence of endothelial dysfunction/endotheliitis/endotheliopathy.
Endothelial function and dysfunction
The endothelium, the widely-distributed organ of the human body, is essential for maintaining tissue homeostasis by producing a variety of vasoactive molecules. These vasoactive molecules tightly control the fine balance between vasodilatory and vasoconstrictory, pro-proliferative and anti-proliferative, pro-thrombotic and anti-thrombotic, pro-oxidant and antioxidant, fibrinolytic and anti-fibrinolytic, and pro-inflammatory and anti-inflammatory responses (Fig. 1). Physiological functions of the vascular endothelium include: (1) maintenance of barrier integrity; (2) regulation of vascular tone; (3) regulation of hemostasis; (4) maintenance of an anti-inflammatory, anti-oxidant and anti-thrombotic interface; (5) regulation of anti-proliferative properties, and (6) regulation of cellular metabolism of ATP, glucose, amino acids, etc. Among these physiological functions, nitric oxide (NO) represents the key mechanism for maintaining endothelial homeostasis [2, 15].
NO is one of the most important vasodilatory substances produced by the vascular endothelium with the action of the endothelial NO synthase (eNOS) and several cofactors. NO also has an anti-thrombotic action, by preventing leukocyte and platelet adhesion to activated endothelium, thereby inhibiting immunothrombosis and atherosclerotic plaque development [15].
Endothelial dysfunction is generally defined as decreased NO bioavailability and an increase in vasoconstrictory substances (such as endothelin-1 (ET1), angiotensin II (Ang II) and many others). The decrease of NO bioavailability occurs partially because of a decrease in eNOS-derived NO production and enormous production of reactive oxygen species (ROS), which inactivates eNOS and causes eNOS uncoupling. ECs are also capable of counteracting ROS, by increasing superoxide dismutase (SOD), catalase, glutathione peroxidase, and NRF2-dependent heme-oxygenase 1 expression [2]. Contemporary definition of endothelial dysfunction has been extended to a constellation of cellular events including oxidative stress, inflammation/leukocyte adhesion, EndoMT, mitochondria dysfunction, senescence and deregulated endothelial cell metabolism [15].
Endothelial dysfunction and COVID-19 associated multi-organ injury
COVID-19 can present with multiple manifestations arising from endothelial dysfunction/endotheliopathy as below (Fig. 2) [2, 16].

Direct SARS-CoV-2 infection or indirect effect arising from SARS-CoV-2 infection leads to endothelial dysfunction in pan-vasculature, which results in the development of multi-organ tissue injury. SASP senescence-associated secretory phenotype.
Acute lung injury
According to the American-European Consensus Conference on acute respiratory distress syndrome (ARDS), the ARDS is the most severe form of the acute lung injury [17] as well as ongoing COVID-19 associated lethality [18]. Pulmonary capillary endothelium provides a fertile “soil” for viral entry, replication, thereby facilitating viral entry to the circulating blood [19, 20]. SARS-CoV-2 infects the ECs and epithelial cells in lung tissues via angiotensin-converting enzyme-2 (ACE2) and alternative receptors [21] on host cells [22]. Interestingly, live SARS-CoV-2 virus and sera from COVID-19 patients, but not dead virus or spike protein triggers increased endothelial permeability [20, 23]. Endothelial integrity is essential for maintaining the pulmonary capillary-alveolar barrier and lung homoeostasis. COVID-19 is associated with pervasive ECs injury, increased capillary permeability, infiltration of inflammatory cells into perivascular tissues, interstitial edema and fluid retention in alveolar spaces [19]. In addition, the release of inflammatory cytokines after severe SARS-CoV-2 infection leads to cytokine storm, tight junction barrier disruption, pulmonary hypertension, and lung fibrosis [24]. Therefore, emerging therapies targeting endothelial dysfunction and endotheliopathy are hopeful to ameliorate COVID-19 associated lung injury [25].
Myocardial injury and myocardial infarction
While COVID-19 primarily affects the lungs, it also affects other organs, the heart in particular. Mortality of COVID-19 patients is increased by comorbidities of cardiovascular disease and hypertension in particular. The most common cardiovascular complications of COVID-19 include arrhythmia, cardiac injury (evidenced by elevated troponin I, creatine kinase, NT-proBNP levels), coagulation (evidenced by elevated level of D-dimer), fulminant myocarditis, heart failure and new-onset atherosclerosis [26]. In these cardiovascular complications, endothelial dysfunction plays a fundamental role [27]. Mechanistically, patients with heart failure demonstrate increased ACE2 gene and protein expression, suggesting that if patients with heart failure were infected by the virus, they are more susceptible to severe COVID-19 and develop into a critically-ill conditions [28]. In addition, COVID-19 is an important risk factor for developing acute myocardial infarction [29]. Data from multi-center registry support that ST-segment elevation myocardial infarction (STEMI) patients enrolled during the first-wave of COVID-19 experience longer time of ischemia and a higher rate of adverse events [30, 31], suggesting the need for COVID-19 vaccines. However, data from a small study cohort demonstrate that the majority of patients with acute myocardial infarction developed symptoms after COVID-19 vaccinations [32]. Since atherosclerosis is an important cause for coronary artery disease, it might be of interest to investigate whether COVID-19 can accelerate the development of endothelial dysfunction and new onset atherosclerosis [26]. In addition, we need to screen for atherosclerotic plaque formation in COVID-19 survivors, as there are no actual clinical data providing the causal relationship between COVID-19 and atherosclerosis.
Liver injury
COVID-19 is also associated with liver injury. By using high-resolution confocal microscopy, a recent study has detected the existence of SARS-CoV-2 viral proteins within the liver sinusoidal endothelial cells (LSECs) from COVID-19 patient liver tissues [33]. In addition, C-type lectin receptor L-SIGN, a receptor highly expressed on LSECs and lymphatic endothelial cells, was identified as the receptor for SARS-CoV-2 infection and may contribute to endotheliopathy in the liver [33].
After virus infection, ensuing cytokine storm occurs in severe COVID-19 patients, particularly the elevated secretion of pro-inflammatory cytokine interleukin 6 (IL-6). Recent studies have suggested that LSEC dysfunction is involved in COVID-19 associated liver injury [34]. A recent study [35] has reported that IL-6 trans-signaling in LSECs leads to endotheliopathy and liver injury in COVID-19 patients. The authors observed elevated levels of markers of coagulopathy/endotheliopathy and liver injury (ALT) in COVID-19 patients. Correlation analysis indicates that the level of IL-6 positively correlated with the level of markers of endothelial activation (vWF, factor VIII, and D-dimer). Activation of IL-6 trans-signaling in LSECs leads to coagulopathy, elevation of pro-inflammatory factors, and platelet adhesion to LSECs. These effects were blocked by soluble glycoprotein 130, ruxolitinib, and STAT1/3 depletion. Therefore, IL-6 trans-signaling represents the mechanistic link between the coagulopathy/endotheliopathy and COVID-19 associated liver injury [35]. The effects and molecular mechanism of COVID-19 on chronic liver injury require detailed further studies [36].
Kidney injury
SARS-CoV-2 infection can also cause acute kidney injury (AKI). A recent multi-omics study has revealed that COVID-19 associated AKI resembles AKI induced by sepsis, which involves the mechanism of mitochondria dysfunction, inflammation, necroptosis, capillary congestion and endothelial injury [37]. ACE2 is highly expressed in renal tissues, the injury of which leads to proteinuria, hematuria and abnormal renal radiography [38]. Like other types of organ injury, SARS-CoV-2 infection causes AKI by both direct and indirect mechanisms, including endotheliitis, thrombosis and glucolipid derangement.
Other injuries
In addition to the above-mentioned organ injuries, COVID-19 also leads to neuropathy [39], redox imbalance and mitochondria dysfunction which may underlie neurological complications of COVID-19 [40]. SARS-CoV-2 can cross the blood brain barrier without affecting the expression of tight junctions (claudin5, ZO-1 and occludin) [41]. It is noted that even in convalescent COVID-19 patients undergoing rehabilitation, cognitive impairment and endothelial dysfunction still exist, indicating the necessity to monitor endothelial dysfunction in convalescent patients [42]. COVID-19 is also associated with acute limb ischemia [43], reproductive system injury, such as erectile dysfunction [44], stroke and deep vein thrombosis [11].
Endothelial dysfunction in SARS-CoV-2 infection: evidence
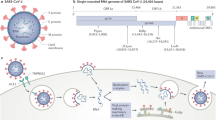
An important question in endothelial dysfunction caused by SARS-Co-V2 is whether SARS-CoV-2 can infect and cause (passively or actively) endothelial dysfunction and long COVID [7]. There are multiple lines of evidences suggesting the involvement of endothelial dysfunction in COVID-19 [45]. The evidence discussed below support both a direct mechanism (virus infection via ACE2, L-SIGN and other receptors) and indirect mechanisms (such as cytokine storm) are involved in SARS-CoV-2 infection associated endothelial dysfunction (Fig. 3).

SARS-CoV-2 or viral proteins can infect endothelial cells and other host cells via reported receptors and the vicious cycle was perpetuated. The infected cell releases danger signals leading to multiple aspects of endothelial dysfunction, which finally leads to impaired vascular activity and multi-organ injury. ACE2 angiotensin-converting enzyme-2, AXL AXL receptor tyrosine kinase, EndoMT endothelial-to-mesenchymal transition, NO nitric oxide, SASP senescence-associated secretory phenotype.
Expression of ACE2, L-SIGN and TMPRSS2
It is appreciated that SARS-CoV-2 infection can trigger systemic vascular injury through binding to ACE2. ACE2 is an important component of the renin-angiotensin-aldosterone system (RAAS) by converting vasoactive AngII into Ang (1–7). ACE2 is described as the first identified receptor responsible for the entry for SARS-CoV-2 into host cells including ECs [46]. Analysis of ACE2 expression in autopsy tissues indicates that high expression of ACE2, transmembrane protease serine 2 (TMPRSS2) and associated endotheliitis in capillaries but less in arterioles/venules from COVID-19 patients, compared with COVID-19-free subjects. Intriguingly, the main coronary arteries have no detectable expression of ACE2, suggesting the occurrence of COVID-19-induced endotheliitis in small vessel like capillaries; however, the culprit in the main coronaries are largely dependent on indirect mechanism arising from SARS-CoV-2 infection [47].
It is reported that under normal conditions, pulmonary ECs express minimal level of ACE2. Heterogeneous ACE2 expression and endothelial damage was observed in COVID-19 autopsy tissues. SARS-CoV-2 infection relies on ACE2 expression in ECs [48]. In addition, with the progress of aging, the expression of ACE2 was increased in the pulmonary vascular ECs with the possible involvement of interleukin 7 via an NF-κB-dependent manner, which can be blocked by Vitamin C [49]. Also, CD209L/L-SIGN was identified as another receptor for mediating SARS-CoV-2 entry into human cells which can also interacts with ACE2 to facilitate SARS-CoV-2 entry [21]. Mechanistically, L-SIGN interacted with high-mannose-type N-glycans on the receptor-binding domain of SARS-CoV-2 spike protein in a Ca2+-dependent manner [33]. Further, removal of the N-glycosylation site at N92 of L-SIGN enhances the binding of S-RBD with L-SIGN [21]. This dual-function mechanisms suggest the important role of L-SIGN as the molecular bridge between ACE2 and SARS-CoV-2 spike protein to allow for virus infection in the patients.
However, there are also reports showing that ACE2 expression is absent from human ECs. For example, one study reported that primary human ECs express minimal level of ACE2 and the protease TMPRSS2, which limits their ability to generate highly infectious viral particles [50]. Based on the evidence presented, there was heterogenous ACE2 expression in ECs from various vascular beds. Further identification of alternative receptors for SARS-CoV-2 is warranted [51].
Viral particles observed in COVID-19 patient tissue endothelium
Several histopathological evidence has supported direct viral infection of endothelial cells, for example, electron microscopy of kidney tissues shows the existence of endotheliitis and viral particles in ECs [52]. Electron microscopy also show coronaviruses and vesicles containing virion particles in venous ECs [53] as well as LSECs from liver autopsy samples from COVID-19 patients [33].
Spike glycoprotein of SARS-CoV-2 cause endothelial cell activation
Numerous reports have reported that infection with the spike protein (S protein) of SARS-CoV-2 virus can elicits profound functional alterations and damage of ECs [7]. Human lung microvascular endothelial cells (HLMVEC) are activated after infection with the S1 protein or S1 infected human macrophages, evidenced by increased expression of pro-coagulant marker (tissue factor), and cytokines/chemokines (ICAM-1, VCAM-1 and MCP1) [54]. Similar effects were observed in ECs infected with SARS-CoV-2 spike pseudovirions (SCV-2-S) [55]. In addition, S1 subunit of SARS-CoV-2 spike protein (S1) decreased endothelial barrier function in cultured human pulmonary microvascular ECs [22]. Similarly, in human aortic ECs (HAECs) treated with recombinant SARS-COV-2 S protein, increased secretion of inflammatory molecules and marker of thrombosis (IL-6, IL-18 and MCP-1 and PAI-1) were observed [56]. Furthermore, spike protein S1 receptor-binding domain (S1-RBD) infection in mouse brain microvascular ECs induced the degradation of endothelial junctional proteins (VE-Cadherin, junctional adhesion molecule-A, Connexin-43 and PECAM-1), thereby impaired endothelial barrier function and caused vascular leakage and endotheliitis in COVID-19 patients [57, 58]. These findings suggest that spike protein interactions with ECs contribute to inflammation, thrombosis, and the severity of COVID-19 and could offer novel mechanistic insights into SARS-CoV-2 induced vascular leakage and the development of targeted therapies [59].
Nucleocapsid protein of SARS-CoV-2 induced endothelial activation
In addition to the spike protein, nucleocapsid protein of SARS-CoV-2 can also promotes endothelial activation via TLR2/NF-κB and MAPK signaling pathways. However, the NPs from other coronaviruses such as Middle East respiratory syndrome coronavirus, SARS-CoV and H1N1 fail to cause endothelial activation, echoing the observation of endotheliitis, vasculopathy and coagulopathy in severe COVID-19 patients [45].
Endothelial dysfunction in COVID-19: mechanism
Since the outbreak of COVID-19 in early 2020, emerging evidence has demonstrated endothelial dysfunction as the unifying and central mechanism of COVID-19 [6]. Direct or indirect mechanisms are operating to collectively indicate the alterations in vascular homeostasis in COVID-19 [54]. Here, we reviewed the potential mechanism of endothelial activation in COVID-19 by overviewing the most recent literature, with the aim to provide targeted therapies (Fig. 3).
Endothelial cell injury and death
COVID-19 is characterized by excessive production of inflammatory mediators (IL-1β, IL-6, IL-8, TNF-α, MCP-1, IP10, RANTES, G-CSF and M-CSF) in a small portion of severe cases due to the severe cytokine storm [60,61,62]. A recent study has shown that markers associated with endothelial inflammation and injury pathway (IL-6, TNF-α, ICAM-1 and caspase-1) were observed in the lung tissues from COVID-19 patients compared with H1N1 subtype 2009 and control cases [63]. Furthermore, it has been demonstrated that exosomes from severe COVID-19 patients trigger the activation of caspase-1 and NLRP3 inflammasome and release of IL-1β in ECs [64]. A recent study has shown that SARS-CoV-2-infection of human brain microvascular ECs showed augmented caspase 3 cleavage and apoptotic cell death of endothelial cells. RNA-sequencing data further revealed the increased expression of markers of endothelial activation such as RELB (p50 subunit) and TNF-α [65]. Thus, hyperinflammation and inflammasome activation associated with SARS-CoV-2 infection will lead to endothelial cell injury and death (such as pyroptosis).
Endothelial glycocalyx damage/degradation
The glycocalyx is a proteoglycan- and glycoprotein-rich microstructure covering ECs essential for maintaining vascular homeostasis via regulating vascular tone, permeability, thrombosis and leukocyte adhesion to endothelium [66]. The glycocalyx consists of highly sulfated proteoglycans with glycosaminoglycan side chains. The intact barrier structure of sulfated glycocalyx of the endothelium could repel SARS-CoV-2. The disrupted glycocalyx structure leads to hyperinflammatory response and oxidative stress, which leads to increased susceptibility to SARS-CoV-2 infection [67]. Using force spectroscopy method, a recent study has revealed the binding of glycocalyx with Spike protein, thus precluding S protein/ACE2 interaction. However, compromised glycocalyx integrity promotes S protein/ACE2 interaction and facilitates viral entry [68]. It has been reported that injury to the endothelial glycocalyx and the release of syndecan-1 (SDC-1) was observed in severe COVID-19 patients [69, 70]. Plasma from COVID-19 patients triggered glycocalyx shedding and disruption in endothelial cells, which can be prevented after treatment with heparin [66].
Even in convalescent COVID-19 patients, the level of SDC-1 levels was significantly elevated compared to healthy controls, demonstrating the existence of persistent endothelial damage after severe COVID-19 progression [71]. These results indicate that the healthy status of glycocalyx is critical for maintaining vascular homeostasis and preventing virus binding. Medications to protect and/or restore the endothelial glycocalyx integrity hold great therapeutic potential for COVID-19 associated glycocalyx disruption.
EndoMT
EndoMT, defined as the loss of endothelial markers/characteristics (CD31, VE-cadherin and Tie2) and gaining of mesenchymal cell markers (FSP-1, α-SMA and vimentin), is central to COVID-19 induced lung fibrosis and pulmonary artery hypertension [72, 73]. A recent study has shown the possible involvement of EndoMT in COVID-19. EndoMT can be induced by cytokine mixture in cultured endothelial cells, for example, the combination of TNF-α and IL-1β, IL-1β and TGFβ1, etc. Interestingly, the secretion of these cytokines is elevated in COVID-19 patients. In terms of the important role of EndoMT in multiple vascular diseases, further mechanistic characterization of EndoMT in COVID-19 patients as well as convalescent patients is urgently needed.
Endothelial hyperpermeability
The existence of cytokine storm could trigger vascular leakage, endothelial permeability in particular. It is possible that these cytokines will disrupt the integrity of various types of junctional proteins, including VE-cadherin, ZO-1, β-catenin and gap junction proteins. The net consequence is the extravasation of inflammatory and immune cell infiltrations [74]. In line with this finding, a recent study has demonstrated that human brain microvascular endothelial cells (hBMECs) infected with SARS-CoV-2 display heightened expression of pro-inflammatory cytokines/chemokines/adhesion molecules (such as TNF-α, IL-1β, MCP-1, CXCL1, CXCL8, CD40, CD44, ICAM1 and VCAM1, etc) and endothelial activation [75]. Pathway enrichment analysis revealed that SARS-CoV-2 infection upregulates expression of genes enriched in signaling pathways relevant to inflammatory response (such as NF-kappa B signaling pathway, TLR signaling pathway, NLRP3 pathway, NOD-like receptor signaling pathway and cytokine–cytokine receptor interaction) [75]. Furthermore, SARS-CoV-2 infection leads to decreased expression of tight junction protein (ZO-1, occluding and claudin5) and blood brain barrier permeability [75].
Endothelial inflammation and leukocyte adhesion to endothelium
Increased endothelial inflammation evidenced by the increased expression of various cytokines/adhesion molecules is a classical instance of endothelial dysfunction. S protein treatment of HAECs leads to increased secretion of inflammatory molecules (IL-6, MCP-1 and IL-18) and PAI-1, which can be attenuated by minerocorticoid receptor antagonists [56]. A recent retrospective study found that the levels of soluble ICAM-1, VCAM-1 and vascular adhesion protein-1 (VAP-1) were elevated in COVID-19 patients and changed during disease progression and regression, raising the possibility that these inflammatory markers are good index of endothelial inflammation and dysfunction in COVID-19 [76]. In addition, spike protein S1-mediated elevation of markers of endothelial inflammation and injury (including E-selectin, ICAM-1, VCAM-1 and PAI-1) and THP-1 monocyte adhesion to ECs was further exacerbated by dihydrotestosterone or TNF-α treatment, but ameliorated by spironolactone treatment [77]. In vivo, SARS-CoV-2-infected K18 mice develop severe COVID-19 and endothelial dysfunction in pulmonary vessels suggested by VCAM-1 and ICAM-1 upregulation and VE-cadherin downregulation [78]. More recently, it is reported that thrombomodulin level was associated with augmented infiltration of immune cells in autopsy lung tissues [79], explaining the existence of thromboembolism in COVID-19 patients. Resistin is peptide hormone derived from adipose tissue which is associated with endothelial injury and inflammatory response. Plasma level of resistin is increased in COVID-19 patients and associated with disease severity as well as the expression of inflammatory cytokines (IL-6, IL-8 and MCP-1) and adhesion molecules (ICAM1 and VCAM1) [80]. One universal mechanism of endothelial inflammation in COVID-19 patients is plausibly associated with the downregulation of KLF2 [81], a master regulator of vascular homeostasis.
Increased angiogenesis
SARS-CoV-2 infection with lung microvascular ECs leads to the release of pro-inflammatory and pro-angiogenic factors [82]. In support of this finding, significantly higher level of angiogenesis was observed in lung tissues from COVID-19 patients, compared with patients with influenza [83]. In addition, plasma profiling study of patients with COVID-19 revealed elevated circulating levels of markers of angiogenesis (such as VEGF-A) in COVID-19 patients [84]. The elevated VEGF-A level further promotes endothelial leakage and inflammatory cell infiltration [19]. The spatio-temporal release of VEGF-A and its effects on endothelial proliferation, migration and capillary-like tube formation warrant further study.
Increased oxidative stress and decreased NO bioavailability
Mitochondria is an important organelle that regulates antioxidant/redox signaling, by fine-tuning mitochondria-derived reactive oxygen species (mtROS) production. Excessive production of mtROS causes oxidative stress that perpetuates ECs inflammation, senescence and dysfunction. A recent review has proposed the detailed mechanism of SARS-CoV-2 induced mtROS production and the consequence of it, including cardio-pulmonary injury associated with COVID-19. In addition to mtROS, other sources of ROS can also be possible, such as ROS derived from NADPH oxidase activation as well as eNOS uncoupling [85]. The burst of ROS after SARS-CoV-2 infection will elicit long-term deleterious effects on endothelial cells, including decreased eNOS expression and NO bioavailability as well as flow-mediated vasodilation in COVID-19 patients.
Virus-induced senescence of endothelial cells
Endothelial senescence is an important aspect of endothelial dysfunction. Infection with various types of viruses, including SARS-CoV-2, can trigger endothelial senescence. Cellular senescence is a primary stress response in virus-infected endothelial cells. Like other types of cell senescence, virus-induced senescence is associated with senescence-associated secretory phenotype (SASP), which is evidenced by increased secretion of pro-inflammatory cytokines, pro-coagulatory factors and VEGF. Elevated level of VEGF in senescent ECs and COVID-19 patients are potent trigger of increased angiogenesis in patient tissues, underlying the clinical utility of anti-VEGF treatment for COVID-19 patients [86, 87]. The net effect of SARS-COV-2 infection induced senescence and angiogenesis is potentially dependent on stage of disease. Combinatorial treatment of human ECs with TNF-α and IFN-γ increased the expression of ACE2, the receptor mediating viral entry via JAK/STAT1 pathway [13]. Moreover, supernatant from virus-infected cells can trigger neutrophil extracellular trap formation and platelet activation [88]. A recent study has shown that increased levels of cellular senescence-associated markers, including PAI-1, p21 and sirtuin-1 in patient serum as well as lung ECs [89]. Cellular senescence was also associated with endothelial inflammation (augmented expression of ICAM-1 and VCAM-1), which is essential for promoting leukocyte adhesion to activated endothelium. Pharmacological inhibition of senescence or SASP can reverse endothelial inflammation and leukocyte adhesion. Therefore, primary endothelial cell senescence or secondary senescence caused by SRAS-CoV-2 infected non-ECs can be exploited as a new therapeutic target for ameliorating COVID-19 associated endotheliitis [90].
Cytokine storm
Besides directly infected by SARS-CoV-2, the ECs also undergo injury by systemic inflammation caused by over-activation of innate immune response, referring to “cytokine storm” [91, 92]. Severe COVID-19-induced cytokine storm (such as IL-6, IL-1β, TNF-α, MCP-1, etc) is a good predictor of the severity of COVID-19, which also aggravates multi-organ injury by propagating the vicious cycle of ECs damage, inflammation and thrombosis [93].
Impairment of mitochondrial function
Mitochondrial dysfunction usually occurs after viral infection, thereby exacerbating tissue damage. A recent study has demonstrated that SARS-CoV-2 infection increased superoxide anion production, and mitochondrial DNA (mtDNA) release, leading to the activation of TLR9 and NF-B, which orchestrates the expression of inflammatory genes [94]. However, blockade of TLR9 significantly mitigated SARS-CoV-2-induced IL-6 release and reversed SARS-CoV-2-induced eNOS downregulation. In addition, mtDNA release also increased vascular reactivity to ET1 [94]. Of translational significance, COVID-19 patients derived serum also increased mtDNA release in ECs, compared to control subjects. This study highlights the novel role of mitochondria dysfunction in SARS-CoV-2 infection-induced endothelial dysfunction and the potential to develop novel therapeutic strategies for COVID-19 based on mtDNA/TLR9 and NF-B activation [94]. Another recent study has demonstrated that, SARS-CoV-2 infection in human brain microvascular ECs increased the secretion of angiogenic factors and altered mitochondrial dynamics, such as increased the expression of mitofusin-2 (a protein involved in a maintenance of an appropriate mitochondrial architecture, metabolism and signaling) and fostered the formation of mitochondrial networks [65].
Complement activation
Recent studies have demonstrated that complement activation is associated with SARS-CoV-2 infection induced inflammation, endothelial injury, hypercoagulability and thrombosis [95]. Consistent with this notion, elevated level of C3a in severe COVID-19 patients induced the activation of CD16+ cytotoxic T cells which promotes endothelial injury and the release of monocyte chemoattractant proteins as well as neutrophil activation [96]. These evidences suggest that inhibition of complement pathway could be an effective strategy to manage endothelial injury/endotheliitis accompanying COVID-19 [97].
Other new mechanisms
It is well-established that SARS-CoV-2 enters host cells including ECs via ACE2 and coreceptor TMPRSS2. Recently, miR-98-5p was identified as a negative regulator of TMPRSS2 gene transcription in human lung and umbilical vein ECs [98]. Internalization of SARS-CoV-2 also needs Neuropilin-1, a transmembrane protein with known angiogenic and immune-modulatory functions. In this regard, miR-24-3p has recently been identified as an essential regulator of Neuropilin-1 gene transcription, thereby maintaining barrier integrity via suppressing VEGF-induced endothelial leakage in human brain ECs [99].
It remains to be investigated whether other mechanisms that are more closely related to COVID-19, such as long non-coding RNA, circular RNA, RNA methylation, microbiota and metabolites, are involved in triggering endothelial dysfunction following SARS-COV-2 infection. In addition, due to the fact that trained immunity in ECs is an important mechanism in propagating endothelial response to inflammatory/immune insults after prior exposure to microbial stimuli [100], detailed mechanisms of epigenetic memory in transducing the SARS-COV-2 infection-induced immune signal needs further studies.
Markers of endothelial dysfunction in COVID-19
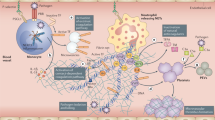
When endothelial dysfunction/endotheliopathy/endotheliitis occurs in COVID-19, several markers of endothelial cell activation are used for assessing endothelial dysfunction in COVID-19 (Figs. 4 and 5) [101].

When endothelial dysfunction occurs, listed markers of endothelial dysfunction related to endothelial inflammation, thrombosis, glycocalyx damage, vascular tone are widely used. Angpt-2 angiopoietin-2, CCL2 chemokine (C-C motif) ligand 2, ECs endothelial cells, FMD flow-mediated dilation, HMWM high-molecular-weight multimers, IL-1β interleukin-1β, IL-6 interleukin 6, PDGF-BB platelet-derived growth factor BB, s-Flt soluble fms-like tyrosine kinase, sICAM1 soluble ICAM1, sVCAM1 soluble VCAM1, sVE-cadherin soluble vascular endothelial cadherin, TF tissue factor, TNF-α tumor necrosis factor, VEGF-A vascular endothelial growth factor A, vWF von Willebrand factor.

ACE2 angiotensin-converting enzyme-2, TMPRSS2 transmembrane protease serine 2, ICAM-1 intercellular adhesion molecule 1, VCAM-1 vascular cell adhesion molecule 1, MCP1 monocyte chemoattractant protein-1, TGF-β transforming growth factor β, VEGF vascular endothelial growth factor, NO nitric oxide, IL-1β interleukin-1β, IL-6 interleukin 6, TNF-α tumor necrosis factor.
Biomarker of endothelial cell activation/injury
It has been reported that the secretion of multiple markers of endothelial activation/dysfunction is elevated in COVID-19 patients, such as D-dimer (marker of coagulopathy and systemic thrombosis), vWF (a primary component of coagulation pathway and mediator of vascular inflammation and thrombo-inflammation released from Weibel-Palade bodies), factor VIII (marker of coagulation), PAI-1 (a marker of endothelial damage and senescence), soluble thrombomodulin (sTM), soluble P-selectin (marker of platelet and endothelial activation), soluble ICAM1 (sICAM1, marker of endothelial inflammation), soluble VCAM1 (sVCAM1, marker of endothelial inflammation), angiopoietin-2 (Ang-2, marker of angiogenesis and thrombosis), soluble E-selectin (sE-selectin, marker of endothelial inflammation), ET1 (a potent vasoconstrictor), VEGF-A (marker of angiogenesis and endothelial hyperpermeability), IL-6 and IL-8 (markers of endothelial inflammation), MCP-1 (marker of endothelial inflammation), resistin (an adipokine associated with endothelial damage and vasoconstriction), nitrosylhemoglobin (HbNO), lactate, and syndecan-1 (marker of endothelial glycocalyx damage) [19, 23, 80, 102,103,104,105,106]. In addition, the levels of these endothelial markers are elevated in intensive care units (ICU) non-survivors compared to survivors. The levels of biomarkers of endothelial cell activation/injury well correlate with the expression level of pro-inflammatory cytokines and chemokines [103]. These evidences signify their potential prognostic value to predict severity and mortality of COVID-19 [103, 107]. In addition, reduced flow-mediated dilation (FMD, an easily obtainable method to assess endothelial dysfunction) was observed in COVID-19 patients, thus offering additional markers to serve as the proxy of endothelial cell activation [108]. The combination of multiple markers could afford better discriminative ability for diagnosis of coagulopathy and thromboembolism and are predictive of ICU admission in COVID-19 patients.
Syndecan-1 and heparanase: markers of glycocalyx damage
The glycocalyx, a protective microstructure layer on the vascular endothelium, consists of glycoproteins and regulates capillary homeostasis by controlling vascular inflammation [109]. Glycocalyx layer regulates vascular barrier integrity, leukocyte adhesion, mechanosensing, mechanotransduction, anti-inflammatory and anti-thrombotic functions [109]. Emerging evidence has suggested that thinning of endothelial glycocalyx layer is associated with COVID-19, and thus the glycocalyx integrity was perceived as an important therapeutic target in COVID-19 [109, 110]. Glycocalyx degradation and shedding disrupts endothelial junctional stability and plays a pivotal role in various forms of cardiovascular diseases [111]. Syndecan-1, an important vascular component of glycocalyx released after vasculitis and injury, well correlates with the marker of coagulation (D-dimer) in particular. It represents a potential biomarker for monitoring disease progression in COVID-19 patients [112]. Syndecan-1 level in patients correlates with the levels of thrombomodulin, TNF-α and IL-6 and signify higher level of endothelial inflammatory reactions. Glycocalyx protein component can be degraded by degrading enzyme such as heparinase. Increased heparanase activity and heparan sulphate level have been observed in plasma derived from COVID-19 patients [113]. However, COVID-19 serum induced glycocalyx destruction was reversed by a non-anticoagulant heparin fragment [113]. Therefore, maintaining the integrity of glycocalyx offers new strategies to combat COVID-19 associated endothelial dysfunction [114].
Circulating endothelial cells (CEC)
Endothelial cell number is determined by the balance of cell proliferation and cell death. Under physiological conditions, ECs undergoing apoptotic process are released into circulating blood. However, after SARS-CoV-2 infection, ECs are detached more quickly without efficient regeneration [20]. EC depletion from the luminal surface reduces NO production and impair endothelium-dependent vasorelaxation [20]. It is reported that COVID-19-patients had higher number of CECs than COVID-19-free subjects. Anti-coagulatory or anti-hypertensive drugs treatment before admission leads to reduced number of CECs, indicating that COVID-19-associated coagulopathy and endotheliopathy could be ameliorated by anti-coagulatory or anti-hypertensive therapy [115]. Likewise, in a retrospective study involving ninety-nine patients with COVID-19, the degree of endothelial damage was correlated with disease severity in COVID-19, suggesting that number of CEC is a good predictor of disease severity [116].
Overall, a plethora of biomarkers of endothelial cell activation and injury have been proposed and validated preclinically and in human patients, offering effective diagnostic tools for monitoring endothelial function in COVID-19. The relationship between mechanisms and biomarkers of endothelial dysfunction in COVID-19 is provided in Fig. 5.
Therapeutic agents targeting endothelial dysfunction in COVID-19
In addition to rehabilitation and exercise-based approaches [6], several categories of endothelium-targeted therapies have potential to ameliorate endothelial dysfunction in COVID-19 patients. Evidently, based on the cytokine storm in severe or critically ill COVID-19 patients, targeted inhibition of pro-inflammatory cytokines, such as IL-1β, IL-6 and downstream signal transduction pathways appears to be important avenues [93].
Statins
Statins are prescribed as the first-choice treatment for patients with hypercholesterolemia and coronary artery disease due to their lipid-lowering and pleiotropic anti-inflammatory, antioxidant, anti-thrombotic and immune-modulatory effects. Based on these protective effects, statin may be used as an adjunctive therapy to mitigate endothelial dysfunction accompanying SARS-CoV-2 infection. However, statin use can also induce the expression of ACE2, which may potentially increase virus entry [117]. It remains elusive whether COVID-19 patient should continue or initiate statin therapy. A meta-analysis revealed the favorable effect of statins in COVID-19 patients [118]. These data agree with recent report of the clinical benefits of statin therapy in lowering the risk of mortality of COVID-19 [119]. Mechanistic studies in cultured human ECs suggest that COVID-19 induced endothelial inflammation and monocyte adhesion was ameliorated by atorvastatin and KLF2 overexpression, suggesting the possible utility of KLF2 activator in suppressing COVID-19 associated endothelial dysfunction [120]. In another study, nucleocapsid protein (NP) of SARS-CoV-2 promotes endothelial cell activation via the pro-inflammatory TLR2/NF-κB and MAPK signaling pathways, which can be attenuated by simvastatin treatment. These results suggest that statins can be exploited to treat COVID-19 patients by mitigating endotheliopathy [45, 121].
Metformin
It is well-recognized that patients with type 2 diabetes mellitus (T2DM) present with increased COVID-19 severity and poorer clinical outcomes compared with non-diabetic subjects [122]. Diabetes/hyperglycemia further exacerbate pre-existing endothelial dysfunction and hyperinflammation in COVID-19 patients. Metformin represents the first-line therapy for T2DM [123]. The pleiotropic effects of metformin help to control hyperglycemia, inhibit viral entry, and reduce inflammation following SARS-CoV-2 infection. The polypharmacological profile of metformin makes it a promising candidate drug to be repurposed for controlling inflammation tsunami in diabetic COVID-19 patients [124]. Thus, metformin could be beneficial in reducing the mortality and composite outcomes in COVID-19 patients with T2DM [125]. A retrospective study in COVID-19 patients with pre-existing T2DM indicated that metformin treatment was associated with the occurrence of acidosis as well as reduced heart failure and inflammation. This study provides additional clinical evidence supporting continuation of metformin use in COVID-19 patients with pre-existing T2DM by paying close attention to kidney function and acidosis [126].
SGLT2 inhibitors
SGLT2 inhibitors are rising stars in cardiovascular and diabetic arena due to prominent cardiorenal benefits in several large-scale clinical trials [127]. SGLT2 inhibitors can reduce the composite endpoint of cardiovascular death and HF hospitalizations in heart failure patients either with reduced ejection fraction or preserved ejection fraction. In COVID-19 patients, heart failure and myocardial injury are frequent complications, underscoring the clinical utility of SGLT2 inhibitors [128]. In light of the multiple endothelial protective effects exerted by SGLT2 inhibitors [127], this type of drug hold promises to treat COVID-19 patients with T2DM. Large-scale clinical trials are warranted to evaluate whether the use of SGLT2 inhibitors can reduce the mortality and hospitalizations for heart failure in COVID-19 patients with or without T2DM.
Heparin
A recent randomized clinical trial has shown that heparin treatment was not significantly associated with reduction of primary outcome, but associated with decreased odds of death at 4 weeks [129]. The primary pharmacological target of heparin could possibly be the endothelial glycocalyx, which is an important microstructure in endothelial cells, essential for maintaining vascular homeostasis by regulating vascular tone, barrier integrity, preventing leukocyte adhesion and thrombosis. Increased glycocalyx components (after damage or destruction) were observed in COVID-19 patients compared with control subjects. In cultured endothelial cells, patient plasma also induced glycocalyx shedding and ROS production, which can be prevented by low molecular weight heparin [66]. Inhibition of heparanase by a non-anticoagulant heparin fragment prevented glycocalyx destruction in response to COVID-19 serum treatment [113]. In addition, heparan sulphate, as the major component in the glycocalyx, can also regenerate glycocalyx and promote the effective restoration of homeostatic EC gap junction [111]. All the above evidence pinpoints the protective effect of heparin in COVID-19 could largely be attributable to glycocalyx-stabilizing effect.
ACEIs/ARBs/ET-1 receptor blocker/ACE2 agonists
Both SARS-CoV and SARS-CoV-2 utilizes ACE2 and membrane-bound co-factors for virus entry. ACE2 is highly expressed in many major organs/tissues, including the heart, lung, kidneys. ACE2 can also undergo shedding and the soluble form of ACE2 (sACE2) can be released into circulating blood. ACE2 is a key regulator of RAAS by catalyzing the production of Ang-(1–7) from AngII, and the Ang-(1–7) can act on MAS receptor to combat the harmful effects caused by activation of RAAS [87, 130]. In this regard, ACE2 downregulation and the disrupted balance between the RAAS and ACE2/Ang-(1–7)/MAS axis may also contribute to multiple organ injury in COVID-19 [87, 130]. SARS-CoV-2 causes ACE/ACE2 balance disruption and RAAS activation, which leads ultimately to COVID-19 progression, especially in patients with comorbidities, such as hypertension, diabetes mellitus, and cardiovascular disease. Therefore, ACE2 expression may have paradoxical effects, aiding SARS-CoV-2 pathogenicity, yet conversely limiting viral infection [87, 130].
It is initially conceived that ACE inhibitors (ACEIs) or Ang-II receptor blockers (ARBs), two widely-used anti-hypertensive drugs targeting the renin-angiotensin system (RAS), could increase the vulnerability to SARS-CoV-2 by upregulating the expression of ACE-2. Unexpectedly, propensity score-weighted analysis showed that treatment with ACEI/ARB was not significantly associated with the occurrence of defined end-points. Instead, ACEIs/ARBs discontinuation is associated with poorer clinical outcomes. In another translational study, treatment of human pluripotent stem cell-derived endothelial cells (hECs) with SARS-CoV-2 leads to enriched gene program involved in immunity, inflammation and viral response (such as TNF-α, IFNs and NF-κB signaling pathway). However, pre-treatment of ECs with losartan (belonging to ARB) and lisinopril (belonging to ACEI), fail to affect the susceptibility of hEC to SARS-CoV-2 infection [131]. These findings agree with a recent retrospective analysis by Zhang et al. [132] and the expert recommendations from the professional cardiovascular societies, supporting that ACEIs and ARBs does not alter SARS-CoV-2 infection and should not be discontinued in COVID-19 patients [133]. In addition, a recent study has shown that circulating level of ET-1, a potent vasoconstrictive peptide, was elevated in hospitalized patients with acute phase of COVID-19, indicating that ET-1 receptor blockers could potentially offer clinical benefits for COVID-19 patients [104].
It has been recently reviewed that restoration of balanced effects between the RAAS and ACE2/Ang-(1–7)/MAS could be a promising way to ameliorate multi-organ injury associated with COVID-19 [130]. Treatment of virus-infected human lung microvascular endothelial cells (HMVECs) with diminazene aceturate (an ACE2 agonist) reverses SARS-CoV-2 infection-induced hyperpermeability, indicating the possibility that ACE2 agonism indeed stabilizes endothelial barrier integrity without affecting viral uptake into ECs [23]. ACE2 agonism also blocks VEGF-A mediated pro-angiogenic signaling endothelial hyperpermeability. The protective role of ACE2 agonism in suppressing angiogenesis and maintaining endothelial integrity in COVID-19 warrant further investigation [19].
Glucocorticoids (e.g., dexamethasone)
Several large-scale clinical trials have suggested that glucocorticoid drug dexamethasone treatment is beneficial for COVID-19 patients [134]. Severe COVID-19 patients had significantly higher levels of glycocalyx disruption (endocan and syndecan-1), endothelial damage (angiopoietin-2 and vWF), and inflammation (upregulation of soluble receptor for advanced glycation end-products, IL-6, ICAM-1 and VCAM-1). Dexamethasone treated group exhibited a significantly decreased levels of various markers associated with endothelial dysfunction, including Ang-2, ICAM-1, and sRAGE [135]. The effect of glucocorticoids on COVID-19 might be multifactorial, and their endothelium-stabilizing properties by direct activation of endothelial glucocorticoid receptors to block production of IL-6 and VEGF might be the main operating mechanisms [19].
Tocilizumab
Activated cytokine storm and IL-6 signaling has been observed in endothelial dysfunction during bacterial infection and SARS-CoV-2 infection [92]. IL-6 directly impacts vascular ECs by promoting the production of numerous cytokines/chemokines/adhesion molecules essential for promoting leukocyte adhesion, vascular leakage and activating the coagulation cascade [136]. The PAI-1 level in COVID-19 patients were as highly elevated compared with other cytokine release syndrome (sepsis or ARDS). Treatment with a humanized anti-IL-6 receptor antibody-tocilizumab, decreased the PAI-1 level and alleviated critical illness in severe COVID-19 patients. This study highlights the crucial role of IL-6 trans-signaling in endothelial dysfunction/endotheliopathy in COVID-19 [137]. Further studies revealed that tocilizumab inhibited the expression of senescence markers (p21 and p16), ROS generation as well as endothelial adhesion molecule mediated leukocyte adhesion [90]. Tocilizumab also protects against endothelial dysfunction by increasing glycocalyx thickness and reducing the burden of inflammation and oxidative stress. All these reported effects could justify the curative effects of tocilizumab on COVID-19 [138].
Anakinra
IL-1β is an important cytokine released during cytokine storm in COVID-19 as well as its post-acute sequelae [91, 139]. It is well-known that IL-1β induces the expression of itself and other pro-inflammatory and pro-adhesive molecules, such as TNF-α, leading to the amplification of cytokine storm. A recent study has shown that SARS-CoV-2 infection in humanized K18-hACE-2 mice activates the NLRP3 inflammasome, followed by caspase-1 and IL-1β activation [140]. This leads to decreased expression of VE-cadherin in lung endothelial cells. Anakinra, by blocking interleukin 1 receptor, prevented VE-cadherin downregulation and lung vascular leakage. By doing so, anakinra significantly increased the survival rate of infected mice [140]. Anakinra treatment reduced both the need for mechanical ventilation in patients admitted to ICU and mortality of severe COVID-19 patients, with good safety profile [141], especially in patients with CRP concentrations >100 mg/L [142].
Colchicine
Colchicine is an ancient and low-cost drug isolated from Chinese herbal medicine. Colchicine is an anti-inflammatory drug traditionally used in gout and familial Mediterranean fever [143, 144]. Recent studied have shown that colchicine is able to reduce the length of stay in hospitalized COVID-19 patients with the possible mechanism of inhibition of NLRP3 inflammasome and resultant IL-1β production [143, 144].
JAK inhibitors
JAK/STAT pathway is a canonical pathway in driving inflammation. Inflammatory cytokines, such as IL-6, promotes JAK and STATs phosphorylation [145]. After that, STATs translocate into cell nucleus to orchestrate the expression of inflammatory cytokines, further instigating the cytokine storm feedback loop. The SARS-CoV-2 structural and non-structural proteins promote virus entry and its survival in host cells. Cytokine storm in COVID-19 can trigger inflammation via the JAK/STAT pathway, which results in increased recruitment of leukocytes/immune cells [146]. However, several JAK/STAT inhibitors such as ruxolitinib, tofacitinib and baricitinib can suppress cytokine signaling cascade. Several clinical trials are ongoing to evaluate the safety and efficacy of JAK/STAT inhibitors [146].
Senolytics
Virus-induced senescence is a pathogenic trigger of endothelial dysfunction. The levels of senescent markers, such as PAI-1, p21 and sirtuin-1 in the plasma and lung ECs are elevated. Senolytic drugs such as navitoclax and quercetin/dasatinib combination selectively eradicated senescent cells and reduced inflammation in SARS-CoV-2-infected animals [89]. The therapeutic potential of senolytics in COVID-19 patients warrants studies from clinical trials.
L-Arginine
COVID-19 is an endothelial disease in which endothelial dysfunction played a major role. L-arginine improves endothelial dysfunction by being the substrate of NO generation in endothelial cells. In a randomized clinical trial, L-arginine add-on therapy significantly reduces the length of hospitalization in severe COVID-19 patients and reduces the need of respiratory support, with no serious adverse events [147].
Fluvoxamine
Recent randomized clinical trials revealed that treatment with fluvoxamine (a selective serotonin reuptake inhibitors, SSRI) reduced the need for COVID-19 hospitalization, reduced mortality and improved outcome over 15 days [148,149,150]. Mechanistically, fluvoxamine is a sigma-1 receptor (S1R) agonist which, on the one hand, reduces the expression of IL-6, while increasing that of eNOS. On the other hand, S1R agonism by fluvoxamine activates Akt-eNOS signaling in mouse aorta in a S1R-dependent manner. These findings suggest that fluvoxamine can be repurposed as novel anti-COVID-19 drugs although further studies are warranted to assess the therapeutic potential of fluvoxamine in patients [151].
Vitamin C
Vitamin C is an essential, safe and inexpensive nutrient [152] that has anti-oxidant, anti-infectious, anti-inflammatory, anti-thrombotic and immune-modulatory effects [153]. In a single-center observational study, 82% of critically ill COVID-19 patients have significantly lower plasma level of vitamin C [154]. Therefore, supplementation with high dose intravenous vitamin C (HIVC) could hold therapeutic potential for COVID-19 patients. HIVC improves myocardial injury via decreasing biomarkers associated with inflammation in critically ill COVID-19 patients [155]. HIVC also protect against severe COVID-19 by decreasing the rates of mechanical ventilation and cardiac arrest in hospitalized severe patients [156]. Vitamin C consumption significantly reduces mortality risk with COVID-19 patients [157]. Furthermore, HIVC in combination with other drugs such as giammonium glycyrrhizinate, decreased the incidence rate of ARDS in COVID-19 patients [158]. Despite the observed benefits of HIVC, conflicting results have been reported in severe COVID-19 patients [159]. Data from randomized controlled clinical trials are scarce. Therefore, the therapeutic role of JIVC in treating severe COVID-19 patients warrants further investigation [160].
Traditional Chinese medicine (TCM)
TCM has a well-documented safety profile in protecting against COVID-19 on the basis of standard care. TCM could significantly relieve clinical symptoms, reduce disease severity, reduce the need for mechanical ventilation, shortening the duration of hospitalization, accelerate symptom recovery, and ultimately reduce mortality rate [161,162,163,164]. Till now, several TCM have shown good therapeutic effects in COVID-19, such as Lianhua Qingwen, Xuebijing Injection, Shuanghuanglian, Jinyinhua and Qingfei Paidu Decoction [161,162,163,164]. These studies illustrated that TCM in combination with standard care might be safe and potentially effective for COVID-19. However, conclusions need to be analyzed with caution due to small sample size [165]. It remains to be investigated further whether TCM ameliorates COVID-19 partially by improving endothelial function.
Others
In addition to the above drugs discussed, there are several other reports showing that serine protease inhibitors (camostat mesylate), KLF2 activators [120], RIPK3 inhibitors [166], spironolactone [77], glycocalyx repairing drugs [67], purified glycosaminoglycan mixture sulodexide [167], CCR5 blockers (Maraviroc), anti-VEGF (bevacizumab [168]), adrecizumab [169], mesenchymal stem cell therapy [170], estrogen [171], melatonin [172] and NO donor [173] could also be beneficial (Fig. 6).

Based on the important role of endothelial dysfunction in COVID-19, several categories of endothelial protective drugs are possible to ameliorate endothelial dysfunction in COVID-19. These drugs include lipid-lowering drugs, anti-hypertensive drugs, anti-diabetic drugs, anti-VEGF agents, anti-coagulatory drugs, antioxidants, anti-inflammatory drugs and others. ACE2 angiotensin-converting enzyme-2, ACEI angiotensin converting enzyme inhibitors, ARB angiotensin receptor blockers, BRD4i bromodomain-containing protein 4 inhibitors, JAK janus kinase, SGLT2i sodium-glucose cotransporter-2 inhibitors.
Concluding remarks and future perspectives
It has been increasingly appreciated that COVID-19 is not only an infectious disease involving the lung; but also, a vascular disease affecting extrapulmonary organs [174]. Endothelial dysfunction-induced endotheliitis/endothelialitis/endotheliopathy following SARS-CoV-2 infection arises from a plethora of physiopathological mechanisms, including both direct mechanism of virus infection or indirect mechanisms such as paracrine effects of infected cells [2, 68]. Studies in the past two years altogether provide important mechanistic insights into the pathogenesis of COVID-19 and yield promising new therapeutic targets (Fig. 7). Although current pharmacotherapies against acute and post-acute COVID-19 mainly centered on blocking viral replication and limiting inflammation/inflammasome activation, it is likely that novel therapeutic approaches targeting endothelial dysfunction could represent a promising strategy to cardiovascular sequelae in COVID-19 convalescent patients [6] in light of elevated circulating level of biomarker soluble P-selectin in COVID-19 convalescent donors compared to healthy controls [175]. Antiviral therapies and effective vaccination reduce viral load and could potentially offer endothelial protection in perivascular spaces [19]. Further outstanding questions and research directions in the realm of endothelial dysfunction and COVID-19 include the following:
-
(i)
The development of assays of assessing endothelial function in long COVID-19 patients and convalescents, such as brachial artery flow-mediated dilation (FMD) and arterial stiffness [carotid-femoral pulse wave velocity (cfPWV)]; This aspect is important considering the recent observation showing the decreased FMD in patients with COVID-19 stemming from expression of inflammatory cytokines/chemokines [176];
-
(ii)
Cellular and animal models of evaluating endothelial dysfunction in COVID-19 to accelerate drug discovery;
-
(iii)
The therapeutic potential of specialized pro-resolving lipid mediators, such as resolvin D1, resolvin E1, aspirin-triggered resolvin D1 in resolving cytokine storm induced inflammatory responses can be pursued;
-
(iv)
The identification of alternative receptors for SARS-CoV-2 infection into different vascular beds beyond known ones (such as ACE2, AXL and L-SIGN) remain to be identified;
-
(v)
Drug repurposing or high-throughput drug screening to identify new drugs targeting endothelial dysfunction in COVID-19;
-
(vi)
The role of epigenetic modification arising from DNA methylation and histone modification and long-lasting epigenetic memory effects caused by SARS-CoV2 infection in long COVID (post–acute COVID-19 syndrome) remain to be evaluated [7];
-
(vii)
Metabolic disturbance has been shown to be associated with the pathogenesis of COVID-19 [177]. To this end, lactate production from glycolysis pathway serves as a useful biomarker of EC barrier dysfunction [106]. Molecular underpinnings of metabolic alterations caused by SARS-CoV-2 infection warrants further studies;
-
(viii)
Lastly, considering the evolving mutations of SARS-CoV-2 (such as Delta variant and Omicron variant), effect and mechanism of these variants in viral entry and endothelial functionality warrant further studies.

These targets are directed at improving oxidative stress, endothelial inflammation/inflammasome, senescence, fibrosis, cell death, thrombosis, coagulopathy, angiogenesis, EndoMT and immunity mechanisms. BRD4 bromodomain-containing protein 4, CD31 cluster of differentiation 31, CXCLs chemokine (C-X-C motif) ligands, EndoMT endothelial-to-mesenchymal transition, eNOS endothelial nitric oxide synthase, ET-1 endothelin 1, FN fibronectin, GCLC glutamate-cysteine ligase catalytic subunit, GCLM glutamate-cysteine ligase modifier subunit, HO-1 heme oxygenase-1, IL-1β interleukin-1β, IL-6 interleukin 6, JAK janus kinase, KLF2 krüppel-like factor 2, MCP-1 monocyte chemoattractant protein-1, NF-κB nuclear factor-kappa B, NLRP3 NLR family pyrin domain containing 3, NO nitric oxide, NQO1 NAD(P)H quinone oxidoreductase, Nrf2 nuclear factor erythroid 2-related factor 2, PAI-1 plasminogen activator inhibitor 1, RIG-I retinoic acid-inducible gene I, RIPK3 receptor-interacting protein kinase 3, SMA smooth muscle actin, STAT3 signal transducer and activator of transcription 3, TLR toll-like receptor, TLR9 toll-like receptor 9, TNF-α tumor necrosis factor, VCAM1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
Collectively, based on the multifaceted nature of endothelial dysfunction and complex patho-mechanisms of COVID-19, it warrants to be evaluated whether directly targeting endothelial dysfunction could result in a clinically significant improvement in outcome of COVID-19 patients.
References
Yuen KS, Ye ZW, Fung SY, Chan CP, Jin DY. SARS-CoV-2 and COVID-19: The most important research questions. Cell Biosci. 2020;10:40.
Fodor A, Tiperciuc B, Login C, Orasan OH, Lazar AL, Buchman C, et al. Endothelial dysfunction, inflammation, and oxidative stress in COVID-19-mechanisms and therapeutic targets. Oxid Med Cell Longev. 2021;2021:8671713.
Schnaubelt S, Oppenauer J, Tihanyi D, Mueller M, Maldonado-Gonzalez E, Zejnilovic S, et al. Arterial stiffness in acute COVID-19 and potential associations with clinical outcome. J Intern Med. 2021;290:437–43.
Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41:3038–44.
Mezoh G, Crowther NJ. Endothelial dysfunction as a primary consequence of SARS-CoV-2 Infection. Adv Exp Med Biol. 2021;1321:33–43.
Ambrosino P, Calcaterra IL, Mosella M, Formisano R, D’Anna SE, Bachetti T, et al. Endothelial dysfunction in COVID-19: a unifying mechanism and a potential therapeutic target. Biomedicines. 2022;10:812.
de Rooij L, Becker LM, Carmeliet P. A role for the vascular endothelium in post-acute COVID-19? Circulation. 2022;145:1503–5.
Gladka MM, Maack C. The endothelium as Achilles’ heel in COVID-19 patients. Cardiovasc Res. 2020;116:e195–e7.
Lowenstein CJ, Solomon SD. Severe COVID-19 is a microvascular disease. Circulation. 2020;142:1609–11.
Ma Z, Yang KY, Huang Y, Lui KO. Endothelial contribution to COVID-19: an update on mechanisms and therapeutic implications. J Mol Cell Cardiol. 2021;164:69–82.
Flaumenhaft R, Enjyoji K, Schmaier AA. Vasculopathy in COVID-19. Blood. 2022;140:222–35.
Basta G. Direct or indirect endothelial damage? An unresolved question. EBioMedicine. 2021;64:103215.
Kandhaya-Pillai R, Yang X, Tchkonia T, Martin GM, Kirkland JL, Oshima J. TNF-α/IFN-γ synergy amplifies senescence-associated inflammation and SARS-CoV-2 receptor expression via hyper-activated JAK/STAT1. Aging Cell. 2022;21:e13646.
Nägele MP, Haubner B, Tanner FC, Ruschitzka F, Flammer AJ. Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis. 2020;314:58–62.
Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73:924–67.
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med. 2020;26:1017–32.
Villar J, Kacmarek RM. The American-European Consensus Conference definition of the acute respiratory distress syndrome is dead, long live positive end-expiratory pressure! Med Intensiv. 2012;36:571–5.
Hussain M, Khurram Syed S, Fatima M, Shaukat S, Saadullah M, Alqahtani AM, et al. Acute respiratory distress syndrome and COVID-19: a literature review. J Inflamm Res. 2021;14:7225–42.
Filbin MR. Insights into endotheliopathy in COVID-19. Am J Respir Crit Care Med. 2022. https://doi.org/10.1164/rccm.202207-1258ED.
Six I, Guillaume N, Jacob V, Mentaverri R, Kamel S, Boullier A, et al. The endothelium and COVID-19: an increasingly clear link brief title: endotheliopathy in COVID-19. Int J Mol Sci. 2022;23:6196.
Amraei R, Yin W, Napoleon MA, Suder EL, Berrigan J, Zhao Q, et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent Sci. 2021;7:1156–65.
Colunga Biancatelli RML, Solopov PA, Sharlow ER, Lazo JS, Marik PE, Catravas JD. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Κ18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2021;321:L477–l84.
Joffre J, Rodriguez L, Matthay ZA, Lloyd E, Fields AT, Bainton RJ, et al. COVID-19-Associated lung microvascular endotheliopathy: a “from the bench” perspective. Am J Respir Crit Care Med. 2022. https://doi.org/10.1164/rccm.202107-1774OC.
Ashour L. Roles of the ACE/Ang II/AT1R pathway, cytokine release, and alteration of tight junctions in COVID-19 pathogenesis. Tissue Barriers. 2022; 2090792. https://doi.org/10.1080/21688370.2022.2090792.
Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial damage in acute respiratory distress syndrome. Int J Mol Sci. 2020;21:8793.
Liu Y, Zhang HG. Vigilance on new-onset atherosclerosis following SARS-CoV-2 infection. Front Med. 2020;7:629413.
Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116:1666–87.
Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020;116:1097–100.
Katsoularis I, Fonseca-Rodríguez O, Farrington P, Lindmark K, Fors Connolly AM. Risk of acute myocardial infarction and ischaemic stroke following COVID-19 in Sweden: a self-controlled case series and matched cohort study. Lancet (Lond, Engl). 2021;398:599–607.
Fardman A, Zahger D, Orvin K, Oren D, Kofman N, Mohsen J, et al. Acute myocardial infarction in the Covid-19 era: Incidence, clinical characteristics and in-hospital outcomes-A multicenter registry. PLoS One. 2021;16:e0253524.
Toscano O, Cosentino N, Campodonico J, Bartorelli AL, Marenzi G. Acute myocardial infarction during the COVID-19 pandemic: an update on clinical characteristics and outcomes. Front Cardiovasc Med. 2021;8:648290.
Aye YN, Mai AS, Zhang A, Lim OZH, Lin N, Ng CH, et al. Acute myocardial infarction and myocarditis following COVID-19 vaccination. QJM. 2021. https://doi.org/10.1093/qjmed/hcab252.
Kondo Y, Larabee JL, Gao L, Shi H, Shao B, Hoover CM, et al. L-SIGN is a receptor on liver sinusoidal endothelial cells for SARS-CoV-2 virus. JCI insight. 2021;6:e148999.
Saviano A, Baumert TF. Unraveling the role of liver sinusoidal endothelial cells in COVID-19 liver injury. J Hepatol. 2021;75:503–5.
McConnell MJ, Kawaguchi N, Kondo R, Sonzogni A, Licini L, Valle C, et al. Liver injury in COVID-19 and IL-6 trans-signaling-induced endotheliopathy. J Hepatol. 2021;75:647–58.
Jothimani D, Venugopal R, Abedin MF, Kaliamoorthy I, Rela M. COVID-19 and the liver. J Hepatol. 2020;73:1231–40.
Alexander MP, Mangalaparthi KK, Madugundu AK, Moyer AM, Adam BA, Mengel M, et al. Acute kidney injury in severe COVID-19 has similarities to sepsis-associated kidney injury: a multi-omics study. Mayo Clin Proc. 2021;96:2561–75.
Henry BM, de Oliveira MHS, Cheruiyot I, Benoit JL, Cooper DS, Lippi G, et al. Circulating level of Angiopoietin-2 is associated with acute kidney injury in coronavirus disease 2019 (COVID-19). Angiogenesis. 2021;24:403–6.
Poloni TE, Medici V, Moretti M, Visonà SD, Cirrincione A, Carlos AF, et al. COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol (Zur, Switz). 2021;31:e12997.
Kaundal RK, Kalvala AK, Kumar A. Neurological implications of COVID-19: role of redox imbalance and mitochondrial dysfunction. Mol Neurobiol. 2021;58:4575–87.
Zhang L, Zhou L, Bao L, Liu J, Zhu H, Lv Q, et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther. 2021;6:337.
Moretta P, Maniscalco M, Papa A, Lanzillo A, Trojano L, Ambrosino P. Cognitive impairment and endothelial dysfunction in convalescent COVID-19 patients undergoing rehabilitation. Eur J Clin Invest. 2022;52:e13726.
Ilonzo N, Judelson D, Al-Jundi W, Etkin Y, O’Banion LA, Rivera A, et al. A review of acute limb ischemia in COVID-positive patients. Semin Vasc Surg. 2021;34:8–12.
Sansone A, Jannini EA. COVID-19 and erectile dysfunction: endothelial dysfunction and beyond. world J men’s health. 2021;39:820–1.
Qian Y, Lei T, Patel PS, Lee CH, Monaghan-Nichols P, Xin HB, et al. Direct activation of endothelial cells by SARS-CoV-2 nucleocapsid protein is blocked by simvastatin. J Virol. 2021;95:e0139621.
Barbosa LC, Gonçalves TL, de Araujo LP, Rosario LVO, Ferrer VP. Endothelial cells and SARS-CoV-2: An intimate relationship. Vasc Pharmacol. 2021;137:106829.
Maccio U, Zinkernagel AS, Shambat SM, Zeng X, Cathomas G, Ruschitzka F, et al. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine. 2021;63:103182.
Klouda T, Hao Y, Kim H, Kim J, Olejnik J, Hume AJ, et al. Interferon-alpha or -beta facilitates SARS-CoV-2 pulmonary vascular infection by inducing ACE2. Angiogenesis. 2022;25:225–40.
Ma S, Sun S, Li J, Fan Y, Qu J, Sun L, et al. Single-cell transcriptomic atlas of primate cardiopulmonary aging. Cell Res. 2021;31:415–32.
Schimmel L, Chew KY, Stocks CJ, Yordanov TE, Essebier P, Kulasinghe A, et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin Transl Immunol. 2021;10:e1350.
Amraei R, Rahimi N. COVID-19, Renin-Angiotensin system and endothelial dysfunction. Cells. 2020;9:1652.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet (Lond, Engl). 2020;395:1417–8.
Stahl K, Bräsen JH, Hoeper MM, David S. Direct evidence of SARS-CoV-2 in gut endothelium. Intensive Care Med. 2020;46:2081–2.
Rotoli BM, Barilli A, Visigalli R, Ferrari F, Dall’Asta V. Endothelial cell activation by SARS-CoV-2 Spike S1 protein: a crosstalk between endothelium and innate immune cells. Biomedicines. 2021;9:1220.
Li F, Li J, Wang PH, Yang N, Huang J, Ou J, et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochimica et Biophysica Acta Mol Basis Dis. 2021;1867:166260.
Jover E, Matilla L, Garaikoetxea M, Fernández-Celis A, Muntendam P, Jaisser F, et al. Beneficial effects of mineralocorticoid receptor pathway blockade against endothelial inflammation induced by SARS-CoV-2 spike protein. Biomedicines. 2021;9:639.
Raghavan S, Kenchappa DB, Leo MD. SARS-CoV-2 spike protein induces degradation of junctional proteins that maintain endothelial barrier integrity. Front Cardiovasc Med. 2021;8:687783.
Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, et al. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res. 2021;128:1323–6.
Biering SB, de Sousa FTG, Tjang LV, Pahmeier F, Ruan R, Blanc SF, et al. SARS-CoV-2 Spike triggers barrier dysfunction and vascular leak via integrins and TGF-β signaling. bioRxiv: the preprint server for biology 2021. https://doi.org/10.1101/2021.12.10.472112.
Frisoni P, Neri M, D’Errico S, Alfieri L, Bonuccelli D, Cingolani M, et al. Cytokine storm and histopathological findings in 60 cases of COVID-19-related death: from viral load research to immunohistochemical quantification of major players IL-1β, IL-6, IL-15 and TNF-α. Forensic Sci Med Pathol. 2021: 1-15.
Li S, Jiang L, Li X, Lin F, Wang Y, Li B, et al. Clinical and pathological investigation of patients with severe COVID-19. JCI insight. 2020;5:e138070.
Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255–73.
Nagashima S, Mendes MC, Camargo Martins AP, Borges NH, Godoy TM, Miggiolaro A, et al. Endothelial dysfunction and thrombosis in patients with COVID-19-brief report. Arteriosclerosis Thrombosis Vasc Biol. 2020;40:2404–7.
Sur S, Steele R, Isbell TS, Ray R, Ray RB. Circulatory exosomes from COVID-19 patients trigger NLRP3 inflammasome in endothelial cells. mBio. 2022: e0095122.
Torices S, Motta C, da Rosa B, Marcos A, Alvarez-Rosa L, Siqueira M, et al. SARS-CoV-2 infection of human brain microvascular endothelial cells leads to inflammatory activation through NF-κB non-canonical pathway and mitochondrial remodeling. Res Square. 2022. https://doi.org/10.21203/rs.3.rs-1762855/v1.
Potje SR, Costa TJ, Fraga-Silva TFC, Martins RB, Benatti MN, Almado CEL, et al. Heparin prevents in vitro glycocalyx shedding induced by plasma from COVID-19 patients. Life Sci. 2021;276:119376.
du Preez HN, Aldous C, Hayden MR, Kruger HG, Lin J. Pathogenesis of COVID-19 described through the lens of an undersulfated and degraded epithelial and endothelial glycocalyx. FASEB J. 2022;36:e22052.
Targosz-Korecka M, Kubisiak A, Kloska D, Kopacz A, Grochot-Przeczek A, Szymonski M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci Rep. 2021;11:12157.
Stahl K, Gronski PA, Kiyan Y, Seeliger B, Bertram A, Pape T, et al. Injury to the endothelial glycocalyx in critically Ill patients with COVID-19. Am J Respiratory Crit Care Med. 2020;202:1178–81.
Yamaoka-Tojo M. Vascular endothelial glycocalyx damage in COVID-19. Int J Mol Sci. 2020;21:9712.
Vollenberg R, Tepasse PR, Ochs K, Floer M, Strauss M, Rennebaum F, et al. Indications of persistent glycocalyx damage in convalescent COVID-19 patients: a prospective multicenter study and hypothesis. Viruses. 2021;13:23424.
Eapen MS, Lu W, Gaikwad AV, Bhattarai P, Chia C, Hardikar A, et al. Endothelial to mesenchymal transition: a precursor to post-COVID-19 interstitial pulmonary fibrosis and vascular obliteration? Eur Respir J. 2020;56:2003167.
Falleni M, Tosi D, Savi F, Chiumello D, Bulfamante G. Endothelial-mesenchymal transition in COVID-19 lung lesions. Pathol Res Pract. 2021;221:153419.
Rauti R, Shahoha M, Leichtmann-Bardoogo Y, Nasser R, Paz E, Tamir R, et al. Effect of SARS-CoV-2 proteins on vascular permeability. eLife. 2021;10:e69314.
Yang RC, Huang K, Zhang HP, Li L, Zhang YF, Tan C, et al. SARS-CoV-2 productively infects human brain microvascular endothelial cells. J Neuroinflammation. 2022;19:149.
Tong M, Jiang Y, Xia D, Xiong Y, Zheng Q, Chen F, et al. Elevated expression of serum endothelial cell adhesion molecules in COVID-19 patients. J Infect Dis. 2020;222:894–8.
Kumar N, Zuo Y, Yalavarthi S, Hunker KL, Knight JS, Kanthi Y, et al. SARS-CoV-2 spike protein S1-mediated endothelial injury and pro-inflammatory state is amplified by dihydrotestosterone and prevented by mineralocorticoid antagonism. Viruses. 2021;13:2209.
Qin Z, Liu F, Blair R, Wang C, Yang H, Mudd J, et al. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics. 2021;11:8076–91.
Won T, Wood MK, Hughes DM, Talor MV, Ma Z, Schneider J, et al. Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. EBioMedicine. 2022;75:103812.
Ebihara T, Matsumoto H, Matsubara T, Togami Y, Nakao S, Matsuura H, et al. Resistin associated with cytokines and endothelial cell adhesion molecules is related to worse outcome in COVID-19. Front Immunol. 2022;13:830061.
Wu D, Lee TH, Huang RT, D Guzy R, Schoettler N, Adegunsoye A, et al. SARS-CoV-2 infection is associated with reduced krüppel-like factor 2 in human lung autopsy. Am J Respiratory Cell Mol Biol. 2021;65:222–6.
Caccuri F, Bugatti A, Zani A, De Palma A, Di Silvestre D, Manocha E, et al. SARS-CoV-2 infection remodels the phenotype and promotes angiogenesis of primary human lung endothelial cells. Microorganisms. 2021;9:1438.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–8.
Pine AB, Meizlish ML, Goshua G, Chang CH, Zhang H, Bishai J, et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm Circ. 2020;10:2045894020966547.
Chang R, Mamun A, Dominic A, Le NT. SARS-CoV-2 mediated endothelial dysfunction: the potential role of chronic oxidative stress. Front Physiol. 2020;11:605908.
Maldonado F, Morales D, Díaz-Papapietro C, Valdés C, Fernandez C, Valls N, et al. Relationship between endothelial and angiogenesis biomarkers envisage mortality in a prospective cohort of COVID-19 patients requiring respiratory support. Front Med. 2022;9:826218.
Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. 2019;117:15–22.
Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature. 2021;599:283–9.
D’Agnillo F, Walters KA, Xiao Y, Sheng ZM, Scherler K, Park J, et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci Transl Med. 2021;13:eabj7790.
Meyer K, Patra T, Vijayamahantesh, Ray R. SARS-CoV-2 spike protein induces paracrine senescence and leukocyte adhesion in endothelial cells. J Virol. 2021;95:e0079421.
Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. 2021;93:250–6.
Huang Q, Wu X, Zheng X, Luo S, Xu S, Weng J. Targeting inflammation and cytokine storm in COVID-19. Pharmacol Res. 2020;159:105051.
Huang P, Zuo Q, Li Y, Oduro PK, Tan F, Wang Y, et al. A vicious cycle: in severe and critically Ill COVID-19 patients. Front Immunol. 2022;13:930673.
Costa TJ, Potje SR, Fraga-Silva TFC, da Silva-Neto JA, Barros PR, Rodrigues D, et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vasc Pharmacol. 2021;142:106946.
Ma L, Sahu SK, Cano M, Kuppuswamy V, Bajwa J, McPhatter J, et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci Immunol. 2021;6:eabh2259.
Georg P, Astaburuaga-García R, Bonaguro L, Brumhard S, Michalick L, Lippert LJ, et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell. 2022;185:493–12.
Noris M, Benigni A, Remuzzi G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020;98:314–22.
Matarese A, Gambardella J, Sardu C, Santulli G. miR-98 regulates TMPRSS2 expression in human endothelial cells: key implications for COVID-19. Biomedicines. 2020;8:462.
Mone P, Gambardella J, Wang X, Jankauskas SS, Matarese A, Santulli G. miR-24 targets the transmembrane glycoprotein neuropilin-1 in human brain microvascular endothelial cells. Non-coding RNA. 2021;7:9.
Shao Y, Saredy J, Xu K, Sun Y, Saaoud F, Drummer CT, et al. Endothelial immunity trained by coronavirus infections, DAMP stimulations and regulated by anti-oxidant NRF2 may contribute to inflammations, myelopoiesis, COVID-19 cytokine storms and thromboembolism. Front Immunol. 2021;12:653110.
Choudhary S, Sharma K, Singh PK. Von Willebrand factor: A key glycoprotein involved in thrombo-inflammatory complications of COVID-19. Chem-Biol Interact. 2021;348:109657.
Dupont A, Rauch A, Staessens S, Moussa M, Rosa M, Corseaux D, et al. Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arteriosclerosis Thrombosis Vasc Biol. 2021;41:1760–73.
Osburn WO, Smith K, Yanek L, Amat-Alcaron N, Thiemann DR, Cox AL, et al. Markers of endothelial cell activation are associated with the severity of pulmonary disease in COVID-19. PLoS One. 2022;17:e0268296.
Abraham GR, Kuc RE, Althage M, Greasley PJ, Ambery P, Maguire JJ, et al. Endothelin-1 is increased in the plasma of patients hospitalised with Covid-19. J Mol Cell Cardiol. 2022;167:92–6.
Nogueira RC, Minnion M, Clark AD, Dyson A, Tanus-Santos JE, Feelisch M. On the origin of nitrosylated hemoglobin in COVID-19: Endothelial NO capture or redox conversion of nitrite?: Experimental results and a cautionary note on challenges in translational research. Redox Biol. 2022;54:102362.
Yang K, Holt M, Fan M, Lam V, Yang Y, Ha T, et al. Cardiovascular dysfunction in COVID-19: association between endothelial cell injury and lactate. Front Immunol. 2022;13:868679.
Vassiliou AG, Keskinidou C, Jahaj E, Gallos P, Dimopoulou I, Kotanidou A, et al. ICU admission levels of endothelial biomarkers as predictors of mortality in critically Ill COVID-19 patients. Cells. 2021;10:186.
Oliveira MR, Back GD, da Luz Goulart C, Domingos BC, Arena R, Borghi-Silva A. Endothelial function provides early prognostic information in patients with COVID-19: A cohort study. Respir Med. 2021;185:106469.
Zha D, Fu M, Qian Y. Vascular endothelial glycocalyx damage and potential targeted therapy in COVID-19. Cells. 2022;11:1972.
Okada H, Yoshida S, Hara A, Ogura S, Tomita H. Vascular endothelial injury exacerbates coronavirus disease 2019: The role of endothelial glycocalyx protection. Microcirculation (N. Y, N. Y: 1994). 2021;28:e12654.
Mensah SA, Cheng MJ, Homayoni H, Plouffe BD, Coury AJ, Ebong EE. Regeneration of glycocalyx by heparan sulfate and sphingosine 1-phosphate restores inter-endothelial communication. PLoS One. 2017;12:e0186116.
Suzuki K, Okada H, Tomita H, Sumi K, Kakino Y, Yasuda R, et al. Possible involvement of Syndecan-1 in the state of COVID-19 related to endothelial injury. Thrombosis J. 2021;19:5.
Drost CC, Rovas A, Osiaevi I, Rauen M, van der Vlag J, Buijsers B, et al. Heparanase is a putative mediator of endothelial glycocalyx damage in COVID-19 - A proof-of-concept study. Front Immunol. 2022;13:916512.
Zhang D, Li L, Chen Y, Ma J, Yang Y, Aodeng S, et al. Syndecan-1, an indicator of endothelial glycocalyx degradation, predicts outcome of patients admitted to an ICU with COVID-19. Mol Med (Camb, Mass). 2021;27:151.
Khider L, Gendron N, Goudot G, Chocron R, Hauw-Berlemont C, Cheng C, et al. Curative anticoagulation prevents endothelial lesion in COVID-19 patients. J Thrombosis Haemost. 2020;18:2391–9.
Guervilly C, Burtey S, Sabatier F, Cauchois R, Lano G, Abdili E, et al. Circulating endothelial cells as a marker of endothelial injury in severe COVID -19. J Infect Dis. 2020;222:1789–93.
Lee KCH, Sewa DW, Phua GC. Potential role of statins in COVID-19. Int J Infect Dis. 2020;96:615–7.
Onorato D, Pucci M, Carpene G, Henry BM, Sanchis-Gomar F, Lippi G. Protective effects of statins administration in European and North American patients infected with COVID-19: a meta-analysis. Semin Thrombosis Hemost. 2021;47:392–9.
Zhang XJ, Qin JJ, Cheng X, Shen L, Zhao YC, Yuan Y, et al. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab. 2020;32:176–87.
Xu S, Liu Y, Ding Y, Luo S, Zheng X, Wu X, et al. The zinc finger transcription factor, KLF2, protects against COVID-19 associated endothelial dysfunction. Signal Transduct Target Ther. 2021;6:266.
Pawlos A, Niedzielski M, Gorzelak-Pabiś P, Broncel M, Woźniak E. COVID-19: direct and indirect mechanisms of statins. Int J Mol Sci. 2021;22:4177.
Corrao S, Pinelli K, Vacca M, Raspanti M, Argano C. Type 2 diabetes mellitus and COVID-19: a narrative review. Front Endocrinol. 2021;12:609470.
Ding Y, Zhou Y, Ling P, Feng X, Luo S, Zheng X, et al. Metformin in cardiovascular diabetology: a focused review of its impact on endothelial function. Theranostics. 2021;11:9376–96.
Samuel SM, Varghese E, Büsselberg D. Therapeutic potential of metformin in COVID-19: reasoning for its protective role. Trends Microbiol. 2021;29:894–907.
Han T, Ma S, Sun C, Zhang H, Qu G, Chen Y, et al. The association between anti-diabetic agents and clinical outcomes of COVID-19 in patients with diabetes: a systematic review and meta-analysis. Arch Med Res. 2021;53:186–95.
Cheng X, Liu YM, Li H, Zhang X, Lei F, Qin JJ, et al. Metformin is associated with higher incidence of acidosis, but not mortality, in individuals with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 2020;32:537–47.
Liu Z, Ma X, Ilyas I, Zheng X, Luo S, Little PJ, et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: from pharmacology to pre-clinical and clinical therapeutics. Theranostics. 2021;11:4502–15.
Bauersachs J, de Boer RA, Lindenfeld J, Bozkurt B. The year in cardiovascular medicine 2021: heart failure and cardiomyopathies. Eur Heart J. 2022;43:367–76.
Sholzberg M, Tang GH, Rahhal H, AlHamzah M, Kreuziger LB, Áinle FN, et al. Effectiveness of therapeutic heparin versus prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with covid-19 admitted to hospital: RAPID randomised clinical trial. BMJ. 2021;375:n2400.
Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care (Lond, Engl). 2020;24:422.
Iwanski J, Kazmouz SG, Li S, Stansfield B, Salem TT, Perez-Miller S, et al. Antihypertensive drug treatment and susceptibility to SARS-CoV-2 infection in human PSC-derived cardiomyocytes and primary endothelial cells. Stem Cell Rep. 2021;16:2459–72.
Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized With COVID-19. Circ Res. 2020;126:1671–81.
Tetlow S, Segiet-Swiecicka A, O’Sullivan R, O’Halloran S, Kalb K, Brathwaite-Shirley C, et al. ACE inhibitors, angiotensin receptor blockers and endothelial injury in COVID-19. J Intern Med. 2021;289:688–99.
Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384:693–704.
Kim WY, Kweon OJ, Cha MJ, Baek MS, Choi SH. Dexamethasone may improve severe COVID-19 via ameliorating endothelial injury and inflammation: A preliminary pilot study. PLoS One. 2021;16:e0254167.
Kang S, Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. 2021;53:1116–23.
Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci USA. 2020;117:22351–6.
Ikonomidis I, Pavlidis G, Katsimbri P, Lambadiari V, Parissis J, Andreadou I, et al. Tocilizumab improves oxidative stress and endothelial glycocalyx: A mechanism that may explain the effects of biological treatment on COVID-19. Food Chem Toxicol. 2020;145:111694.
Schultheiß C, Willscher E, Paschold L, Gottschick C, Klee B, Henkes SS, et al. The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep Med. 2022;3:100663.
Xiong S, Zhang L, Richner JM, Class J, Rehman J, Malik AB. Interleukin-1RA mitigates SARS-CoV-2-induced inflammatory lung vascular leakage and mortality in humanized K18-hACE-2 mice. Arterioscler Thrombosis Vasc Biol. 2021;41:2773–85.
Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. 2020;2:e393–e400.
Kyriazopoulou E, Huet T, Cavalli G, Gori A, Kyprianou M, Pickkers P, et al. Effect of anakinra on mortality in patients with COVID-19: a systematic review and patient-level meta-analysis. Lancet Rheumatol. 2021;3:e690–e7.
Schattner A. Colchicine-new horizons for an ancient drug. Eur J Intern Med. 2022;96:34–41.
Zhang FS, He QZ, Qin CH, Little PJ, Weng JP, Xu SW. Therapeutic potential of colchicine in cardiovascular medicine: a pharmacological review. Acta Pharmacol Sin. 2022;43:2173–90.
Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6:402.
Satarker S, Tom AA, Shaji RA, Alosious A, Luvis M, Nampoothiri M. JAK-STAT pathway inhibition and their implications in COVID-19 therapy. Postgrad Med. 2021;133:489–507.
Fiorentino G, Coppola A, Izzo R, Annunziata A, Bernardo M, Lombardi A, et al. Effects of adding L-arginine orally to standard therapy in patients with COVID-19: A randomized, double-blind, placebo-controlled, parallel-group trial. Results of the first interim analysis. EClinicalMedicine. 2021;40:101125.
Oskotsky T, Maric I, Tang A, Oskotsky B, Wong RJ, Aghaeepour N, et al. Mortality risk among patients with COVID-19 prescribed selective serotonin reuptake inhibitor antidepressants. JAMA Netw Open. 2021;4:e2133090.
Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, et al. Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: a randomized clinical trial. JAMA. 2020;324:2292–300.
Reis G, Dos Santos Moreira-Silva EA, Silva DCM, Thabane L, Milagres AC, Ferreira TS, et al. Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the TOGETHER randomised, platform clinical trial. Lancet Glob Health. 2022;10:e42–e51.
Papadopoulos KI, Sutheesophon W, Aw TC. Anti-SARS-CoV-2 action of fluvoxamine may be mediated by endothelial nitric oxide synthase. Pharmacopsychiatry. 2022;55:57.
Hemilä H, de Man AME. Vitamin C and COVID-19. Front Med. 2020;7:559811.
May CN, Bellomo R, Lankadeva YR. Therapeutic potential of megadose vitamin C to reverse organ dysfunction in sepsis and COVID-19. Br J Pharmacol. 2021;178:3864–8.
Tomasa-Irriguible TM, Bielsa-Berrocal L. COVID-19: Up to 82% critically ill patients had low vitamin C values. Nutr J. 2021;20:66.
Xia G, Qin B, Ma C, Zhu Y, Zheng Q. High-dose vitamin C ameliorates cardiac injury in COVID-19 pandemic: a retrospective cohort study. Aging. 2021;13:20906–14.
Hess AL, Halalau A, Dokter JJ, Paydawy TS, Karabon P, Bastani A, et al. High-dose intravenous vitamin C decreases rates of mechanical ventilation and cardiac arrest in severe COVID-19. Intern Emerg Med. 2022: 1-10.
Bhowmik KK, Barek MA, Aziz MA, Islam MS. Impact of high-dose vitamin C on the mortality, severity, and duration of hospital stay in COVID-19 patients: a meta-analysis. Health Sci Rep. 2022;5:e762.
Tan R, Xiang X, Chen W, Yang Z, Hu W, Qu H, et al. Efficacy of diammonium glycyrrhizinate combined with vitamin C for treating hospitalized COVID-19 patients: a retrospective, observational study. QJM. 2022;115:77–83.
Chen S, Zheng C, Chen T, Huang D, Pan Y, Chen S. Relationship between plasma vitamin C and COVID-19 susceptibility and severity: a two-sample mendelian randomization study. Front Med. 2022;9:844228.
Milani GP, Macchi M, Guz-Mark A. Vitamin C in the treatment of COVID-19. Nutrients. 2021;13:1172.
Huang N, Li S High-quality trials and pharmacological studies needed as translational evidence for the application of traditional Chinese medicine Lianhua Qingwen against COVID-19. Phytother Res. 2022, https://doi.org/10.1002/ptr.7574.
Kang X, Jin D, Jiang L, Zhang Y, Zhang Y, An X, et al. Efficacy and mechanisms of traditional Chinese medicine for COVID-19: a systematic review. Chin Med. 2022;17:30.
Xing D, Liu Z. Effectiveness and safety of traditional chinese medicine in treating COVID-19: clinical evidence from China. Aging Dis. 2021;12:1850–6.
Ni L, Wen Z, Hu X, Tang W, Wang H, Zhou L, et al. Effects of Shuanghuanglian oral liquids on patients with COVID-19: a randomized, open-label, parallel-controlled, multicenter clinical trial. Front Med. 2021;15:704–17.
Li M, Zhu H, Liu Y, Lu Y, Sun M, Zhang Y, et al. Role of traditional chinese medicine in treating severe or critical covid-19: a systematic review of randomized controlled trials and observational studies. Front Pharmacol. 2022;13:926189.
Wenzel J, Lampe J, Müller-Fielitz H, Schuster R, Zille M, Müller K, et al. The SARS-CoV-2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat Neurosci. 2021;24:1522–33.
Charfeddine S, Ibnhadjamor H, Jdidi J, Torjmen S, Kraiem S, Bahloul A, et al. Sulodexide significantly improves endothelial dysfunction and alleviates chest pain and palpitations in patients with long-COVID-19: Insights From TUN-EndCOV study. Front Cardiovasc Med. 2022;9:866113.
Pang J, Xu F, Aondio G, Li Y, Fumagalli A, Lu M, et al. Efficacy and tolerability of bevacizumab in patients with severe Covid-19. Nat Commun. 2021;12:814.
Karakas M, Jarczak D, Becker M, Roedl K, Addo MM, Hein F, et al. Targeting endothelial dysfunction in eight extreme-critically ill patients with COVID-19 using the anti-adrenomedullin antibody adrecizumab (HAM8101). Biomolecules. 2020;10:1171.
Qin H, Zhao A. Mesenchymal stem cell therapy for acute respiratory distress syndrome: from basic to clinics. Protein Cell. 2020;11:707–22.
Breithaupt-Faloppa AC, Correia CJ, Prado CM, Stilhano RS, Ureshino RP, Moreira LFP. 17β-Estradiol, a potential ally to alleviate SARS-CoV-2 infection. Clin (Sao Paulo, Braz). 2020;75:e1980.
Cecon E, Fernandois D, Renault N, Coelho CFF, Wenzel J, Bedart C, et al. Melatonin drugs inhibit SARS-CoV-2 entry into the brain and virus-induced damage of cerebral small vessels. Cell Mol Life Sci. 2022;79:361.
Fang W, Jiang J, Su L, Shu T, Liu H, Lai S, et al. The role of NO in COVID-19 and potential therapeutic strategies. Free Radic Biol Med. 2021;163:153–62.
Zhang X, Jiang M, Yang J. Potential value of circulating endothelial cells for the diagnosis and treatment of COVID-19. Int J Infect Dis. 2021;107:232–3.
Müller R, Rink G, Uzun G, Bakchoul T, Wuchter P, Klüter H, et al. Increased plasma level of soluble P-selectin in non-hospitalized COVID-19 convalescent donors. Thrombosis Res. 2022;216:120–4.
Mansiroglu AK, Seymen H, Sincer I, Gunes Y. Evaluation of endothelial dysfunction in COVID-19 with flow-mediated dilatation. Arq Bras Cardiol. 2022;119:319–25.
Batabyal R, Freishtat N, Hill E, Rehman M, Freishtat R, Koutroulis I. Metabolic dysfunction and immunometabolism in COVID-19 pathophysiology and therapeutics. Int J Obes (2005). 2021;45:1163–9.
Acknowledgements
This study was supported by grants from National Key R&D Program of China (Grant No. 2021YFC2500500), National Natural Science Foundation of China (Grant No. 82070464, 81941022, 81530025) and Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB38010100). This work was also supported by Program for Innovative Research Team of The First Affiliated Hospital of USTC (CXGG02), Anhui Provincial Key Research and Development Program (Grant No. 202104j07020051), Anhui Province Science Fund for Distinguished Young Scholars (Grant No. 2208085J08) and Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (Grant No. 2017BT01S131), Hefei Comprehensive National Science Center (Grant No. BJ9100000005), and Hefei Municipal Development and Reform Commission Emergency Funding for COVID-19 disease.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, Sw., Ilyas, I. & Weng, Jp. Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol Sin 44, 695–709 (2023). https://doi.org/10.1038/s41401-022-00998-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00998-0
Keywords
This article is cited by
-
Association of circulating biomarkers with illness severity measures differentiates myalgic encephalomyelitis/chronic fatigue syndrome and post-COVID-19 condition: a prospective pilot cohort study
Journal of Translational Medicine (2024)
-
Impaired retinal oxygen metabolism and perfusion are accompanied by plasma protein and lipid alterations in recovered COVID-19 patients
Scientific Reports (2024)
-
IL-6 and IL-17 as potential links between pre-existing hypertension and long-term COVID sequelae in patients undergoing hemodialysis: a multicenter cross-sectional study
Scientific Reports (2024)
-
Association Between Periodontitis and COVID-19
Current Oral Health Reports (2024)
-
Serum from COVID-19 patients promotes endothelial cell dysfunction through protease-activated receptor 2
Inflammation Research (2024)
