
Abstract
Small-cell lung cancer (SCLC) is an aggressive high-grade neuroendocrine carcinoma of the lung associated with early metastasis and an exceptionally poor prognosis. Little progress has been made in developing efficacious targeted therapy for this recalcitrant disease. Herein, we showed that H3.3, encoded by two genes (H3F3A and H3F3B), was prominently overexpressed in SCLC. Darinaparsin (ZIO-101), a derivative of arsenic trioxide, dose- and time-dependently inhibited the viability of SCLC cells in an H3.3-dependent manner. More importantly, ZIO-101 treatment resulted in substantial accumulation of H3.3 and PARP1 besides induction of G2/M cell cycle arrest and apoptosis in SCLC cells. Through integrative analysis of the RNA-seq data from Cancer Cell Line Encyclopedia dataset, JNCI and Genomics of Drug Sensitivity in Cancer 2 datasets, we found that H3F3A expression was negatively correlated with the IC50 values of PARP inhibitors (PARPi). Furthermore, co-targeting H3.3 and PARP1 by ZIO-101 and BMN673/olaparib achieved synergistic growth inhibition against SCLC in vitro and in vivo. In conclusion, it is feasible to target H3.3 by ZIO-101 to potentiate the response rate of PARPi in SCLC patients.
Similar content being viewed by others


Introduction
Lung cancer remains the leading cause of cancer death worldwide in 2020, representing 18.0% of all cancer death [1, 2]. Small-cell lung cancer (SCLC), which makes up about 15% of lung cancer cases, is a highly aggressive neuroendocrine lung tumor with an exceptionally poor prognosis due to its high metastatic potential [3, 4]. No breakthrough has been made in the therapeutic strategies for treating SCLC in the past three decades [5]. So far, platinum-based chemotherapy remains the standard for both limited-stage SCLC (LS-SCLC) and extensive-stage disease (ES-SCLC). The 2-year overall survival rates are 21% in LS-SCLC and 7% in ES-SCLC [6], owing to a quick cancer relapse or incurability. Although several drug combinations or the addition of third-line drugs have demonstrated efficacy in the preclinical investigation [7, 8], more clinical trials need to further proceed. Recently, although the inhibitors of the programmed cell death protein and its ligand (PD-1/PD-L1) combined with chemotherapy have been approved by the Food and Drug Administration (FDA) to treat ES-SCLC owing to improving survival in patients with ED-SCLC, the beneficiaries are still limited [6, 9]. Therefore, there is a pressing need for new treatment options to improve SCLC patients’ survival.
Poly-(ADP)-ribose polymerase 1 (PARP1), a critical enzyme for DNA damage repair, has emerged as a promising therapeutic target in multiple cancer types [10,11,12,13]. Several PARP inhibitors (PARPi) as single agents or in combination with chemotherapeutic drugs have been approved by the FDA for ovarian, breast, and pancreatic cancer patients [10, 11]. PARP1 is highly expressed at both the mRNA and protein levels in SCLC cell lines and clinical specimens [14,15,16]. PARPi niraparib as monotherapy modestly delays disease progression and prolongs survival with an acceptable tolerability profile in patients with ES-SCLC [17]. PARPi combined with other inhibitors has been exploited in the clinic and showed some improvement in SCLC patients [18, 19]. However, PARPi is not currently part of the treatment armamentarium for SCLC.
The histone variant H3.3, encoded by two genes (H3F3A and H3F3B), is highly evolutionarily conserved from yeast to humans and dynamically incorporated into chromatin with the independence of DNA replication [20, 21]. When cells are suffered UV irradiation, newly synthesized H3.3 is deposited at DNA damage sites prior to repair. H3.3 knockout (KO) cells are susceptible to UV light [22]. These results indicate that H3.3 might play a role in DNA damage response. Both mutations and misregulation in H3F3A and H3F3B have been increasingly recognized in a variety of malignancies [23,24,25]. For instance, the overexpression of H3F3A activates G-protein-coupled receptor 87 (GPR87) expression and promotes migration through intronic regulation in lung cancer cells [26]. Metastatic stimuli increase H3.3 incorporation into chromatin, leading to chromatin remodeling and expression of metastatic genes in breast and NSCLC cancer cells [27]. However, the role of the H3.3 genes in SCLC remains elusive.
Darinaparsin (ZIO-101, S-dimethylarsino-glutathione), an organic arsenical derivative of arsenic trioxide (As2O3), exerts a wide range of potency against many cancer cells and is well-tolerated when administered in humans [28,29,30,31]. Interestingly, ZIO-101 has radiosensitizing activity against solid tumor cells but not normal bone marrow and intestinal crypts [32]. In addition, a recent study elucidates that ZIO-101 induces apoptosis and exhibits a therapeutic effect in leukemia cells by targeting H3.3 [33]. Nevertheless, the anti-tumor effects of ZIO-101 in SCLC have not been explored so far.
The current study finds that H3.3 is significantly overexpressed in SCLC. ZIO-101 displays a distinct inhibitory effect on SCLC cells, likely owing to a G2/M cell cycle arrest and apoptosis. Integrative analysis of the RNA-seq data from Cancer Cell Line Encyclopedia (CCLE), Genomics of Drug Sensitivity in Cancer 2 (GDSC2), and JNCI datasets suggests that H3.3 expression has a considerable impact on the sensitivity of many chemotherapeutic drugs, including PARPi. Moreover, the ZIO-101 treatment elicits a substantial accumulation of H3.3 and PARP1. Combining ZIO-101 and PARPi BMN673 causes striking synergistic effects on tumor growth in vitro and in vivo. Collectively, targeting epigenetic factors via H3.3 inhibition enhances the sensitivity of SCLC to PARPi.
Materials and methods
Reagents and antibodies
BMN673 and olaparib were purchased from Selleck Chemical (Shanghai, China). Darinaparsin (ZIO-101) was obtained from Glpbio (Montclair, CA, USA). Antibodies against PARP, cleaved caspase-3, phospho-histone H2A.X, Bcl-xL, and Rad51 were purchased from Cell Signaling Technology (Danvers, MA, USA). Cyclin B1 and H3.3 antibodies were obtained from Abcam (Abcam, Cambridge, UK). Anti-PARP1 was purchased from Active Motif (Carlsbad, CA, USA). Anti-β-Actin was purchased from TransBionovo (Beijing, China).
Cell line maintenance
Human SCLC cell lines (DMS273 and SHP77) and human lung adenocarcinoma (LUAD) cell lines (PC9 and A549) were routinely maintained in RPMI-1640 medium (Cellgro, Manassas, VA, USA). A human small airway epithelial cell line, HSAEC, was cultivated in DMEM medium (Cellgro, Manassas, VA, USA). All cells culture was supplemented with 10% fetal bovine serum (VivaCell, Shanghai, China) and 1% penicillin/streptomycin (Servicebio, Wuhan, China) in a 37 °C incubator containing 5% CO2. In addition, all cell lines were routinely tested for mycoplasma contamination using a mycoplasma detection kit (Beyotime, Shanghai, China). Exponential growing cells were harvested for the experiments.
Analysis of CCLE, GDSC, and JNCI datasets
RNA-seq data of 1393 cancer cell lines, including 50 SCLC and 75 LUAD cell lines, and general information for these cell lines from the CCLE were downloaded from the CCLE database (https://portals.broadinstitute.org/ccle/data). The GDSC dataset was downloaded from https://www.cancerrxgene.org/downloads/anova. The JNCI dataset was obtained from Polley et al. [34]. The normalized gene expression from microarray analysis was downloaded from the Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo. accession number: GDS4794). Expression data for H3F3A, H3F3B, and PARP1 and IC50 values from SCLC cell lines were retrieved, analyzed, and displayed in scatter plots.
Construction of stable H3F3A knockdown and H3F3A overexpressing cell lines
The short hairpin RNA (shRNA) target sequences were designed using the AsiDesigner software (http://sysbio.kribb.re.kr:8080/AsiDesigner/). The shRNA targeting sequences were as followed: shH3F3A #1: 5′-GCGGAGACGTAAAGCATTAAT-3′ (targeting region: 3′ UTR); shH3F3A #2: 5′-CAGGAAGCAACTGGCTACAAA-3′ (targeting region: CDS); shControl: 5′-TACGCCACCAATTTCGTTTCA-3′. The shH3F3A #1 and shH3F3A #2 sequences were subcloned into the pLKO.1-puro (lentiviral vector) and pLKO.1-puro-tet on (Doxycycline (Dox)-inducible lentiviral vector), respectively. For H3F3A overexpressing stable cell lines, the H3F3A gene was amplified by PCR and inserted into the pTRIPZ, a Dox-inducible lentiviral vector. For viral particle generation, cloned plasmid DNAs, psPAX2 (Addgene plasmid #12260), and pMD2.G (Addgene plasmid #12259) were co-transfected into HEK293T cells using Effectene Transfection Reagent (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Eight hours post transfection, the tissue culture media was removed and replaced with fresh RPMI- 1640. The viral supernatant was harvested 48 h after transfection. A total of 1.5 × 105 cells were added to each well of a 6-well plate for viral infection and incubated overnight. The lentiviral particles were added to each well in the presence of 8 mg/mL polybrene (Sigma–Aldrich, Saint Louis, MO, USA). The selection of resistant colonies was initiated 48 h later using 2 mg/mL puromycin (Sigma–Aldrich, Saint Louis, MO, USA). For cells carrying an inducible transgene, the cells were treated daily with 1 mg/mL doxycycline (Sigma–Aldrich, Saint Louis, MO, USA) for the timepoints indicated to induce transgene expression.
RNA isolation and quantitative RT-qPCR
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN, Hilden, Germany) and subsequently synthesized into single-stranded cDNA using M-MLV reverse transcriptase (Promega, Madison, WI, USA) following the manufacturer’s instructions. Quantitative real-time PCR (RT-qPCR) reactions were conducted in triplicate on a LightCycler 96 machine (Roche, Mannheim, Germany) using the LightCycler 96 SYBR Green I Master Mix (Roche, Mannheim, Germany). Relative gene expression levels were normalized to β-Actin expression. The primers were listed as follows: H3F3A-F: 5′-CTTTCAATTGTGTTCGCAGCCGCCG-3′, H3F3A-R: 5′-TTGCGGGCAGTCTGCTTTGTACGAG-3′; H3F3B-F: 5′-CCAAGCAGACTGCTCGTAAGTC-3′, H3F3B-R: 5′-CGATGGCTGCGCTCTGAAACC-3′; β-Actin-F: 5′-TCCCTGGAGAAGAGCTACGA-3′, β-Actin-R: 5′-AGCACTGTGTTGGCGTACAG-3′.
Cell viability assays
Cells were seeded into 96-well plates (Costar, Kennebunk, ME, USA) at a density of 3000 cells per well. Then, 24 h later, cells were treated with corresponding drugs for 72 h in triplicate. Cell viability was determined using the CellTiter-Glo® luminescent assay (Promega, Madison, WI, USA). The luminescence signals were recorded using a multilabel plate reader (Envision PerkinElmer, USA). The percentage of cell viability at each concentration was calculated against the control. The IC50 values were determined from the sigmoidal dose–response curves. Drug synergy was determined quantitatively using Chou and Talalay’s combination index (CI) method [35]. The CI was calculated using the formula: CI = D1/DX1 + D2/DX2, in which D1 and D2 are the doses used to achieve a specific growth inhibition when two drugs are combined, and DX1 represents the dose of drug 1 alone required to reach inhibition and DX2 is the dose of drug 2 alone required to reach inhibition. The parameters were calculated in CalcuSyn software (Biosoft, Cambridge, UK). In general, “CI < 1” indicates synergism, whereas “CI > 1” indicates antagonism.
Colony-formation assays
The long-term effects of drug treatment were assessed by colony-formation assay after exposure of the cells to a drug-containing medium. Seven to ten days later, cell colonies were washed with PBS twice, fixed with methanol, and stained with 1% crystal violet solution. Colonies were scored based on standard morphological criteria. The surviving fraction at each concentration was calculated after normalization to untreated samples.
Western blotting
After treatment, cell lysates were collected for Western blotting. Total protein samples were prepared in lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 8.0, 1% Triton X-100, and 1 mM EDTA) supplemented with protease and phosphatase inhibitors (Roche, Mannheim, Germany), subsequently quantified using the Pierce BCA Protein Assay Kit (BI, Shanghai, China). Twenty micrograms of protein were used for SDS-PAGE to separate and then transferred to an Immobilon-P PVDF membrane (Millipore, Saint Louis, MO, USA) and probed with the primary antibody incubation at 4 °C overnight. HRP-conjugated anti-mouse or anti-rabbit IgG antibody was used as the secondary antibody at a final concentration of 1:5000 in 5% milk-TBST for 1 h at room temperature (RT). Membranes were washed three times for 5 min each in TBST, and antibody binding was detected using the ECL Western blotting kit (Thermo Fisher Scientific, USA).
Cell cycle and apoptosis analysis
After treatment, the cells (~1 × 105) were collected by centrifugation at 500 × g for 5 min and then fixed in 75% cold ethanol and incubated at −20 °C overnight. Fixed cells were washed once with PBS and stained with PI/RNase staining buffer (Beckman Coulter, Brea, CA, USA) for 10 min at RT. Flow cytometry was performed using a FACSCalibur system (Beckman Coulter, Brea, CA, USA). The cell-cycle profiles were determined using FlowJo software (Beckman Coulter, Brea, CA, USA). The cells were stained with FITC Annexin V and PI using a FITC Annexin V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA) to measure apoptosis. The luminescent signals were then monitored in a FACSCalibur system (Beckman Coulter, Brea, CA, USA). At least 10,000 events were counted. All results were further analyzed using FlowJo software (Beckman Coulter, Brea, CA, USA).
Xenograft tumor model
Animal experiments were carried out according to a protocol approved by the Institutional Animal Care and Use Committee of Hefei Institutes of Physical Science. Briefly, female nu/nu athymic mice at the age of 4–6 weeks were purchased from Vital River (Shanghai, China) and maintained in the research animal facility of Hefei Institutes of Physical Science, Chinese Academy of Sciences, under standard environmental conditions. A nude mice model with xenograft tumor was established by the subcutaneous inoculation of 2 × 106 DMS273 cells. The tumor-bearing mice were randomly assigned to four groups (Vehicle, ZIO-101, BMN673, ZIO-101 plus BMN673 combination group). Before administration, ZIO-101 and BMN673 were dissolved in PBS, then administered via intraperitoneal injection at a dose of 50 and 330 mg/kg every other day, respectively. The Vehicle group received the same volume of PBS, and the combination group received the same dose. Tumor sizes were measured using a caliper every other day, and tumor volumes were calculated using the following equation: tumor volume [mm3] = (tumor length × tumor width2) / 2.
H&E staining and immunohistochemistry
Tumors were harvested from euthanized mice, fixed in 4% paraformaldehyde for 24 h, and subsequently embedded in paraffin wax. After sections were cut in 6 μm, tissue slices were dewaxed, rehydrated, and stained with Mayer’s hematoxylin and eosin Y solution. Subsequently, the sections were dehydrated and mounted. Immunohistochemistry was performed as previously described [14]. For the quantification of immunohistochemistry staining, the H3.3, PARP1, Ki67, Bcl-xL, cyclin B1, and cleaved caspase-3 staining were quantified by integrated optical density equaled to optical density by area using image-pro plus software.
Statistical analysis
All data were analyzed using Prism 6 Software (GraphPad Software, Inc., La Jolla, CA, USA). All values were expressed as mean ± SEM, and statistical significance was calculated and assessed using two-tailed unpaired Student’s t-tests. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Results
H3.3 is highly expressed in SCLC
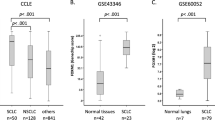
Misregulation of H3.3 has also been implicated in various cancers besides recurrent mutation of H3F3A in pediatric glioblastoma and other cancer types [26, 27, 36]. However, the roles of H3.3 in SCLC remain exclusive. To characterize H3.3 mutation status in SCLC, the somatic mutation profiles of H3F3A and H3F3B were first performed based on 249 SCLC clinical samples obtained from the cBioPortal database (https://www.cbioportal.org/). As depicted in Supplementary Fig. S1, H3F3A harbors a mutation in 0.4% of cases, and no mutation was observed in H3F3B. When H3F3A and H3F3B mRNA expression levels among over 1000 cancer cell lines from the CCLE were analyzed, we found that SCLC cells had almost the highest H3F3A and H3F3B expression among all investigated cancer cell lines (Fig. 1a, b). The expression of canonical H3 and its variants were then evaluated. The analysis showed that H3F3A and H3F3B were significantly higher than their canonical counterparts H3.1 and H3.2, H3.1t, H3.5, and CENPA genes in SCLC cells (Fig. 1c). Notably, Analysis of mRNA expression of H3.3 using the RNA-seq data from CCLE demonstrated that H3F3A and H3F3B expression levels were also strongly increased in SCLC compared with LUAD cells (Fig. 1d, e). In addition, we revealed that H3F3A was highly expressed in human SCLC specimens compared with normal tissues, with 73.9% (17/23) samples showing at least a 1.5-fold increase (Fig. 1f). RT-qPCR and Western blotting assays confirmed that the mRNA levels of H3F3A and H3F3B, and protein level of H3.3 were higher in SCLC cells than in LUAD cells (Fig. 1g, h). These results indicate that H3.3 is prominently overexpressed in SCLC.

a, b mRNA expression level of H3F3A (a) and H3F3B (b) in cell lines representing 39 distinct cancer types from the Cancer Cell Line Encyclopedia (CCLE) database. c Expression levels of classical histone H3 (H3.1 and H3.2) genes and all corresponding variants, including H3.1t, H3.3, H3.5, and CENPA genes in SCLC from CCLE data (n = 50). d, e Scatter plot comparison of H3F3A (d) and H3F3B (e) expression in SCLC cell lines (n = 50) relative to LUAD cell lines (n = 75) from CCLE. f Relative H3F3A expression in human SCLC tissues compared to the normal lung tissues from GDS4794 dataset (n = 23, the red line means more than a 1.5-fold increase). g RT-qPCR analysis of H3F3A and H3F3B mRNA in SCLC cells and LUAD cells. β-Actin was used as an internal control. For statistical analysis, PC9 cell line was used as a control. h Western blotting detection of H3.3 expression in SCLC cells (DMS273 and SHP77), LUAD cells (PC9 and A549), and human small airway epithelial cells (HSAEC). β-Actin was used as a loading control. The differences were analyzed by two-tailed unpaired Student’s t test in (d), (e), and (g). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns no significance.
ZIO-101 induces apoptosis in SCLC cells
We hypothesized that H3.3 could be used as a therapeutic target in SCLC since H3.3 expression is upregulated. A recent study reported that H3.3 is a target of ZIO-101 [33]. First, H3.3 was predicted to associate with ZIO-101 using molecular docking-based binding energy (Fig. 2a). Subsequently, we assessed the anti-tumor activities of ZIO-101. Cell viability assays showed that the ZIO-101 had a significant anti-tumor effect and its half lethal dose (IC50) value was time-dependent in DMS273 and SHP77 cells (Fig. 2b and Supplementary Fig. S2a). Notably, DMS273 cells were more sensitive than SHP77 cells in response to ZIO-101 treatment (Fig. 2b and Supplementary Fig. S2a). Since H3F3A and H3F3B expression were higher in DMS273 cells than in SHP77 cells (Fig. 1g, h), H3.3 expression might affect the sensitivity of SCLC cells to ZIO-101. Therefore, we constructed H3.3 knockdown and H3.3-overexpressing stable cell lines, respectively (Fig. 2c and Supplementary Fig. S2b), and then the SCLC cells were treated with a range of concentrations of ZIO-101 for 24, 48, and 72 h. After being treated with ZIO-101 for 48 h, IC50 values of H3.3-overexpressing cells were significantly lower than control cells. On the contrary, silencing of H3F3A in DMS273 and SHP77 cells induced resistance to ZIO-101 (Fig. 2d). In accordance with the results upon 48 h treatment, cells treated with ZIO-101 for 24 and 72 h showed a similar pattern (Supplementary Fig. S2c). Interestingly, cell viability assays demonstrated that H3.3 depletion significantly inhibited cell proliferation; on the contrary, H3.3 overexpression distinctly promoted the cell proliferation ability of SCLC cells (Supplementary Fig. S2d). In addition, colony-formation assays also showed that the expression level of H3F3A significantly promoted the sensitivity of ZIO-101 in DMS273 and SHP77 cells (Fig. 2e). Together, these observations suggest that H3.3 expression could be a predictive factor in the sensitivity of ZIO-101 in SCLC cells. Furthermore, evaluation of the effect of ZIO-101 on apoptosis by flow cytometry demonstrated that ZIO-101 induced dose- and time-dependent apoptosis in SCLC cells (Fig. 2f, g and Supplementary Fig. S2e–h). Western blotting analysis of cleaved PARP and cleaved caspase-3, two well-recognized apoptotic markers, showed that ZIO-101 treatment caused dose-dependent accumulation in DMS273 and SHP77 cells. Moreover, Bcl-xL, an anti-apoptotic protein [37], was dose-dependently diminished upon treatment with ZIO-101 (Fig. 2h and Supplementary Fig. S2i), reinforcing the notion that ZIO-101 triggers apoptotic phenotypes. Altogether, these results suggest that ZIO-101 has an H3.3-dependent inhibitory effect on SCLC cells primarily through induction of apoptosis.

a Molecular docking prediction of the interaction between ZIO-101 and H3.3. b Growth inhibition curves of ZIO-101 in DMS273 and SHP77 cells. The cells were treated with different concentrations of ZIO-101 for 48 h. The cell viability was then assessed by the CellTiter-Glo assay. The Half lethal dose (IC50) values were determined from the sigmoidal dose–response curves. c Western blotting detection of H3.3 in H3F3A knockdown and H3F3A overexpressing DMS273 and SHP77 cells. SCR is a control for shH3F3A #1. shH3F3A #2 -Dox and H3F3A OV -Dox are the controls of shH3F3A #2 +Dox and H3F3A OV +Dox, respectively. β-Actin was used as a loading control. d Growth inhibition curves of ZIO-101 in H3F3A knockdown and H3F3A overexpressing DMS273 and SHP77 cells for 48 h. The cell viability was assessed by the CellTiter-Glo assay and IC50 values were determined from the sigmoidal dose–response curves. e Clonogenic assay illustrating the effect of H3.3 expression on the sensitivity of ZIO-101 in DMS273 and SHP77 cells. Cells were exposed to a range of concentrations of ZIO-101 for a period of 7–10 days. Quantification of the colony-formation assay was shown in the right panel. f, g Apoptosis was detected by flow cytometry in DMS273 and SHP77 cells treated with a range of concentrations of ZIO-101 for 48 h (f) and quantification of apoptotic rates upon ZIO-101 treatment (g). h Western blotting analysis of Bcl-xL, cleaved PARP, caspase-3, and cleaved caspase-3 in DMS273 and SHP77 cells treated as shown in (f). β-Actin was used as a loading control. The control groups were treated with the same volume of DMSO. The differences were analyzed by two-tailed unpaired Student’s t test in (g). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns no significance.
Treatment with ZIO-101 induces a G2/M cell cycle arrest and substantial accumulation of H3.3 in SCLC
Surprisingly, we found that ZIO-101 treatment induced dose- and time-dependent accumulation of H3.3 (Fig. 3a and Supplementary Fig. S3a). To elucidate whether the position of the cell cycle influences H3.3 expression, DMS273 cells were synchronized in the G1, S, G2, and M phases of the cell cycle, and H3.3 expression was analyzed by Western blotting. In the G1 and S phases of the cell cycle, H3.3 expression was typically expressed at relatively low levels. However, as the cell cycle progressed, H3.3 protein levels increased, reaching high levels at the G2/M phases (Fig. 3b), suggesting a cell cycle-dependent expression of H3.3. Then, we assessed the effect of ZIO-101 on cell cycle progression by flow cytometry. The results showed that ZIO-101 treatment could induce a G2/M cell cycle arrest in a dose- and time-dependent manner (Fig. 3c, d and Supplementary Fig. S3b, c, e, f). At the same time, cyclin B1, a surrogate marker of G2/M arrest [38], was also markedly upregulated (Fig. 3e and Supplementary Fig. S3d). As the G2/M checkpoint is required for genomic stability, H3.3 accumulation in G2/M phases prompted us to reason that ZIO-101 treatment could induce DNA damage. To test this hypothesis, DNA damage repair-related proteins were analyzed in cells treated with different concentrations of ZIO-101. The results showed that γH2AX and Rad51 were distinctly accumulated after ZIO-101 treatment (Fig. 3f and Supplementary Fig. S3g). Noticeably, the activation of the DNA damage response signal was significantly diminished in H3.3-overexpressing DMS273 and SHP77 cells treated with different concentrations of ZIO-101 (Supplementary Fig. S3h). These results indicate that H3.3 might play a protective role in genome defense against DNA damage.

a Western blotting analysis of PARP1 and H3.3 after DMS273 and SHP77 cells were incubated with different concentrations of ZIO-101 for 48 h or different times of ZIO-101 at a concentration of 5 μM, respectively. β-Actin was used as a loading control. b Western blotting analysis of H3.3 in DMS273 cell at different phases of the cell cycle. β-Actin was used as a loading control. c, d Cell cycle distribution of DMS273 and SHP77 cells upon treatment with different concentrations of ZIO-101 for 48 h by flow cytometry (c) and quantification of cell population in different phases of the cell cycle was shown (d). e, f Western blotting analysis of Cyclin B1 (e), γH2AX, and Rad51 (f) in DMS273 and SHP77 cells treated as shown in (c). β-Actin was used as a loading control. The control groups were treated with the same volume of DMSO. The differences were analyzed by two-tailed unpaired Student’s t test in (d). **P < 0.01; ****P < 0.0001.
H3.3 expression correlates with the sensitivity of SCLC cells to chemotherapeutic drugs
To envisage the possible role that H3.3 potentiates chemotherapy sensitivity, H3F3A mRNA expression data and IC50 values of chemotherapeutic drugs from the CCLE database, JNCI, and GDSC datasets were retrieved, respectively. The correlation was evaluated by Pearson correlation analysis. The sensitivity analysis showed that high H3F3A expression was positively associated with the sensitivity of several chemotherapeutic drugs in SCLC cells, including commonly used drugs in the clinic (topotecan, cisplatin, SAHA, olaparib, and BMN673) and epigenetic inhibitors (entinostat, ACY1215, olaparib, and BMN673) (Fig. 4). In contrast, there was a weak or no correlation between H3F3B mRNA level and sensitivity to chemotherapy (Supplementary Fig. S4). Together, these data suggest that H3.3 expression correlates with the sensitivity of chemotherapeutic drugs to SCLC cells.

IC50 values of common chemotherapeutic drugs were negatively correlated with H3F3A expression. Pearson correlation analysis of H3F3A mRNA expression and IC50 values of chemotherapeutic drugs (BMN673, olaparib, SAHA, topotecan, entinostat, doxorubicin, ACY1215, MK1775 and cisplatin) from the JNCI and Genomics of Drug Sensitivity in Cancer (GDSC) datasets.
ZIO-101 markedly induces PARP1 expression in SCLC
Given that H3.3 is a promising therapeutic target and a potential chemosensitizer, we determined to explore the possible combinational therapy for SCLC treatment. First, we extracted PARP1 expression data and IC50 values of BMN673 and olaparib from the CCLE database, JNCI, and GDSC2 datasets, respectively. Correlation analysis showed that the transcription level of PARP1 was negatively correlated with the IC50 values of BMN673 and olaparib (Fig. 5a), suggesting that PARP1 expression affects the sensitivity of PARPi in SCLC cells. Intriguingly, treatment with ZIO-101 led to a dose- and time-dependent increase in PARP1 expression (Fig. 3a and Supplementary Fig. S3a). These data remind us of the potential therapeutic value of using ZIO-101 in combination with PARPi in SCLC. Furthermore, we examined the anti-tumor activities of BMN673 and olaparib in stable H3F3A knockdown or H3.3-overexpressing DMS273 and SHP77 cells. Compared with control cells, the IC50 values of H3.3 knockdown cells were significantly higher, whereas H3.3 overexpression sensitized DMS273 and SHP77 cells to PARPi (Fig. 5b), confirming that H3.3 expression potentiates PARPi activity. Notably, when treated with PARPi for 48 h, H3.3 expression was dose-dependently increased even though there was no effect on PARP1 expression (Fig. 5c), implying the potential of ZIO-101 to combine with PARPi. Altogether, H3.3 expression substantially impacts the sensitivity of BMN673 and olaparib, and ZIO-101, as an effective H3.3 inhibitor, has the potential value of combination therapy with BMN673 or olaparib in SCLC.

a IC50 values of BMN673 and olaparib were negatively correlated with PARP1 expression. Pearson correlation analysis of PARP1 expression and IC50 values of BMN673 and olaparib from GDSC2 dataset. b Growth inhibition curves of BMN673 and olaparib in H3F3A knockdown and H3F3A overexpressing DMS273 and SHP77 cells for 48 h. The IC50 values were determined from the sigmoidal dose–response curves. c Western blotting analysis of H3.3 and PARP1 in DMS273 and SHP77 cells treated with different concentrations BMN673 and olaparib for 48 h. β-Actin was used as a loading control. The control groups were treated with the same volume of DMSO.
Synergistically therapeutic effects of ZIO-101 and PARP inhibitors in vitro
Based on the above findings, we evaluated whether co-targeting H3.3 using ZIO-101 and PARP1 using BMN673 or olaparib could be a potential therapeutic strategy for SCLC treatment. The DMS273 and SHP77 cells were incubated with ZIO-101, BMN673, or olaparib, alone or in combination, for 48 h. Cell viability was measured using the CellTiter-Glo luminescent assay. As shown in Fig. 6a, combination treatment with ZIO-101 and BMN673/olaparib yielded synergistic growth inhibition in DMS273 and SHP77 cells. Western blotting analysis showed that H3.3 expression was significantly increased in the combined treatment group compared with the ZIO-101, BMN673, or olaparib alone groups. However, no noticeable change in PARP1 expression was observed (Fig. 6b). Flow cytometry assays and Western blotting analysis demonstrated that ZIO-101 combined with PARPi exhibited a stronger and long-lasting effect on G2/M arrest than ZIO-101, BMN673, or olaparib alone groups (Fig. 6c–e). To further investigate the cytotoxic activity of ZIO-101 in combination with PARPi, we also sought to evaluate the contribution of apoptosis. FACS analysis showed that combined treatment with ZIO-101 and BMN673 or olaparib resulted in a much greater apoptosis rate than treatment with ZIO-101, BMN673, or olaparib alone in SCLC cells. In particular, ZIO-101 combined with PARPi treatment in H3.3-overexpressing cells showed a stronger apoptosis rate than the control cells. Furthermore, the induction of apoptosis by ZIO-101 combined with PARPi was significantly attenuated by H3.3 knockdown (Fig. 6f and Fig. S5). Notably, the colony-formation assay also showed a significantly anti-tumor long-term effect in the combined treatment groups with ZIO-101 and BMN673 or olaparib compared with the ZIO-101, BMN673 or olaparib alone groups (Fig. 6g). At the same time, Western blotting analysis of cleaved PARP and cleaved caspase-3 showed marked high expression in the combination group. Concomitantly, Bcl-xL expression was markedly reduced once ZIO-101 was combined with PARPi (Fig. 6h). These results indicate that the combined use of ZIO-101 and BMN673/olaparib achieved striking synergy in SCLC cells through apoptosis induction.

a The CellTiter-Glo Luminescent assays demonstrating the combination effects of ZIO-101 and BMN673 or olaparib in DMS273 and SHP77 cells. A mathematical model was applied to calculate the combination index using the CalcuSyn software program. b Western blotting analysis of H3.3 and PARP1 in SCLC cells treated with ZIO-101 and BMN673 or olaparib alone or in combination for 48 h. β-Actin was used as a loading control. c, d Cell cycle distribution of DMS273 and SHP77 cells treated as shown in (b) for 48 h by flow cytometry (c) and quantification of cell population in different phases of the cell cycle was shown (d). e Western blotting analysis of Cyclin B1 in SCLC cells treated as shown in (b). f Apoptosis was detected by flow cytometry in H3F3A knockdown and H3F3A overexpressing DMS273 cells treated as shown in (b). Quantification of the apoptosis rate was shown in the right panel. g Clonogenic assay illustrating that ZIO-101 potentiated the inhibiting effect of BMN673 and olaparib. DMS273 and SHP77 cells were exposed to ZIO-101, BMN673, or olaparib, alone or in combination for a period of 7–10 days. h Western blotting analysis of BcL-xL, cleaved PARP, caspase-3, and cleaved caspase-3 in DMS273 and SHP77 cells treated as shown in (b). β-Actin was used as a loading control. The control groups were treated with the same volume of DMSO. The differences were analyzed by two-tailed unpaired Student’s t test in (d) and (f). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns no significance.
Dual inhibitions of H3.3 and PARP are synergistic in vivo
The synergistic effects induced by H3.3 inhibition and PARP inhibition in vitro led us to evaluate the therapeutic efficacy of combining ZIO-101 with BMN673 in SCLC xenograft models. After the successful establishment of the xenograft model using DMS273 cells, the tumor-bearing mice were treated with vehicle control, ZIO-101, BMN673, or combination (Fig. 7a). After 2-week treatment, ZIO-101 or BMN673 alone modestly suppressed tumor growth (Fig. 7b, c) and reduced tumor weight compared with the vehicle group (Fig. 7d). Importantly, ZIO-101 and BMN673 combination synergistically attenuated tumor growth and reduced tumor weight to a much greater extent than either drug alone (Fig. 7b–d). Notably, all treatments did not induce significant body weight loss, indicating minimal toxicity of ZIO-101 or BMN673 alone or the combination in vivo (Fig. 7e). Furthermore, H&E staining showed that the tumor tissues became looser and the space around the cells became larger in the groups treated with ZIO-101 or BMN673, particularly in the combination of ZIO-101 and BMN673 group, than in the vehicle group, which might be due to nuclear condensation caused by apoptosis (Fig. 7f). The immunohistochemical analysis delineated that the Ki67- and Bcl-xL-positive cells were substantially decreased. Concomitantly, cleaved caspase-3- and cyclin B1-positive tumor cells significantly increased in the group treated with ZIO-101 combined with BMN673 compared with the vehicle and monotherapy groups (Fig. 7f). Remarkably, although there was a slight increase in the monotherapy group, the combination of ZIO-101 and BMN673 showed the most remarkable upregulation of H3.3 expression, which may be due to a cell cycle arrest. Meanwhile, consistent with our previous results, upregulation of PARP1 was also observed, especially in the ZIO-101 monotherapy group (Fig. 7f). Collectively, the in vivo study demonstrated that combined administration of ZIO-101 and BMN673 induced synergistic anti-tumor effects against SCLC xenografts.

a Graphical abstract of the xenograft mice and drugs treatment. DMS273 cell was injected into the dorsal flanks of nude mice to form xenograft tumors, and then the tumor-bearing mice were treated with ZIO-101, BMN673, or a combination of ZIO-101 and BMN673 for 14 days (n = 6 for each group). b Tumor volume curves of xenograft mice treated with ZIO-101, BMN673, or a combination of ZIO-101 and BMN673 for 14 days in vivo. c Images of representative xenograft tumors (n = 6). d Scatter plot representing tumor weights of xenograft mice treated with ZIO-101, BMN673, or a combination of ZIO-101 and BMN673 for 14 days at the endpoint of experiments (n = 6). e Body weight curves of xenograft mice during the course of treatment with ZIO-101, BMN673, or a combination of ZIO-101 and BMN673 for 14 days. f Representative H&E and immunohistochemistry (IHC) images of H3.3, Cyclin B1, Bcl-xL, Ki67, cleaved caspase-3, and PARP1 on tumors. Quantification of the IHC was shown in the right panel. Scale bar, 40 and 100 μm inside and outside the black box, respectively. g Schematic of the proposed synergistic anti-tumor effect of ZIO-101 and PARPi. ZIO-101 targets H3.3 and causes a significant accumulation of H3.3 and PARP1. PARPi also results in an increase of H3.3. The combination of ZIO-101 and PARPi leads to substantial cell apoptosis and cell cycle arrest, exerting synergistic anti-tumor effects. The differences were analyzed by two-tailed unpaired Student’s t test in (b), (d), and (f). ZIO, BMN, and CC3 are abbreviations for ZIO-101, BMN673, and CC3, respectively, in (b), (d), and (f). *P < 0.05; **P < 0.01; ***P < 0.001; ns no significance.
Discussion
The US National Cancer Institute has singled out SCLC as a designated “recalcitrant” cancer based on incidence rate, exceptionally high lethality, and the lack of substantial therapeutic progress made over several decades [4, 39]. The epigenetic regulatory network has become a matter of intense investigation from a pathological perspective since epigenetic aberrations are more amenable to reversal and are considered better targets for therapeutic intervention than genetic changes [40]. Thus, the development of small molecules for enhanced therapeutic benefit against SCLC, specifically targeted epigenetic factors, is pressingly needed. Although distinct and recurring mutations of H3.3 have been associated with a high proportion of malignant pediatric brain cancers, chondroblastoma, and giant-cell tumors of the bone [24, 41], H3.3 has also been reported to be misregulated in various cancers [25, 26, 36, 42]. Previous studies have reported that the stimuli of metastasis increases incorporation of H3.3, which in turn induces chromatin reprogramming and expression of metastatic genes in breast and non-SCLC cells [27]. Moreover, H3F3A overexpression directly activates GPR87 transcription via intronic regulation and facilitates lung cancer cell migration [26]. However, H3.3 is not always elevated in tumor tissue. For example, the MLL5-mediated H3.3 loss in adult glioma has been attributed to a trademark of self-renewing cells [42]. Furthermore, a decline of H3.3 is also observed in HCC tissue samples [43]. These results indicate that the role of H3.3 in the tumor is context-dependent and warrants further investigation. Nevertheless, few studies have addressed the roles of H3.3 in SCLC. In this study, H3.3 has been shown to overexpress at both the mRNA and protein levels in SCLC cell lines and clinical specimens. We then reported that ZIO-101 was effective in inhibiting SCLC cells at low micromolar concentrations in a dose- and time-dependent manner and ZIO-101 activity depends on the H3.3 expression level. More importantly, when cells were treated with different concentrations of ZIO-101, the expression levels of H3.3 and PARP1 were significantly upregulated, which provides a rationale for combined therapy with ZIO-101 and PARPi. Indeed, our results demonstrated that targeting H3.3 inhibition potentiated the sensitivity of SCLC to the PARPi in vitro and in vivo. PARPi frequently elicits an excellent initial response. However, most patients develop resistance to these agents and relapse [44,45,46]. Acquired resistance represents a major cause of treatment failure and relapse. PARPi-induced accumulation of H3.3 might be one of the acquired resistance mechanisms that limit responsiveness to PARPi treatment in SCLC and possibly in other cancer types. Various combination strategies have been proposed to overcome resistance and improve clinical efficacy. The potential activity of olaparib, talazoparib, and rucaparib, combined with cytotoxic agents, has been demonstrated in preclinical models in several types of cancer [15]. Targeting p97 ATPase using CB5083 enhances the cytotoxicity of talazoparib in homologous recombination-defective colorectal cancer cells and patient-derived breast cancer organoids [47]. Selinexor, an XPO1 inhibitor, synergizes with PARPi to improve the efficacy and tolerability of PARPi in SCLC [16]. Herein, we propose that combining ZIO-101 and PARPi is another therapeutic strategy to overcome resistance and enhance the anti-tumor activity of PARPi in SCLC and beyond.
Although the expression of H3.3 is present throughout the cell cycle in a DNA synthesis-independent manner [41, 48], we found that H3.3 expression was dynamically changed during cell cycle progression, with the highest expression at the G2 and M phases. Intriguingly, our investigation confirmed that ZIO-101 tends to induce G2/M cell cycle arrest and apoptosis. Concomitantly, ZIO-101 treatment could induce a dose-and time-dependent accumulation of H3.3 in SCLC cells. The co-occurrence of G2/M arrest and accumulation of H3.3 indicates that H3.3 might function at the G2/M transition or G2/M DNA damage checkpoint. Indeed, our data suggest that perturbation of H3.3 causes a marked increase of Rad51 and substantial accumulation of DNA damage, which lays a reliable theoretical basis for the combined use of ZIO-101 and BMN673 through modulation of DNA repair capacity. Although the link between H3.3 and the cell cycle is confirmable, how the progression of the cell cycle regulates H3.3 remains to be fully elucidated.
Integrative analysis of gene expression data from CCLE and drug sensitivity datasets from GDSC demonstrated that PARP1 expression is positively associated with PARPi sensitivity, raising the possibility that manipulating PARP1 expression could enhance PARPi activity. However, few studies have addressed the upstream regulatory factors of PARP1 expression. Our investigation showed that ZIO-101 could trigger a dose-dependent increase of PARP1, suggesting that ZIO-101 could potentiate PARPi activity by increasing PARP1 expression. Moreover, ZIO-101 did induce H3.3 accumulation in a dose- and time-dependent manner. Therefore, an increase in PARP1 and H3.3 could both contribute to the synergistic effect of the combined use of ZIO-101 and BMN673. On the other hand, PARPi treatment also led to a marked increase of H3.3, which might sensitize ZIO-101 activity in SCLC cells. Notably, the observation of co-accumulation of H3.3 and PARP1 implies that there might be a functional interplay between H3.3 and PARP1. Therefore, it is worth further illustrating the functional connection between H3.3 and PARP1. Our bioinformatics analysis also suggested that targeting H3.3 (H3F3A) could also be used as a chemosensitizing agent for other clinically applied drugs, including topotecan, cisplatin, and SAHA. Future investigation of the potential sensitization by targeting H3.3 with ZIO-101 might maximize the clinical efficacy of PARPi and improve the therapeutic effects of these drugs in patients with SCLC and beyond.
Although the FDA approves chemotherapy plus immunotherapy as a first-line treatment for SCLC patients, the treatment outcome and the patient’s prognosis are not ideal [9]. Therefore, it still is of great significance to identify novel therapeutic targets and to develop new drug combination strategies to improve clinical outcomes for SCLC patients. We nominate H3.3 as a potential target in SCLC and show that targeting epigenetic factor H3.3 by ZIO-101 potentiates the sensitivity of SCLC to the PARPi in vitro and in vivo. Further clinical trials are needed to assess the therapeutic efficacy of combination therapies that inhibit H3.3 and PARP1, as this strategy shows excellent potential to extend PARPi application in SCLC and beyond.
Overall, our data indicate that the H3.3 is prominently overexpressed in SCLC, and the H3.3 mRNA level is positively associated with the sensitivity of several chemotherapeutic drugs in SCLC cells, including PARPi. Moreover, we show that ZIO-101 displays dose- and time-dependent inhibitory effects on SCLC cells in an H3.3-dependent manner, and we present evidence to demonstrate that ZIO-101 treatment results in substantial accumulation of H3.3 and PARP1 besides induction of a G2/M cell cycle arrest and apoptosis in SCLC cells, whose expressions are proved to be negatively correlated with the IC50 values of PARPi. Last but not least, we show that co-targeting H3.3 and PARP1 by ZIO-101 and BMN673/olaparib exhibits synergistic growth inhibition against SCLC in vitro and in vivo (Fig. 7g). Collectively, our results, for the first time, illustrate a previously uncharacterized function of ZIO-101 that could significantly improve the anti-cancer effects of PARPi, holding the promise of developing ZIO-101 as possibly potential chemosensitizers in cancer treatment in the future.
Change history
13 November 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41401-022-01019-w
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398:535–54.
Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Prim. 2021;7:3.
Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 2017;17:725–37.
Hiddinga BI, Raskin J, Janssens A, Pauwels P, Van Meerbeeck JP. Recent developments in the treatment of small cell lung cancer. Eur Respir Rev. 2021;30:210079.
Remon J, Aldea M, Besse B, Planchard D, Reck M, Giaccone G, et al. Small cell lung cancer: a slightly less orphan disease after immunotherapy. Ann Oncol. 2021;32:698–709.
Yin YP, Ma LY, Cao GZ, Hua JH, Lv XT, Lin WC. FK228 potentiates topotecan activity against small cell lung cancer cells via induction of SLFN11. Acta Pharmacol Sin. 2022;43:2119–27.
Yin YP, Shi WH, Deng K, Liu XL, Li H, Lv XT, et al. Combinations of proteasome inhibitors with obatoclax are effective for small cell lung cancer. Acta Pharmacol Sin. 2021;42:1298–310.
Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–9.
Chan CY, Tan KV, Cornelissen B. PARP inhibitors in cancer diagnosis and therapy. Clin Cancer Res. 2021;27:1585–94.
Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol. 2021;18:773–91.
Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. 2020;383:2345–57.
Passiglia F, Reale ML, Cetoretta V, Parlagreco E, Jacobs F, Listi A, et al. Repositioning PARP inhibitors in the treatment of thoracic malignancies. Cancer Treat Rev. 2021;99:102256.
Bian X, Wang X, Zhang Q, Ma L, Cao G, Xu A, et al. The MYC paralog-PARP1 axis as a potential therapeutic target in MYC paralog-activated small cell lung cancer. Front Oncol. 2020;10:565820.
Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2:798–811.
Wang J, Sun T, Meng Z, Wang L, Li M, Chen J, et al. XPO1 inhibition synergizes with PARP1 inhibition in small cell lung cancer by targeting nuclear transport of FOXO3a. Cancer Lett. 2021;503:197–212.
Ai X, Pan Y, Shi J, Yang N, Liu C, Zhou J, et al. Efficacy and safety of niraparib as maintenance treatment in patients with extensive-stage SCLC after first-line chemotherapy: a randomized, double-blind, phase 3 study. J Thorac Oncol. 2021;16:1403–14.
Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, et al. Randomized, double-blind, phase II study of temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. J Clin Oncol. 2018;36:2386–94.
Byers LA, Bentsion D, Gans S, Penkov K, Son C, Sibille A, et al. Veliparib in combination with carboplatin and etoposide in patients with treatment-naive extensive-stage small cell lung cancer: a phase 2 randomized study. Clin Cancer Res. 2021;27:3884–95.
Martire S, Banaszynski LA. The roles of histone variants in fine-tuning chromatin organization and function. Nat Rev Mol Cell Biol. 2020;21:522–41.
Talbert PB, Henikoff S. Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol. 2017;18:115–26.
Frey A, Listovsky T, Guilbaud G, Sarkies P, Sale JE. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr Biol. 2014;24:2195–201.
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82.
Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24:567–74.
Filipescu D, Muller S, Almouzni G. Histone H3 variants and their chaperones during development and disease: contributing to epigenetic control. Annu Rev Cell Dev Biol. 2014;30:615–46.
Park SM, Choi EY, Bae M, Kim S, Park JB, Yoo H, et al. Histone variant H3F3A promotes lung cancer cell migration through intronic regulation. Nat Commun. 2016;7:12914.
Gomes AP, Ilter D, Low V, Rosenzweig A, Shen ZJ, Schild T, et al. Dynamic incorporation of histone H3 variants into chromatin is essential for acquisition of aggressive traits and metastatic colonization. Cancer Cell. 2019;36:402–17. e13
Costa M. Review of arsenic toxicity, speciation and polyadenylation of canonical histones. Toxicol Appl Pharmacol. 2019;375:1–4.
Matulis SM, Morales AA, Yehiayan L, Croutch C, Gutman D, Cai Y, et al. Darinaparsin induces a unique cellular response and is active in an arsenic trioxide-resistant myeloma cell line. Mol Cancer Ther. 2009;8:1197–206.
Zhou Y, Wang H, Tse E, Li H, Sun H. Cell cycle-dependent uptake and cytotoxicity of arsenic-based drugs in single leukemia cells. Anal Chem. 2018;90:10465–71.
Bansal N, Farley NJ, Wu L, Lewis J, Youssoufian H, Bertino JR. Darinaparsin inhibits prostate tumor-initiating cells and Du145 xenografts and is an inhibitor of hedgehog signaling. Mol Cancer Ther. 2015;14:23–30.
Tian J, Zhao H, Nolley R, Reese SW, Young SR, Li X, et al. Darinaparsin: solid tumor hypoxic cytotoxin and radiosensitizer. Clin Cancer Res. 2012;18:3366–76.
Xu X, Wang H, Li H, Hu X, Zhang Y, Guan X, et al. S-Dimethylarsino-glutathione (darinaparsin(R)) targets histone H3.3, leading to TRAIL-induced apoptosis in leukemia cells. Chem Commun (Camb). 2019;55:13120–3.
Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, et al. Small cell lung cancer screen of oncology drugs, investigational agents, and gene and microRNA expression. J Natl Cancer Inst. 2016;108:djw122.
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6.
Reddy D, Bhattacharya S, Shah S, Rashid M, Gupta S. DNA methylation mediated downregulation of histone H3 variant H3.3 affects cell proliferation contributing to the development of HCC. Biochim Biophys Acta Mol Basis Dis. 2022;1868:166284.
Li MX, Wang D, He JH, Chen LX, Li H. Bcl-X-L: a multifunctional anti-apoptotic protein. Pharmacol Res. 2020;151:104547.
Wang ZQ, Fan M, Candas D, Zhang TQ, Qin LL, Eldridge A, et al. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell. 2014;29:217–32.
Chan JM, Quintanal-Villalonga A, Gao VR, Xie Y, Allaj V, Chaudhary O, et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell. 2021;39:1479–96. e18.
Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene. 2012;31:4257–65.
Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421–34.
Gallo M, Coutinho FJ, Vanner RJ, Gayden T, Mack SC, Murison A, et al. MLL5 orchestrates a cancer self-renewal state by repressing the histone variant H3.3 and globally reorganizing chromatin. Cancer Cell. 2015;28:715–29.
Tvardovskiy A, Schwammle V, Kempf SJ, Rogowska-Wrzesinska A, Jensen ON. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 2017;45:9272–89.
Barayan R, Ran X, Lok BH. PARP inhibitors for small cell lung cancer and their potential for integration into current treatment approaches. J Thorac Dis. 2020;12:6240–52.
Knelson EH, Patel SA, Sands JM. PARP inhibitors in small-cell lung cancer: rational combinations to improve responses. Cancers (Basel). 2021;13:727.
Huang D, Kraus WL. The expanding universe of PARP1-mediated molecular and therapeutic mechanisms. Mol Cell. 2022:82:2315–34.
Krastev DB, Li S, Sun Y, Wicks AJ, Hoslett G, Weekes D, et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nat Cell Biol. 2022;24:62–73.
Filipescu D, Szenker E, Almouzni G. Developmental roles of histone H3 variants and their chaperones. Trends Genet. 2013;29:630–40.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (No. 81972191 and No. 81672647, China), and a portion of this work was supported by the High Magnetic Field Laboratory of Anhui Province.
Author information
Authors and Affiliations
Contributions
GZC and WCL conceived and designed the project. GZC, LYM, ZHZ, and JHZ performed most experiments and interpreted the data. YL, JHH, and SBZ performed part of in vivo experiments. GZC, XLW, and JO analyzed the data and prepared the figures and manuscript draft. WCL revised the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
The original online version of this article was revised: After check of our published article entitled “Darinaparsin (ZIO-101) enhances the sensitivity of small-cell lung cancer to PARP inhibitors” in Acta Pharmacologica Sinica, we found that there were misspellings in Fig. 2, Fig. 6, Supplementary Fig. S2 and Supplementary Fig. S5. FTIC-A in these figures need to be modified to FITC-A. In addition, Fig.S1a and Fig.S5a should be changed to Fig.S1 and Fig.S5 in the text and the “a” should be deleted in the supplementary figures Fig.S1 and Fig.S5 and corresponding figure legends.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cao, Gz., Ma, Ly., Zhang, Zh. et al. Darinaparsin (ZIO-101) enhances the sensitivity of small-cell lung cancer to PARP inhibitors. Acta Pharmacol Sin 44, 841–852 (2023). https://doi.org/10.1038/s41401-022-00994-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00994-4
Keywords
This article is cited by
-
HMGN1 loss sensitizes lung cancer cells to chemotherapy
Scientific Reports (2024)
-
Chemotherapy-induced metastasis: molecular mechanisms and clinical therapies
Acta Pharmacologica Sinica (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}